

Abstract
A novel 1,4-N→O migration of a disubstituted phenyl ring was observed during N-methylation of a bicalutamide derivative, (2S)-2-(tert-butyldimethylsilanyloxy)-N-(4-cyano-3-trifluoromethylphenyl)-3-(4-fluorophenoxy)-2-methylpropionamide, in the presence of CsF-Celite/acetonitrile and desilylation of (2S)-2-(tert-butyldimethylsilanyloxy)-N-(4-cyano-3-trifluoromethylphenyl)-3-(4-fluorophenoxy)-2,N-dimethylpropionamide in tetra-n-butylammonium fluoride/THF. Both NMR and X-ray analysis confirmed the structure of the 1,4-N→O disubstituted phenyl ring migrated product.
Keywords: 1,4-N→O migration; CsF mediated rearrangement; Tetra-n-butylammonium fluoride mediated rearrangement
Bicalutamide (Casodex) is a well-known drug for the treatment of prostate cancer. Our group has been synthesizing several bicalutamide derivatives that showed androgen receptor (AR) agonist activity.1–4 In continuation of our work to prepare such compounds, we planned to synthesize the prodrug by following the reaction steps shown in Scheme 1 (reaction of the substituted aniline 1 with acid 25 to give amide 3,6 followed by the formation of epoxide 47 that subsequently reacts with p-fluorophenol to give ether 57), and then by replacing the amide hydrogen of 57 by diethylphosphonate. The diethylphosphate of 5 was proposed to be a prodrug of 5. In an attempt at preparing the prodrug, we protected the hydroxyl group of compound 57 using tert-butyldimethylsilyl trifluoromethane sulfonate with 2,6-lutidine in DCM to get (2S)-2-(tert-butyldimethyl-silanyloxy)-N-(4-cyano-3-trifluoromethylphenyl)-3-(4-fluorophenoxy)-2-methylpropionamide 68 (Scheme 2), and then tried to substitute the amide hydrogen of compound 68 by diethylphosphonate using diethylchlorophosphonate in the presence of various bases like NaOH, NaH, and NaNH2 under stirring conditions at room temperature. In all these cases, we did not get the N-substituted derivative of 6.8 Then, we changed our interest towards preparing N-methyl and N-benzyl derivatives of compound 68 via N-alkylation using methyl iodide or benzyl bromide under stirring conditions at room temperature in a K2CO3/acetone or KOH/THF medium. Surprisingly, no N-alkylated compounds were obtained. Recently, Bayer and co-workers9 described a CsF-Celite/alkyl halide/acetonitrile combination method to alkylate carboxamides and several nitrogen heterocycles. This report encouraged us to use CsF-Celite as a solid base that enforces N-methylation of 68 with methyl iodide in anhydrous acetonitrile.
Scheme 1.
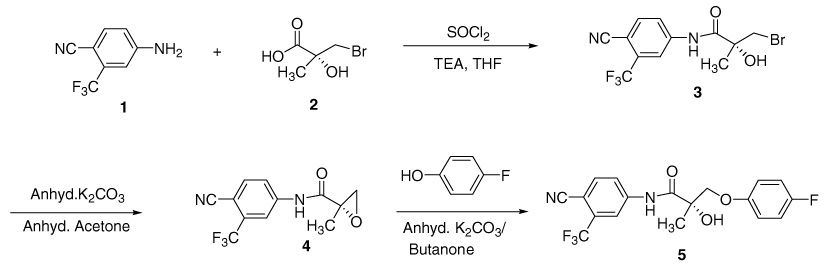
Schematic route for the synthesis of compound 5.
Scheme 2.

Schematic route for the synthesis of compounds 6–10.
As a result, we observed methylation at the amide coupled with 1,4-N→O migration of the disubstituted phenyl ring of compound 68 thus yielding (2S)-2-(4-cyano-3-trifluoromethylphenoxy)-3-(4-fluorophenoxy)-2,N-dimethylpropionamide 1010 as the major product (37%). The actual target compound 710 was obtained only in 16% yield along with the desilylated alcohol 810 (10%), the methoxyether 910 (11%), and compound 57 (22%) (Scheme 2). All these compounds were separated by column chromatography (1:9 to 6:4 ethyl acetate/hexane). In addition, when compound 710 was desilylated using tetra-n-butylammonium fluoride in dry THF, the same 1,4-N→O shift of the disubstituted phenyl ring was observed exclusively thus yielding compound 1011 (55%) rather than the expected compound 810 (Scheme 3).
Scheme 3.

Schematic route for the synthesis of compound 10 from desilylation of compound 7. Compound 10 is the only product from this reaction.
Initially, we had difficulties assigning the correct structure to compound 10.10,11 Desilylation and single methylation were evident from the analysis of both mass spectrometry and NMR data. In the NMR, the NH proton appeared as a quartet indicating a scalar coupling constant of 4.5 Hz with the added methyl group. Since we had never observed bond breaking for the amide group in the synthesis of many analogs of 7,10 we initially attributed the NH quartet to a scalar coupling across the hydrogen bond (3hJHH), formed by NH…O–CH3 in a structure similar to that of compound 9.10 However, density functional theory (DFT) calculations,12–15 utilizing the coupled perturbed DFT (CP-DFT) approach for NMR spin–spin coupling constants published by Cremer and co-workers,15 indicated at most 0.1 Hz for the 3hJHH spin–spin coupling constant, clearly ruling out this type of structure.12–15 The MS did not lead to characteristic fragmentation peaks to make an unambiguous structure assignment for compound 10.10,11 In the end, we were able to grow crystals successfully in benzene suitable for X-ray structural analysis. The result of the X-ray analysis revealed the actual structure for compound 10,10,11 clearly indicating a 1,4-N→O migration of the disubstituted phenyl ring of the amide group (Fig. 1).16,17 The structure of 1010,11 explained the appearance of the NMR spectra. Remaining compounds were confirmed by spectroscopy methods without difficulty.
Figure 1.

X-ray crystal structure of compound 10.
In conclusion, we have found that the CsF-Celite and tetra-n-butylammonium fluoride mediated reactions of compounds 68 and 7,10 respectively, initiates a novel 1,4-N→O shift of disubstituted phenyl. The novel structure of compound 1010,11 was confirmed by detailed spectroscopic studies, quantum mechanics calculations, and X-ray structural analysis.
Acknowledgements
Financial support of this work by the National Institutes of Health grant R01 DK59800, and The GTx, Inc. are gratefully acknowledged. Support of this research by the National Science Foundation (DMR-9708246) and, in part, by Cancer Center Support CORE Grant, P30 CA 21765 is gratefully acknowledged as is that of the American Lebanese Syrian Associated Charities (ALSAC) by C.R.R.
References and notes
- 1.Dalton JT, Mukherjee A, Zhu Z, Kirkovsky L, Miller DD. Biochem. Biophys. Res. Commun. 1998;244:1–4. doi: 10.1006/bbrc.1998.8209. [DOI] [PubMed] [Google Scholar]
- 2.Marhefka CA, Gao W, Chung K, Kim J, He Y, Yin D, Bohl C, Dalton JT, Miller DD. J. Med. Chem. 2004;47:993–998. doi: 10.1021/jm030336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He Y, Yin D, Perera M, Kirkovsky L, Stourman N, Li W, Dalton JT, Miller DD. Eur. J. Med. Chem. 2002;37:619–634. doi: 10.1016/s0223-5234(02)01335-1. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Hwang DJ, Bohl CE, Miller DD, Dalton JT. J. Pharmacol. Exp. Ther. 2005;312:546–553. doi: 10.1124/jpet.104.075424. [DOI] [PubMed] [Google Scholar]
- 5.Kirkovsky L, Mukherjee A, Yin D, Dalton JT, Miller DD. J. Med. Chem. 2000;43:581–590. doi: 10.1021/jm990027x. [DOI] [PubMed] [Google Scholar]
- 6.(2R)-3-Bromo-N-(4-cyano-3-trifluoromethylphenyl)-2-hydroxy-2-methylpropionamide. Thionyl chloride (4.60 g, 38.71 mmol) was added drop wise to a solution of (2R)-3-bromo-2-hydroxy-2-methylpropanoic acid 2 (7.1 g, 38.71 mmol) in anhydrous THF (25 mL) under nitrogen atmosphere at 0 to −5 °C, and stirred at the same temperature for 2 h to give the corresponding acid chloride, which was treated in situ with a solution of compound 1 (6.0 g, 32.26 mmol) in anhydrous THF (20 mL) followed by triethyl amine (8.16 g, 80.65 mmol) at 0 to −5 °C. The reaction mixture was allowed to attain room temperature, and then refluxed overnight. The reaction mixture was concentrated under reduced pressure, and extracted with ethyl acetate. The organic layer was washed with 5% HCl, saturated NaHCO3 solution, followed by water. The solvent was dried over anhydrous Na2SO4, evaporated under reduced pressure, and recrystallized with ethyl acetate and hexane to yield 3 as a yellow powder (8.61 g, 76%). Mp 133–134 °C. ¹H NMR (300 MHz, DMSO-d6) δ 10.52 (s, 1H, −NH), 8.54 (s, 1H, ArH), 8.30 (dd, J = 1.8, 1.8 Hz, 1H, ArH), 8.10 (d, J = 8.7 Hz, 1H, ArH), 6.40 (s, 1H, OH), 3.82 (d, J = 10.5 Hz, 1H, −CH2), 3.57 (d, J = 10.2 Hz, 1H, −CH2), 1.48 (s, 3H, −CH3). MS (ESI−) m/z 350 (M−H)−.
- 7.(2S)-N-(4-Cyano-3-trifluoromethylphenyl)-3-(4-fluorophenoxy)-2-hydroxy-2-methylpropionamide. Anhydrous K2CO3 (3.94 g, 28.4 mmol) was added to a solution of compound 3 (5.00 g, 14.2 mmol) in acetone (25 mL), and the reaction mixture was refluxed for 2 h. The mixture was concentrated to dryness, the obtained residue was dissolved in water, and extracted with ethylacetate. The organic layer was dried over anhydrous Na2SO4, and removed under reduced pressure to give epoxide 4 as oil which was used in the next step without further purification. A solution of the above epoxide in 2-butanone (25 mL) was treated with anhydrous K2CO3 (3.94 g, 28.4 mmol) and 4-fluorophenol (1.600 g, 14.2 mmol), the mixture was stirred at reflux overnight, and evaporated to dryness. The residue was dissolved in water and extracted with ethylacetate, washed with brine solution, dried over anhydrous Na2SO4, and the solvents were removed under reduced pressure. The crude residue was purified by flash column chromatography (1:9 ethyl acetate/hexane) to give 3.0 g (55%) of 5 as a yellow oil, which was recrystallized from DCM to furnish white colored crystals. Mp 128–129 °C. ¹H NMR (300 MHz, DMSO-d6) δ 10.57 (br s, 1H, −NH), 8.56 (s, 1H, ArH), 8.31 (d, J = 8.7 Hz, 1H, ArH), 8.10 (d, J = 8.7 Hz, 1H, ArH), 7.11–7.05 (m, 2H, ArH), 6.95–6.91 (m, 2H, ArH), 6.26 (br s, 1H, OH), 4.20 (d, J = 9.6 Hz, 1H, −CH2), 3.96 (d, J = 9.6 Hz, 1H, −CH2), 1.43 (s, 3H, −CH3). MS (ESI−) m/z 381 (M−H)−.
- 8.(2S)-2-(tert-Butyldimethylsilanyloxy)-N-(4-cyano-3-trifluoromethylphenyl)-3-(4-fluorophenoxy)-2-methylpropionamide. To a solution of compound 5 (4.00 g, 10.46 mmol) in dichloromethane (75 mL) was added tert-butyldimethylsilyl trifluoromethane sulfonate (3.32 g, 12.56 mmol), and 2,6-lutidine (2.24 g, 20.93 mmol) at 0 °C. The mixture was stirred at ambient temperature overnight, and diluted with water. The water layer was extracted with ethyl acetate, the combined organic layers were dried with anhydrous Na2SO4, and the solvents were evaporated under reduced pressure. The resulting crude residue was purified by flash column chromatography (5:95 ethyl acetate/hexane) to afford 3.85 g (99%) of 6 as a colorless oil. (Recovered starting material yield 1.00 g, 25%.) ¹H NMR (300 MHz, DMSO-d6) δ 9.76 (s, 1H, −NH), 8.32 (s, 1H, ArH), 8.16 (d, J = 8.4 Hz, 1H, ArH), 8.08 (d, J = 8.4 Hz, 1H, ArH), 7.14–7.08 (m, 2H, ArH), 6.97–6.92 (m, 2H, ArH), 4.26 (d, J = 10.2 Hz, 1H, −CH2), 4.11 (d, J = 10.2 Hz, 1H, −CH2), 1.55 (s, 3H, −CH3), 0.91 (s, 9H, 3 × −CH3), 0.14 (d, J = 7.8 Hz, 6H, 2 ×−CH3). MS (ESI+) m/z 519 (M+Na)+.
- 9.Hayat S, Rahman A-U, Iqbal CM, Khan KM, Schumann W, Bayer E. Tetrahedron. 2001;57:9951–9957. [Google Scholar]
- 10.To a stirred solution of compound 6 (0.946 g, 1.94 mmol) in anhydrous acetonitrile (20 mL) was added CsF-Celite (0.442 g, 2.91 mmol) and methyl iodide (0.551 g, 3.88 mmol). The mixture was heated for 5 h, cooled to room temperature, and concentrated under reduced pressure. The residue was dissolved in ethyl acetate, filtered, and the precipitate was washed with ethyl acetate. The combined filtrates were evaporated under reduced pressure, and the crude residue was purified by flash column chromatography (1:9 to 6:4 ethyl acetate/hexane) afforded compounds 5 (0.149 g, 22%), 7 (0.144 g, 16%), 8 (0.070 g, 10%), 9 (0.077 g, 11%), and 10 (0.259 g, 37%). (Recovered starting material yield 0.087 g, 9%.) Data for 7: ¹H NMR (300 MHz, DMSO-d6) δ 8.24 (d, J = 8.1 Hz, 1H, ArH), 7.91 (s, 1H, ArH), 7.82 (d, J = 8.1 Hz, 1H, ArH), 7.18–7.12 (m, 2H, ArH), 7.00–6.96 (m, 2H, ArH), 4.25 (d, J = 9.9 Hz, 1H, −CH2), 4.01 (d, J = 9.9 Hz, 1H, −CH2), 3.50 (s, 3H, −NCH3), 1.60 (s, 3H, −CH3), 0.79 (s, 9H, 3 × −CH3), 0.02 (d, J = 4.8 Hz, 6H, 2 × −CH3); MS (ESI+) m/z 533 (M+Na)+. 8: Mp 85–86 C ¹H NMR (DMSO-d6) δ 7.91 (d, J = 8.4 Hz, 1H, ArH), 7.28 (d, J = 1.8 Hz, 1H, ArH), 7.18–7.09 (m, 3H, ArH), 6.99–6.93 (m, 2H, ArH), 5.38 (s, 1H, OH), 4.10 (d, J = 9.6 Hz, 1H, −CH2), 3.81 (d, J = 9.9 Hz, 1H, −CH2), 3.69 (s, 3H, −NCH3), 1.42 (s, 3H, −CH3); MS (ESI+) m/z 419 (M+Na)+. 9: Mp 96–97 °C ¹H NMR (300 MHz, CDCl3-d6) δ 9.09 (s, 1H, −NH), 8.09 (d, J = 2.1 Hz, 1H, ArH), 7.98 (dd, J = 2.1, 1.8 Hz, 1H, ArH), 7.80 (d, J = 8.4 Hz, 1H, ArH), 7.00–6.92 (m, 2H, ArH), 6.87–6.77 (m, 2H, ArH), 4.27 (d, J = 9.6 Hz, 1H, −CH2), 4.11 (d, J = 10.2 Hz, 1H, −CH2), 3.49 (s, 3H, −OCH3), 1.53 (s, 3H, −CH3); MS (ES+) m/z 419 (M+Na)+. 10: Mp 88–89 °C ¹H NMR (300 MHz, DMSO-d6) δ 8.37 (q, J = 4.5 Hz, 1H, −NH), 8.09 (d, J = 8.4 Hz, 1H, ArH), 7.52 (d, J = 2.4 Hz, 1H, ArH), 7.39 (dd, J = 2.4, 2.4 Hz, 1H, ArH), 7.15–7.06 (m, 2H, ArH), 7.00–6.91 (m, 2H, ArH), 4.31 (q, J = 10.8 Hz, 2H, −CH2), 2.66 (d, J = 4.5 Hz, 3H, −NHCH3), 1.60 (s, 3H, −CH3); MS (ESI+) m/z 419 (M+Na)+.
- 11.To a solution of 7 (0.063 g, 0.123 mmol) in anhydrous THF (10 mL) was added TBAF 1M solution in THF (0.064 g, 0.247 mmol) at 0 °C. The mixture was stirred at room temperature for 1 h, diluted with ethyl acetate, and extracted with water. The organic layer was dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The crude residue was dissolved in DCM, and evaporated to give compound 10 (0.027 g, 55%).
- 12.Kohn W, Sham L. Phys. Rev. A. 1965;140:1133–1138. [Google Scholar]
- 13.Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 14.Hariharan PC, Pople JA. Theoret. Chim. Acta. 1973;28:213–222. [Google Scholar]
- 15.Sychrovsky V, Gräfenstein J, Cremer D. J. Chem. Phys. 2000;113:3530–3547. [Google Scholar]
- 16.Farrugia LJ. J. Appl. Cryst. 1997;30:565. [Google Scholar]
- 17.X-ray crystal structure data were collected to a resolution of approximately 0.8 Å on a Bruker Proteum CCD detector mounted on a Nonius FR591 rotating-anode generator using a copper target (λ = 1.54178 Å) (Bruker AXS, Inc., Madison, WI). Raw data were integrated using SAINT and corrected for absorption using SADABS. Direct-methods structure solution and refinement were performed using the SHELX programs. Anisotropic ADPs were refined for all heavy atoms, resulting in the final refinement statistics of R1 = 0.025 for 3015 reflections with Fo > 4σ(Fo), wR2 = 0.068 and S (goodness-of-fit) = 1.046 for 3071 reflections and 253 refined parameters. Supplementary data in the form of CIFs have been deposited with the Cambridge Crystallographic Data Center (CCDC 286917). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [Fax: +44(0)-1223-336033 or e-mail: deposit@ccdc.cam.ac.uk].