

Abstract
Mutations in the human tau gene leading to aberrant splicing have been identified in FTDP-17, an autosomal dominant hereditary neurodegenerative disorder. Molecular mechanisms by which such mutations cause tau aberrant splicing were not understood. We characterized two mutations in exon 10 of the tau gene, N279K and Del280K. Our results revealed an exonic splicing enhancer element located in exon 10. The activity of this AG-rich splicing enhancer was altered by N279K and Del280K mutations. This exonic enhancer element interacts with human Tra2β protein. The interaction between Tra2β and the exonic splicing enhancer correlates with the activity of this enhancer element in stimulating splicing. Biochemical studies including in vitro splicing and RNA interference experiments in transfected cells support a role for Tra2β protein in regulating alternative splicing of human tau gene. Our results implicate the human tau gene as a target gene for the alternative splicing regulator Tra2β, suggesting that Tra2β may play a role in aberrant tau exon 10 alternative splicing and in the pathogenesis of tauopathies.
Microtubule-associated protein tau plays an important role in microtubule assembly and stabilization (1-9). Recent studies have shown that, together with MAP1B, tau acts as a regulator of microtubule organization during axonal growth and neuronal migration (reviewed in Ref. 10). Tau protein is encoded by a single gene located on chromosome 17 (11). In the adult human brain, six isoforms of tau protein are produced as a result of alternative splicing of exons 2, 3, and 10 (11-13). Alternative pre-mRNA splicing of exon 10 in the tau gene, either exclusion or inclusion of exon 10, results in proteins with either three or four microtubule-binding repeats (Tau3R or Tau4R). In the normal human adult brain, the ratio of Tau4R to Tau3R is approximately 1, and this delicate balance between Tau4R and Tau3R isoforms appears to be critical for neuronal function in maintaining learning and memory (14-18).
Filamentous tau protein-containing inclusions have been recognized as a hallmark of neuropathology of several neurodegenerative diseases, such as Alzheimer's disease, Down's syndrome, several variants of Prion diseases, progressive supranuclear palsy, amyotrophic lateral sclerosis, Pick's disease, corticobasal degeneration, and sporadic frontotemporal dementias (reviewed in Ref. 19). Recent genetic studies have revealed mutations in the tau gene associated with frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), an autosomal dominant hereditary neurodegenerative disorder characterized by behavioral and personality changes as well as muscular rigidity similar to that found in Parkinson's disease. A major neuropathological characteristic of FTDP-17 is filamentous inclusions containing hyperphosphorylated tau protein in the absence of β-amyloid plaques. Since 1998, several groups have reported the association between mutations in the tau gene and FTDP-17 (15, 16, 20-39). So far, tau mutations associated with various forms of frontotemporal dementia can be classified as two types (14): missense mutations impairing Tau protein function and splicing mutations altering Tau4R/3R ratio. Most missense mutations occur inside or near the microtubule-binding domain. These missense mutations may affect tau function by reducing tau binding to microtubules and promoting the formation of paired helical filament aggregation (16, 25-28, 35, 36, 39-41). On the other hand, splicing mutations, including all intronic mutations and several exonic mutations, are associated with changes in Tau4R/3R ratio. These observations suggest that a proper ratio of Tau4R/3R is essential for normal function of Tau in the human brain. It remains unclear how alterations in Tau4R/3R ratio lead to neuronal degeneration and, in some cases, also glial dysfunction and death. However, the discovery of these splicing mutations in the tau gene indicates the critical importance of controlling the balance of different Tau isoforms by alternative splicing for normal brain function.
We are interested in understanding molecular mechanisms underlying tau alternative splicing regulation. Our previous work established a tau minigene system (17). Many of the tau intronic mutations described so far are near the splice donor site (5′ splice site) of the intron following exon 10. Our biochemical studies support the model that some intronic mutations near the 5′ splice site following exon 10 destabilize a stem-loop structure and lead to enhanced U1 snRNP1 interaction with the 5′ splice site of exon 10 and thus an increase in splicing between exon 10 and exon 11 and an increase in the ratio of Tau4R to Tau3R (15, 17, 27, 42, 43).
In addition to intronic mutations, several exonic mutations (shown in Fig. 1), including N279K, Del280K, L284L, N296N, N296H, and S305N, have also been found to alter exon 10 splicing (20, 26, 27, 30, 37). Although it was proposed based on sequence analyses and RT-PCR studies that these mutations may change the activity of certain exonic regulatory elements in exon 10, the mechanism by which these mutations affect exon 10 splicing was not clear. In this study, we used in vitro biochemical assays to dissect the cis-elements and trans-splicing factors involved in the regulation of tau Exon 10 inclusion. Our results have revealed an exonic splicing enhancer element located in exon 10. The activity of this AG-rich splicing enhancer is affected by N279K and Del280K mutations. Proteins interacting with this AG-rich sequence were analyzed using UV cross-linking and immunoprecipitation assays. A 40-kDa protein containing a RNA-binding domain and a domain rich in serine and arginine (the SR domain), Tra2β, has been identified among the proteins interacting with this exonic enhancer element. The interaction of Tra2β with the exonic splicing enhancer correlates with the activity of this enhancer element in stimulating tau exon 10 splicing. Finally, down-regulation of Tra2β expression by RNA interference in transfected cells led to a reduction in tau exon 10 inclusion. These observations suggest that human Tra2β may act as an important regulator for facilitating exon 10 inclusion in tau splicing.
Fig.1.
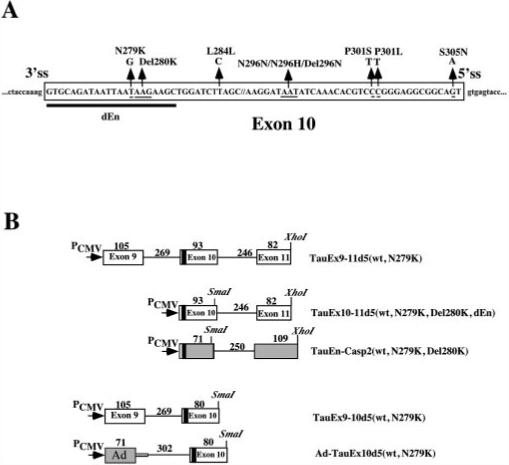
A, positions of reported mutations in exon 10 of the human tau gene. 5′ss, 5′ splice site. 3′ss, 3′ splice site. The sequence deleted from TauEx10−11d5(WT) to generate TauEx10−11d5(dEn) is indicated by the thick black line. B, schematics of a series of tau minigene constructs. The genomic DNA fragments containing exons 9, 10 (WT, N279K, Del280K, and dEn), and 11 as well as intronic sequences flanking exon 10 were inserted in mammalian expression vector pcDNA3 under the control of the cytomegalovirus promoter (PCMV). The AG-rich region in exon 10 is indicated by the black box. The caspase-2 (casp2) exon 9 and 10 splicing unit and first exon region (L1) of the Ad major late transcription unit are shown in gray boxes. The sizes of the corresponding exons and introns are indicated above the respective regions (in base pairs).
EXPERIMENTAL PROCEDURES
Plasmid, Oligonucleotides, and Antibodies
The tau genomic DNA fragments containing exons 9−11 as well as intronic sequences flanking exon 10 were amplified by PCR from the normal adult human or FTDP-17 patient brain genomic DNA to make wild-type or mutant tau constructs. Tau minigene constructs were made by inserting the genomic fragments into a mammalian expression vector pcDNA3 (Invitrogen) between HindIII and XhoI sites under the control of the cytomegalovirus promoter (17). Tau minigene plasmids with N279K and Del280K mutation were made by site-directed mutagenesis with specific oligonucleotides. TauEx10−11d5(dEn) was made by deletion of sequence GTG CAG ATA ATT AAT AAG AAG C as indicated by the thick line in Fig. 1A from TauEx10−11d5, and TauEn-IchEx9−10(WT, N279K, Del280K) were made by inserting this sequence upstream of the caspase-2 exon 9−10 splicing unit (44). Ad-TauEx10d5(WT, N279K) were made by replacing the upstream exon 9 and associated intronic sequences with the first exon region (L1) of the adenovirus (Ad) major late transcription unit. DNA sequence analysis of tau genomic fragments and different expression plasmids was carried out on an ABI 373A automatic sequencer using a PRISM Ready reaction DyeDeoxy Terminator cycle sequencing kit (Applied Biosystems). Oligonucleotides were purchased from Dharmacon and IDT Integrated DNA Technologies, Inc. Anti-SR monoclonal antibody 1H4 was purchased from ATCC. A polyclonal antibody specific for human Tra2β was as described (67).
Transfections and RT-PCR
HeLaRB, HEK293, and N2a cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and were seeded at 2 × 105 cells/well in a six-well dish 24 h prior to transfection. Transfection was done using a standard calcium phosphate precipitation procedure with 1−3 μg of DNA as described (70). Transfection efficiency was routinely around 60%, as evaluated by cotransfection of a green fluorescent protein-expressing plasmid. Cells were harvested 48 h after transfection, and the RNA was extracted using Trizol reagent (Invitrogen). Splicing products derived from the expressed minigenes were detected using RT-PCR as previously described (70). cDNAs were also prepared from the frontal cortex region in human brain tissues obtained at autopsy. RT-PCR was also performed using primers specific for human Tra2β to detect the expression of this gene.
In Vitro Splicing Assays
Splicing substrates were synthesized using T7 RNA polymerase (Promega). Final concentrations of different reagents were as follows in a 10-μl reaction: 500 ng of linearized DNA template, 0.4 mM ATP and CTP, 0.1 mM GTP and UTP, 0.84 mM GpppG cap analogue (Amersham Biosciences), 10 mM dithiothreitol, 0.5 units/μl RNasin (Promega), 1× transcription buffer (Promega), 20 μCi of [α-32P]UTP, and 1 unit/μl RNA polymerase. Samples were then treated with 0.1 unit/μl DNase I (Promega) for 15 min and ethanol-precipitated. The full-length transcripts were gel-purified. Synthesis of cold competitor RNAs was in a scaled-up 100-μl reaction with the following modifications: 0.5 mM ATP, 0.5 mM CTP, 0.5 mM GTP, 0.5 mM UTP, 0.42 mM cap analogue, and a trace amount of [α-32P]UTP for quantification. HeLa cell nuclear extracts were prepared according to previously established protocols and contained 20 mg/ml total proteins (71). Splicing reactions were set up and processed as previously (72) except that some batches of nuclear extracts were supplemented with 1 unit of creatine kinase (Sigma). 2−3 fmol of RNA substrates were added, and the samples were incubated at 30 °C for 2 h unless otherwise mentioned. Splicing products were resolved on 8% polyacrylamide, 8 M urea gels. The identity of lariat molecules was determined by performing a debranching reaction in a S100 extract (73), followed by gel migration alongside molecular weight markers. The intensity of the products was quantified by the PhosphorImager. In some splicing assays, purified SR protein or U2AF65 or cell lysates containing overexpressed Tra2β or SRp40 was added.
UV Cross-linking and Immunoprecipitation Assays
Splicing reactions were set up as described above except using 20 fmol of RNA substrates. 10-μl aliquots were transferred onto a 96-well microtiter plate and irradiated with 1 J in UV Stratalinker 1800 (Stratagene). For site-specific cross-linking, a specific label was put inside the second AAG sequence following a protocol previously described (74, 75). Various amounts of competitive transcripts were added to demonstrate the specifically bound proteins. Samples were then treated for 30 min at 37 °C with equal volume of RNase A (5 mg/ml). Radiolabeled cross-linked proteins were boiled for 5 min in 1× SDS-PAGE loading buffer and separated on 12.5% SDS-PAGE. For immunoprecipitation, following the RNase treatment, samples were incubated with 1H4 anti-SR monoclonal antibody (ATCC). Protein A/G-agarose beads were then added with further incubation and gentle rocking. The RNA-labeled proteins retained on the beads after several washings were eluted and resolved on SDS-PAGE.
Site-specific Label Incorporation and Cross-linking
Human tau exon 10 DNA with N279K mutation was in vitro transcribed into RNA with trace-labeled [α-32P]UTP. 40 pmol of synthesized RNA was annealed with 60 pmol of chimeric oligonucleotide (3′-(UAUUAAU)2′-O-methyl-TCTT(CUUC)2′-O-methyl-5′) and cleaved with RNase H. The 3′-half RNA was treated with calf intestinal phosphatase and labeled with [γ-32P]ATP. The 5′-half RNA was ligated with 3′-half RNA using a DNA ligation kit (Roche Applied Science) by bridge oligonucleotide (5′-CTGGACGTTGCTAAGATCCAGCTTCTTCTTAATTATCTGCACCTTTGG-3′). 30,000 cpm of RNA was incubated with 20−40 μg of HeLa cell nuclear extracts for cross-linking. Various amounts (0, 4, and 40 pmol) of competitor oligonucleotide (5′-GTGCAGATAATTAAGAAGAAGCTGGATC-3′) corresponding to the enhancer element were added.
RNA-Protein Interaction Assays
40 fmol of biotin-conjugated RNAs corresponding to TauEx10−11dEn, TauEx10−11Del280K, TauEx10−11wt, and TauEx10−11N279K were incubated for 30 min on ice with various amounts of cell lysates prepared from HEK293 cells transiently transfected with a Tra2β-Myc expression construct. After affinity selection with streptavidin-agarose beads (Sigma), the beads were washed four times in cold phosphate-buffered saline with 75 mM NaCl and 0.1% Nonidet P-40. Proteins associated with the biotinylated RNAs were dissolved in sample buffer and analyzed by Western blotting using 9E10 anti-Myc monoclonal antibody (Babco).
U snRNP Blocking Assays
Partial inhibition of U snRNAs was achieved by incubation with 2′-O-methyl oligoribonucleotides as described (17). 2′-O-methyl oligonucleotides (U1, 8 μM; U2, 0.3 μM; U5, 12 μM; U6, 13 μM) were added to splicing reactions and incubated at 30 °C for 10 min prior to the addition of RNA substrates. Incubations were then carried on for 1.5 h. Splicing products were resolved on 8% denaturing gels.
U1 snRNP RNase H Protection Assay
The TauEx10−11 RNA substrate (3 fmol) was added to a 12.5 μl of splicing reaction containing mock- or U1 snRNP-partially depleted HeLa cell cytoplasmic extracts S100 (53) and incubated for 15 min at 30 °C. Oligonucleotides (20 pmol) directed against exon 10 5′ splice sites (17) were then added along with 0.2 units of RNase H (Invitrogen), and the incubation continued for 10 min at 37 °C. Resulting RNA fragments were resolved on 6% polyacrylamide, 8 M urea gels (76, 77). SR proteins were prepared as described (78). Tra2β protein-containing cell lysates were prepared from HEK293 cells transfected with Tra2β-Myc expression plasmid.
RNA Interference Experiments
An RNA interference assay was carried out in HEK293 cells as described (79). The oligonucleotides were custom-synthesized by Dharmacon (Boulder, CO).
RESULTS
An Exonic Splicing Enhancer Located in Exon 10 of the Human Tau Pre-mRNA
The N279K mutation in exon 10 of the tau gene was reported to affect exon 10 splicing (Fig. 1A) (20, 27). It was proposed that a T to G change in the N279K mutation might improve an exonic splicing enhancer because of the sequence change from AATAAGAAG to AAGAAGAAG. Consistent with the hypothesis that this AG-rich region acts as a splicing enhancer, is the discovery of the Del280K mutation that changes the sequence from AATAAGAAG to AATAAG in FTDP-17 patients. In these patients, presumably the deletion of one copy of AAG in this enhancer region led to a decrease in exon 10 splicing and therefore decreased formation of Tau4R in the brain (27, 28). However, there was no direct evidence that these exonic mutations affected exon 10 splicing, and published studies only reported Western blotting and RT-PCR results. These studies could not rule out indirect effects other than those directly affecting tau pre-mRNA splicing (27, 28). To test the exonic splicing enhancer hypothesis, we made use of a tau minigene construct containing the upstream exon 9, the 93-bp alternative exon 10, and flanking introns, followed by downstream exon 11 (TauEx9−11d5 in Fig. 1B and Ref. 17). Our previous study demonstrated that this tau minigene was capable of replicating the alternative splicing pattern of tau exon 10 splicing when introduced into human cell lines. To characterize this AG-rich region of exon 10, we made TauEx9−11d5 constructs containing N279K or Del280K mutations by PCR-mediated site-directed mutagenesis (Fig. 1B). The wild-type TauEx9−11d5 (WT) or N279K mutant minigenes were expressed after transient transfection into HeLaRB (17), HEK293, or neuroblastoma N2a cells. The alternatively spliced products (Ex10+ and Ex10−, namely Tau4R and Tau3R) were detected by RT-PCR with primers specific for the tau minigenes as described previously (17). N279K mutation indeed increased the ratio of exon 10+/exon 10− transcripts, suggesting enhanced exon 10 splicing (Fig 2A). To directly examine the mutation effect on tau exon 10 splicing and investigate the molecular mechanism underlying the effect of N279K mutation, we set up an in vitro splicing system using HeLa cell nuclear extracts. Although the three-exon-containing splicing substrate TauEx9−11d5 (either WT or N279K) pre-mRNAs were not spliced efficiently in vitro (Fig. 2B, lanes 1 and 2) (17), TauEx10−11d5 transcripts containing exon 10 and exon 11 underwent efficient splicing in the presence of the same HeLa nuclear extracts under the same conditions (Fig. 2B, lanes 7 and 8). This allowed us to compare the efficiency of the splicing of exon 10 between the wild-type and N279K mutant pre-mRNA transcripts. N279K mutation significantly increased the efficiency of exon 10 splicing in TauEx10−11d5 construct. Similar to TauEx9−11d5 pre-mRNAs, the TauEx9−10d5 (either WT or N279K mutant) substrates also showed a very low efficiency of splicing (Fig. 2B, lanes 9 and 10). This may be due to the weak 3′ splice site upstream of exon 10 as predicted from the nucleotide sequence. Because U2AF65 can stimulate splicing of substrates containing weak 3′ splice sites, we attempted to improve the in vitro splicing efficiency by adding purified recombinant U2AF65 protein in the splicing reaction (Fig. 2B, lanes 3−6). Indeed, the addition of U2AF65 improved the splicing efficiency of TauEx9−11d5 to a level high enough to allow the detection of in vitro splicing products. Using these complemented splicing reactions, we observed that, similar to that in transfected cells and in the brain tissue of FTDP-17 patients, N279K mutation in exon 10 significantly increased the inclusion of exon 10 (Fig. 2B, compare lane 4 with lane 3 and lane 6 with lane 5, with the quantification of the ratio between exon 10-containing and exon 10-skipping mRNA shown in Fig. 2C). These results clearly indicate that the N279K mutation stimulates the inclusion of the exon 10.
Fig.2. N279K mutation enhances exon 10 splicing.
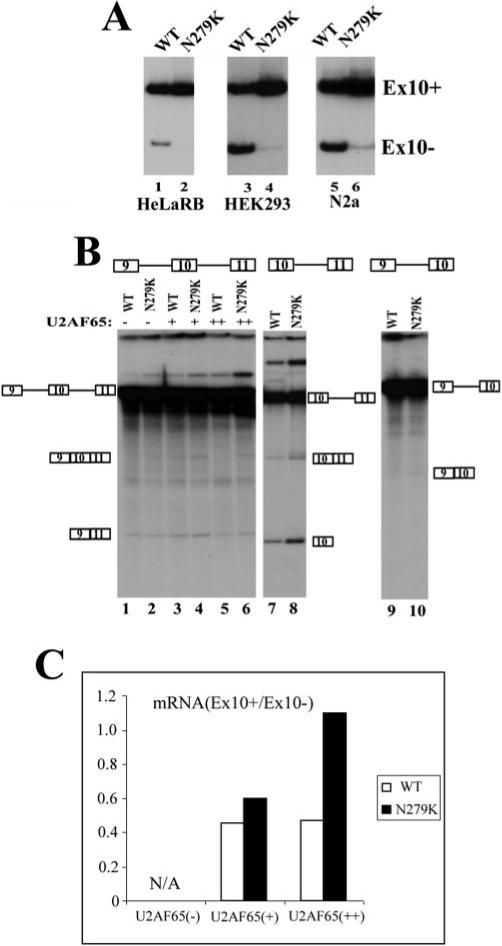
A, alternative splicing of exon 10 from transfected WT or N279K mutant tau minigene. The tau minigene constructs were transfected into HeLaRB, HEK293, and N2a cells. The splicing products expressed from the transfected minigene were detected by RT-PCR with primers specific to the transfected tau minigenes. The positions of exon 10-containing and exon 10-skipping splicing products are as indicated. B, in vitro splicing of TauEx9−11d5 (WT or N279K) (lanes 1−6), TauEx9−10d5 (WT or N279K) (lanes 7 and 8), and TauEx10−11d5 (WT or N279K) (lanes 9 and 10) RNA substrates. 32P-Labeled pre-mRNA substrates were incubated in HeLa nuclear extracts under the splicing conditions. The reactions shown in lanes 3−6 contained different amounts of the exogenous purified U2AF65 protein. Positions of pre-mRNA and splicing products are as indicated. C shows the quantification using a PhosphorImager of splicing products expressed as the ratio of exon 10-containing mRNA (Ex10+) to exon 10-skipping mRNA (Ex10−) in splicing reactions with TauEx9−11d5 substrate (lanes 1−6). In the absence of exogenous U2AF65, the HeLa nuclear extract did not produce a sufficient amount of exon 10-containing splicing products that were detectable by the PhosphorImager; therefore, quantification was only carried out for splicing reactions containing the supplemented U2AF65 protein.
The Mutations in the AG-rich Region of Exon 10 Affect Both Upstream and Downstream Splicing Units
The observation that N279K mutation increases exon 10 splicing supports the presence of an AG-rich exonic splicing enhancer. To gain insights into the mechanism of action of this regulatory element, we constructed several minigenes with this tau exon 10 AG-rich element located in either the 5′ exon or 3′ exon of single-intron pre-mRNA substrates (Fig. 1B). We first tested TauEx9−10d5 minigenes containing wild-type sequence (AATAAGAAG) or the N279K mutation (AAGAAGAAG) in exon 10. As described before, splicing of TauEx9−10d5 transcripts was inefficient, with only a low level of splicing products detectable in N279K mutation (not in WT substrate) when purified U2AF65 protein was not added to the splicing reactions (Fig. 3A, lanes 1 and 2). It was clear that N279K mutation increased the efficiency of splicing between exon 9 and exon 10, as compared with the minigene containing the wild-type sequence (Fig. 3, A and C). When one copy of AAG trinucleotide was deleted as in the Del280K mutant (AATAAG instead of the wild-type AATAAGAAG sequence), the efficiency of splicing between exon 9 and 10 was reduced below a detectable level even in the presence of exogenous U2AF65 protein (data not shown). These results indicate that the AG-rich element in exon 10 acts to promote splicing between exon 9 and exon 10 and that the T to G nucleotide change in N279K mutant further increases the activity of the AG-rich exonic splicing enhancer element. We then tested whether the upstream exon 9 sequence was required for the activity of the AG-rich exonic enhancer present in exon 10. The upstream exon 9 with an associated 5′ splice site was substituted with the first exon region (L1) of the Ad major late transcription unit. In this chimeric splicing substrate lacking the native tau exon 9, N279K mutation also increased the splicing between exon 10 and the upstream heterologous exon (Fig. 3, B and C), demonstrating that the AG-rich element in tau exon 10 was functionally active in a heterologous context. Thus, the AG-rich element, when residing in the downstream exon, acts to stimulate the splicing to the upstream exon either in the native tau exon 9−10 splicing unit or in the heterologous context.
Fig.3. Exonic mutations in the AG-rich region of exon 10 affect the splicing with both upstream and downstream exons.
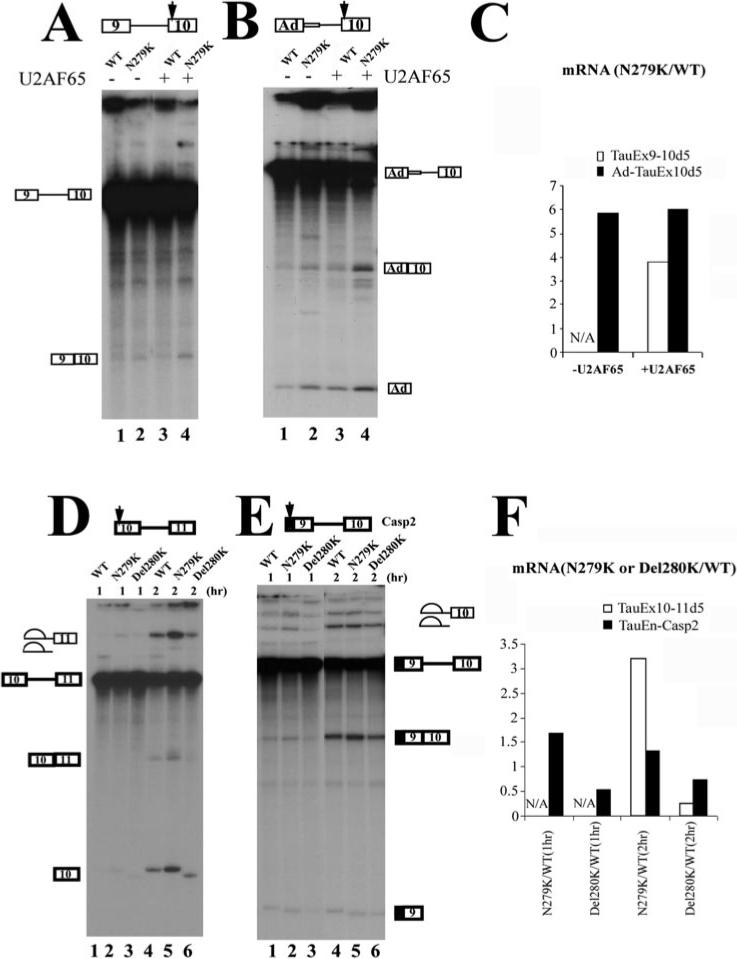
The position of the AG-rich element in each construct is indicated by the arrow. Shown in the figures are in vitro splicing reaction products using pre-mRNA transcripts derived from TauEx9−10 (A), Ad-TauEx10 (B), TauEx10−11 (D), or TauEn-Casp2Ex9−10 (E) plasmids. In A and B, the AG-rich enhancer with WT sequence or N279K mutation is located in the downstream exon. In D and E, the AG-rich element with WT, N279K, or Del280K is located in the upstream exon. The quantification was performed using a PhosphorImager and is expressed as the ratio of corresponding splicing products (ligated exons) produced in the reactions containing either WT or mutant pre-mRNA substrates. N/A, reactions with splicing products below the detection level. The N279K point mutation increases the efficiency of the splicing with both upstream and downstream exons. Del280K reduces the splicing efficiency, as indicated by the quantification shown in C and F.
We then asked whether this AG-rich element could affect splicing to the downstream exon. TauEx10−11d5 pre-mRNA containing N279K mutation in exon 10 showed a significantly increased splicing efficiency as compared with the wild-type TauEx10−11d5 pre-mRNA (Fig. 3, D (lanes 2 and 5 compared with lanes 1 and 4) and F; compare the levels of mRNA products Ex10−11, Ex10, and lariat intermediates). Del280K mutant (with one copy of AAG deleted in the AG-rich element) showed reduced splicing as compared with the wild-type pre-mRNA (Fig. 3, D (lanes 3 and 6) and F). These two mutations showed similar, albeit less dramatic, effects on the splicing to the downstream exon when the AG-rich element was fused to exon 9 of a caspase-2 exon 9−10 splicing cassette (44), namely N279K increasing and Del280K decreasing the splicing efficiency (Fig. 3, E and F). Taken together, these in vitro splicing results indicate that this AG-rich element inside tau exon 10 is an exonic splicing enhancer that functions in a bidirectional manner to facilitate splicing of exon 10 with both the upstream and the downstream exons. This element is also active in certain heterologous contexts. Mutations found in FTDP-17 patients inside this AG-rich element directly affect splicing of both exon 9−10 and exon 10−11 splicing units. Bidirectional splicing enhancers have been described that contain multiple elements with different sequences affecting splicing in different directions. However, to our knowledge, our observation with tau exon 10 splicing provides an example in which a simple AG-rich splicing enhancer acts in a bidirectional manner to promote splicing of both upstream and downstream splicing units.
Trans-acting Splicing Factor Tra2β Interacts with Tau Exon 10 Splicing Enhancer
To identify proteins associated with this exonic splicing enhancer, we made additional plasmids and employed a UV cross-linking assay. As shown in Fig. 1, plasmids were made for synthesizing RNAs containing either WT or mutant exon 10 sequences including N279K and Del280K as described before. In addition, another mutant, dEn, was constructed in which the entire AG-rich enhancer was deleted. α-32P-labeled RNAs corresponding to exon 10 of dEn, Del280K, WT, and N279K constructs were synthesized, and these transcripts contained 0, 1, 2, and 3 copies of AAG in the AG-rich element, respectively (Fig. 1). The wild-type or mutant RNA transcripts were incubated with HeLa nuclear extracts and then subjected to UV cross-linking. After treatment with RNase A, proteins cross-linked to RNA were resolved on SDS-PAGE. Among the cross-linked proteins, a complex of ∼40 kDa was detected with WT and N279K but at a low level in the reaction with Del280K RNA (Fig. 4A). The level of the 40-kDa cross-linking species detected in the UV cross-linking assay correlated with the number of AAG repeats present in exon 10, with the highest level in the reaction containing N279K mutant (three copies of AAG) and in a decreasing order in WT (2 copies), Del280K (1 copy), and almost undetectable in dEn (0 copies of AAG) RNA-containing reactions (Fig. 4A, as marked by the black arrow). A 42-kDa protein and other proteins (as marked by the white arrowheads above the 40-kDa band), however, were present at a similar level in reactions with different RNA substrates (Fig. 4A). Because SR and SR-like proteins are known to bind to exonic sequences where they act to enhance the splicing of the adjacent intron, we focused our search on SR domain-containing splicing regulators. A monoclonal antibody (1H4) was used, which recognized a number of SR domain-containing proteins. Immunoprecipitation experiment with this monoclonal antibody, following the above described UV cross-linking, revealed several proteins, including one of ∼40 kDa in size which interacted with N279K RNA more efficiently than with wild type exon 10 RNA (Fig. 4B). Since the monoclonal antibody 1H4 specifically recognizes SR domain-containing proteins, this result suggests that the 40-kDa protein may contain serine-arginine-rich sequences.
Fig.4. A protein of ∼40 kDa in size specifically interacts with the AG-rich region of tau exon 10.
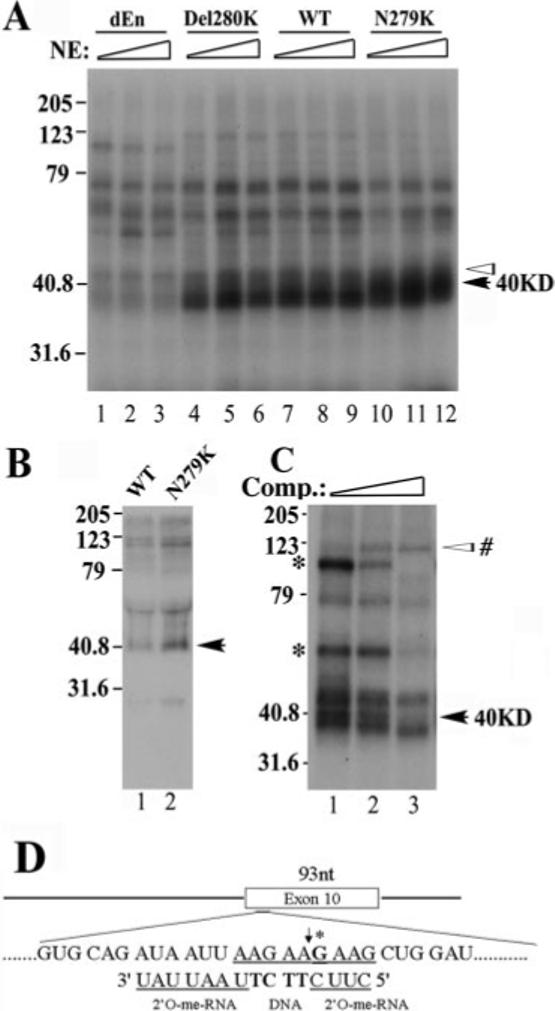
A, cross-linking of protein factors in the HeLa nuclear extract to Tau exon 10 RNA. Splicing reactions were performed as described with 32P-labeled RNA corresponding to exon 10 from dEn, Del280K, WT, and N279K constructs. Various amounts of HeLa nuclear extracts (10, 20, or 30 μg) were incubated with the RNAs under splicing conditions for 15 min. Aliquots of the reactions were UV-irradiated and then treated with RNase A. The cross-linking products were resolved on 12.5% SDS-PAGE and detected by autoradiography. The intensity of a 40-kDa protein (marked with a black arrow) correlates with the copy number of AAG repeats present in the AG-rich region of exon 10, whereas that of several other proteins (as indicated by white arrowheads) does not show such correlation. B, UV cross-linking/immunoprecipitation experiment. Tau exon 10 RNA corresponding to WT (lane 1) and N279K (lane 2) was used in a similar cross-linking experiment except that 1H4 anti-SR monoclonal antibody was added following the RNase A treatment to precipitate SR domain-containing proteins. Immunoprecipitated proteins were resolved on 12.5% SDS-PAGE and detected by autoradiography. C, the UV cross-linking experiments were carried out using N279K mutant RNA as shown in A except that the N279K RNA transcript used contained a site-specific 32P label in the AAG motif. The site-specific labeled G is marked with an asterisk in D. The chimeric oligonucleotide used for RNase H cleavage to generate the half-RNAs is also indicated in D. The cross-linking experiment was done in the presence of increasing amounts of competing oligonucleotide corresponding to the AAG-rich enhancer region. Molecular weights are shown on the left sides of gels (in kDa). The black arrows on the right mark the position of the 40-kDa protein. In C, an asterisk marks the cross-linking species that remain to be investigated.
The UV cross-linking pattern also suggests that the 40-kDa protein may interact with tau RNA at or near the AG-rich region. To confirm that the 40-kDa protein indeed interacted with the AG-rich element, we placed a site-specific label inside the AG-rich sequence element (Fig. 4D, at the G residue marked with an asterisk) and then performed the UV cross-linking experiment as described under “Experimental Procedures.” In the reactions using the tau RNA transcript containing the N279K mutation, a 40-kDa protein was one of the most prominent cross-linking species detected (Fig. 4C), indicating that the 40-kDa protein indeed interacted with this AG-rich region of N279K mutant transcript. In the presence of an increasing concentration of unlabeled cold competing transcripts, the intensity of several cross-linked bands including the 40-kDa protein was reduced, suggesting that these proteins interact specifically with the tau transcript at the AG-rich region. In addition to the 40-kDa species, two other protein species with gel mobility of ∼65 and 100 kDa were also detected to be competed away by the unlabeled competing transcript (as marked by an asterisk on the left side of the gel; Fig. 4C). In addition, another protein of ∼120 kDa in size showed increased binding in the presence of competing transcript. The functional significance of these proteins in tau exon 10 splicing will be further investigated in our future study. We chose to initially follow up the 40-kDa protein because this protein appeared to interact specifically with tau pre-mRNA in a manner dependent on AAG sequence.
Among the known human SR domain-containing proteins, SRp40 and Tra2β are ∼40 kDa in size. SRp40 is a member of a highly conserved family of splicing factors called SR proteins (reviewed in Refs. 54, 55, and 65). Tra2β is a human homologue of Drosophila splicing regulator Tra2, also containing an RNA-binding domain and an RS domain (67). In vitro SELEX studies demonstrated that SRp40 and Tra2β bind to purine-rich exonic sequences (45, 46). We therefore tested whether the 40-kDa protein identified in UV cross-linking experiments was Tra2β or SRp40. Because of the difficulty we encountered in preparing functionally active purified recombinant Tra2β and SRp40 proteins, we used an alternative approach, expressing these proteins in HEK293 cells by transient transfection. Biotinylated TauEx10−11 RNAs corresponding to dEn, Del280K, WT, or N279K RNAs were incubated under splicing conditions with increasing amounts of cell lysates containing transiently expressed Myc-tagged human Tra2β. After affinity selection with streptavidin-agarose beads, the bound proteins were analyzed by Western blotting using specific antibodies against the Myc epitope tag. The amount of Tra2β associated with TauEx10−11 RNA appeared to be proportional to the copy number of AAG present in the RNA transcript (i.e. Tra2β interacted with TauEx10−11 RNAs with increasing efficiency in the order of dEn, Del280K, WT, and N279K (containing zero, one, two, and three copies of AAG, respectively) (Fig. 5A, compare lanes 1, 3, 5, and 7 with lanes 2, 4, 6, and 8). However, when the same experiments were performed using cell lysates containing transiently expressed SRp40, amounts of SRp40 affinity-selected using the same series of TauEx10−11 RNAs did not show any significant differences (Fig. 5B). The level of expression of SRp40 was comparable with that of Tra2β in the lysates of transfected cells (data not shown). These results demonstrate that Tra2β could specifically interact with the AG-rich element inside exon 10 and that mutations found in FTDP-17 patients affect the binding of Tra2β to this element.
Fig.5. Tra2β protein interacts with the Tau exon 10 AG-rich region.
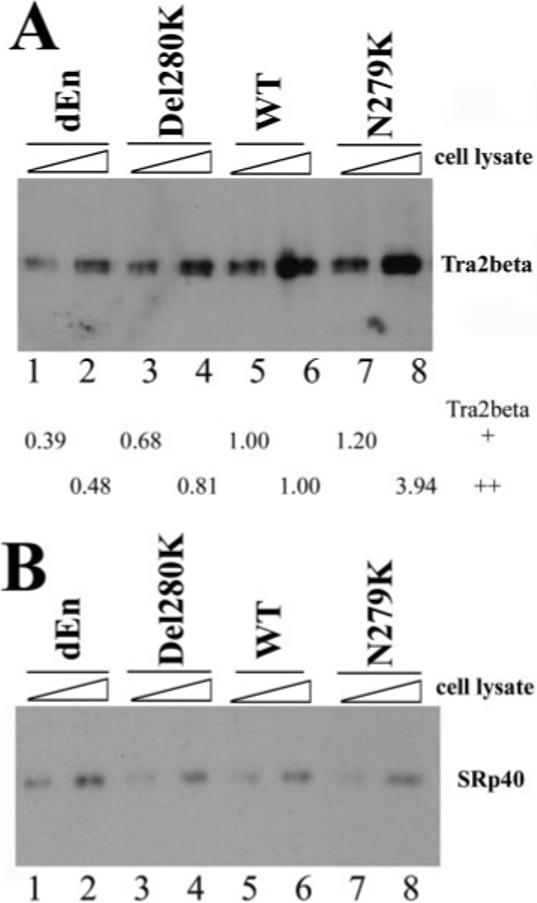
40 fmol of biotinylated TauEx10−11 RNAs corresponding to dEn, Del280K, WT, and N279K (with zero, one, two, and three copies of AAG repeats, respectively) were incubated on ice for 30 min in 25-μl reactions under splicing conditions with increasing amounts of cellular extract (0.5 or 1 μl) containing transiently transfected Myc-tagged Tra2β (A) or HA-tagged SRp40 (B). After affinity selection with streptavidin-agarose beads, the bound proteins were detected by Western blotting using anti-Myc antibody (for Tra2β-Myc) or anti-HA antibody (for SRp40). The intensity of Tra2β protein pulled down by dEn, Del280K, or N279K RNA in A was quantified, respectively, and compared with that pulled down by WT RNA. The numbers shown below the gel in A represent the relative quantification using the band intensity in the wild type (lane 5 or lane 6, respectively) as 1.00 in the presence of either 0.5 μl (lanes 1, 3, 5, and 7) or 1 μl (lanes 2, 4, 6, and 8) of Tra2β-expressing cell lysates.
In order to evaluate the functional significance of Tra2β in tau pre-mRNA splicing, we tested whether Tra2β was able to increase the splicing of exon 10 when added in vitro. It was reported that Tra2β alone was not sufficient to activate the splicing in S100 extracts of a mouse IgM pre-mRNA containing a binding site for Tra2β as a splicing enhancer (46). We saw similar results in S100 extracts using tau minigene constructs (see Fig. 8B, lanes 11 and 12). To overcome this problem, we incubated TauEx10−11 splicing substrates (WT or N279K) with cell lysates containing transiently transfected Myc-tagged Tra2β in the presence of a limiting amount of nuclear extracts. We used exon 10 and 11 splicing substrate to set up the in vitro splicing assay because the in vitro splicing efficiency of either exon 9−11 or exon 9 and 10 transcripts was too low, as described earlier. The addition of Tra2β protein-containing cell lysates further increased splicing efficiency above that of mock cell lysates (Fig. 6A, compare lanes 3 and 4 with lanes 1 and 2 and lanes 9 and 10 with lanes 7 and 8). Furthermore, the difference in splicing efficiency between substrates with WT sequence and N279K mutation was more obvious in the presence of cell lysates containing overexpressed Tra2β protein (Fig. 6, A (compare lanes 7 and 8 with lanes 5 and 6) and B). This is consistent with the higher level of Tra2β associated with N279K transcript (Figs. 4 and 5). Although SRp40 is also able to increase the splicing efficiency (Fig. 6A, compare lanes 5 and 6 with lanes 1 and 2 and lanes 11 and 12 with lanes 7 and 8), it stimulated splicing of substrates with WT sequence and the N279K mutation at almost the same level (Fig. 6B). The results described above indicate that Tra2β protein is a specific splicing activator interacting with the AG-rich element of tau exon 10. The mutations improving or disrupting the exonic splicing enhancer may affect the binding of Tra2β, thereby increasing or decreasing the inclusion of exon 10 as in the case of N279K and Del280K mutations, respectively.
Fig.8. Binding of Tra2β to Tau exon 10 increases the U1 snRNP-dependent complex formation at the 5′ splice site of exon 10.
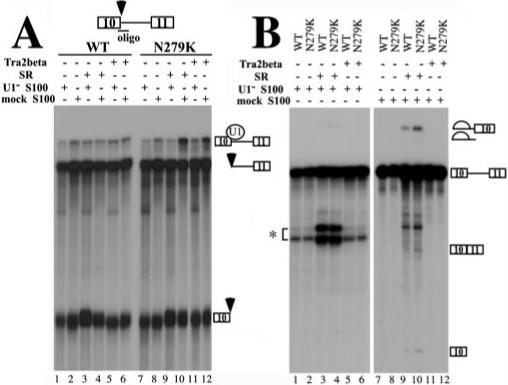
A, U1 protection assays. TauEx10−11 RNA substrate (WT or N279K) was added to a 12.5-μl splicing reaction using mock- or U1 snRNP-partially depleted HeLa cell cytosolic S100, purified SR proteins, and cell lysates with overexpressed Tra2β protein. Following the incubation, the oligonucleotide complementary to the 5′ splice site of exon 10 (17) was added along with RNase H (0.4 units), and the incubation was continued for another 15 min at 37 °C. The RNA cleavage products were then analyzed by gel electrophoresis. Positions of the U1-protected pre-mRNA and the RNase H cleavage products are indicated on the right. Either SR protein mixture or transiently expressed Tra2β protein increased the formation of U1 snRNP-dependent complex when N279K mutant pre-mRNA was used (lanes 10 and 12). B, in vitro splicing of TauEx10−11 RNA substrate (WT or N279K). Mock- or U1 snRNP-partially depleted HeLa cell cytosolic S100, SR proteins, and cell lysates containing overexpressed Tra2β protein were incubated with WT or N279K mutant pre-mRNA transcript under splicing conditions. Splicing products were detected after gel electrophoresis. The asterisk indicates the position of a nonspecific cleavage product generated by hybridization of the U1 RNA-specific oligonucleotide on the tau pre-mRNA.
Fig.6. Tra2β stimulates exon 10 splicing in vitro.
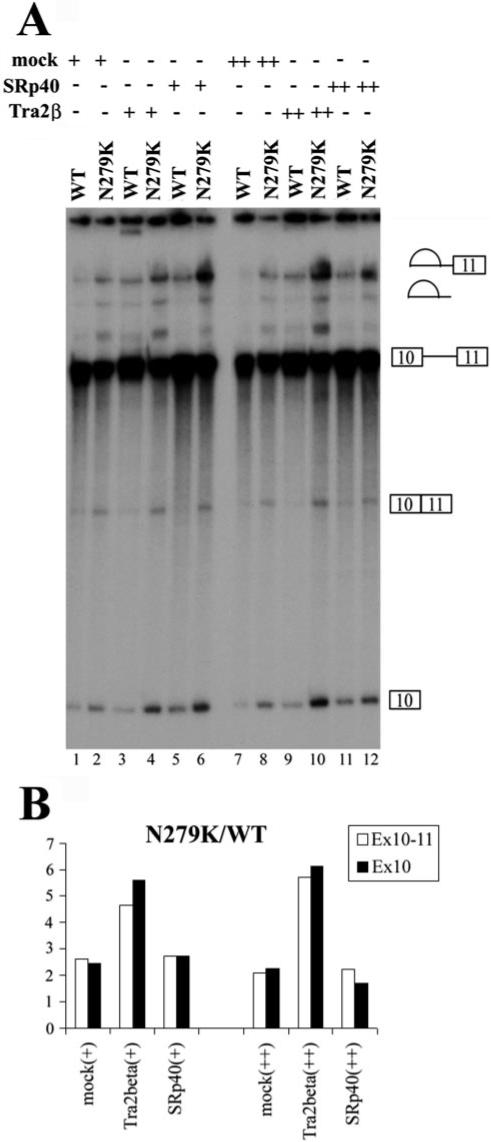
A, TauEx10−11 splicing substrates (WT or N279K) were incubated with different amounts of cell lysates containing vector-transfected mock cell lysates (lanes 1, 2, 7, and 8), transiently transfected Myc-tagged Tra2β (lanes 3, 4, 9, and 10), or HA-tagged SRp40 (lanes 5, 6, 11, and 12) as well as nuclear extracts under normal splicing conditions. Positions of pre-mRNA, splicing intermediate, and splicing products are as indicated. B, the spliced products (i.e. ligated exons) in A were quantified, and the effects of Tra2β and SRp40 on the substrate with N279K mutation were compared with those with WT sequence. The addition of Tra2β-overexpressing cell lysate increases exon 10 splicing in an enhancer-dependent manner. The addition of the control cell lysate (Mock) or SRp40-transfected cell lysate have similar effects on exon 10 splicing, with the ratio of N279K/WT spliced product being ∼2. However, this ratio is increased to higher than 4.5 (4.5−6.3) in the presence of Tra2β-overexpressing cell lysate.
Tra2β Increases the U1 snRNP-dependent Complex Formation at the 5′ Splice Site of Exon 10
To dissect the mechanism by which Tra2β promotes the splicing of tau exon 10, we used U snRNP inactivation assays (17, 47). U1, U2, U5, or U6 snRNPs were partially blocked by using specific individual 2′-O-methyl oligoribonucleotides at appropriate concentrations to treat HeLa nuclear extracts prior to the splicing reactions. When either U2 snRNPs (Fig. 7, lanes 3 and 4), U5 snRNPs (Fig. 7, lanes 5 and 6), or U6 snRNPs (Fig. 7, lanes 7 and 8) were partially blocked, both wild type and N279K RNA splicing were partially inhibited to a similar extent as compared with the splicing reactions containing the mock-treated nuclear extracts (Fig. 7, lanes 9 and 10). However, when U1 snRNP was partially blocked, splicing of wild-type tau was significantly decreased, whereas the splicing of N279K mutation-containing pre-mRNA was barely affected (Fig. 7, lanes 1 and 2). Quantification of the exon 10 splicing under these conditions shows that in the presence of U1 snRNP-partially blocked splicing extract, the ratio of N279K mutant splicing product to that of the wild type splicing product is increased as compared with that in the presence of mock-treated splicing nuclear extract. Partial blocking of other U snRNPs important for splicing, including U2, U5, and U6, did not show the same effect (Fig. 7B). This result suggests that U1 snRNP may play an important role in differential recognition of tau WT and N279K RNA during splicing.
Fig.7. U snRNP inactivation differentially affects wild-type and mutant Tau splicing.
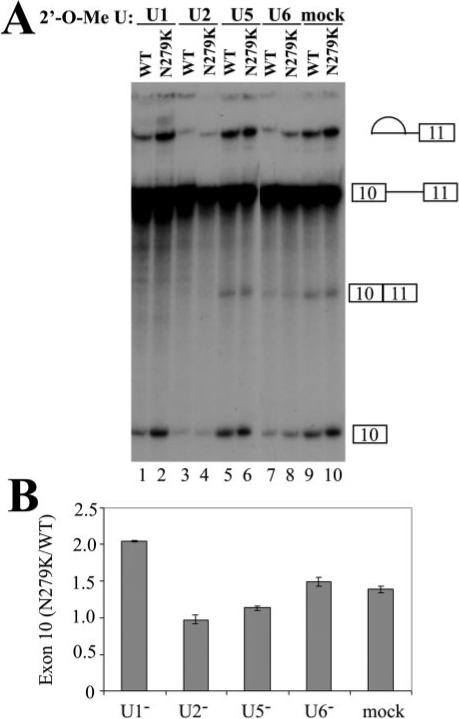
2′-O-Methyl oligoribonucleotides complementary to U1, U2, U5, and U6 snRNAs were added individually to HeLa nuclear extracts, and the splicing reactions were preincubated at 30 °C for 10 min. The concentration of individual 2′-O-methyl oligonucleotides was titrated to give partial inhibition of splicing (U1, 8 μM; U2, 0.3 μM; U5, 12 μM; U6, 13 μM) (17). TauEx10−11d5 WT and N279K mutant pre-mRNAs were then added, and the incubation was continued for 1.5 h. Splicing reaction products were analyzed by urea-polyacrylamide gel electrophoresis. Quantification of three independent experiments is shown in B, showing a consistent and significant increase in the ratio of N279K/WT splicing products as compared with mock-treated or U2, U5, or U6 oligonucleotide-treated reactions.
As described earlier, Tra2β could specifically bind to the AG-rich element of exon 10 and activate tau exon 10 splicing. We further asked whether Tra2β could increase the formation of U1 snRNP-dependent early spliceosome complexes on the 5′ splice site of exon 10 and compared the wild-type with N279K mutant in this assay. A specific U1 protection assay was performed as described before (17). TauEx10−11 substrates with WT sequence or N279K mutation in exon 10 were first incubated with HeLa cell cytosolic S100 extract alone (Fig. 8A, lane 2 or 8), S100 plus purified SR proteins (Fig. 8A, lane 4 or 10), or S100 plus cell lysates containing overexpressed Tra2β protein (Fig. 8A, lane 6 or 12). The S100 extracts used were either mock-treated with a control 2′-O-methyl oligonucleotide or with an U1-specific 2′-O-methyl oligonucleotide to partially inactivate U1 snRNP (17). Then oligodeoxynucleotide complementary to the 5′ splice site and corresponding to the U1 snRNP binding region was added together with RNase H. The cleavage reaction products were resolved on a polyacrylamide gel, and the U1 snRNP-protected as well as cleaved RNA products were detected following autoradiography. Under these conditions, the cleaved and protected products were the result of the oligonucleotide-directed RNase H cleavage at the U1 snRNP binding site of the tau pre-mRNA, thus reflecting U1 snRNP-mediated protection of this 5′ splice site as described previously (17). As shown in Fig. 8, a higher level of protected product was observed with TauEx10−11 RNA containing the N279K mutation than with the wild type RNA in the presence of exogenous Tra2β protein (Fig. 8A; compare lane 12 with lane 6 at the position of the U1 snRNP-protected band) or SR proteins (Fig. 8A, compare lane 10 with lane 4). The increased level of protected band was dependent on the presence of active U1 snRNP because when S100 was treated with U1-specific oligonucleotide to inactivate U1 snRNP, WT and N279K RNA did not show any significant differences (Fig. 8A, compare lane 11 with lane 5 and lane 9 with lane 3). The activities of the mock-treated and U1-depleted S100 extracts used in the experiment were tested by an in vitro splicing assay. In the presence of the SR protein, the mock S100 extract was active in supporting splicing (lanes 9 and 10) and inactivation of U1 snRNP in this S100 extract blocked the activity of the S100 extract to complement splicing (lanes 1−6). As expected, the U1-inactivated S100 extracts did not support the splicing of either wild-type or N279K RNA (Fig. 8B, lanes 1−6), even when SR proteins were added into the reaction. The addition of the SR proteins complemented the splicing activity of the mock-treated S100 extracts, in the presence of either WT or N279K splicing substrates (Fig. 8B, lanes 9 and 10). The addition of Tra2β-overexpressing cell lysate alone, however, did not sufficiently activate the splicing reaction to allow detection of splicing products under these conditions (Fig. 8B, lanes 11 and 12). The same reaction conditions were used in Fig. 8, A and B. Fig. 8B shows that these experiments were carried out under splicing conditions, suggesting that the inability of Tra2β to complement S100 extract for splicing was not due to improper reaction conditions. This observation suggests that Tra2β may work as a splicing activator in a manner dependent on other essential splicing factors. The above results demonstrate that in the presence of the N279K mutation, Tra2β is capable of stimulating the formation of a U1 snRNP-containing complex at the 5′ splice site of exon 10. Enhanced interaction between Tra2β and exon 10 AAGAAGAAG splicing enhancer leading to increased binding of U1 snRNP to the 5′ splice site of exon 10 may provide at least one mechanism by which N279K mutation stimulates exon 10 inclusion.
Down-regulation of Tra2β by RNA Interference Decreases the Inclusion of Exon 10
To further test whether Tra2β plays a role in regulating exon 10 splicing in cells, we tried both antisense and RNA interference (RNAi) approaches to reduce the Tra2β expression in cells. After testing several different antisense and RNAi oligonucleotides, we identified one antisense and one RNAi oligonucleotide that were capable of down-regulating endogenous Tra2β expression when transfected into HEK293 cells. Similar results were obtained, and only RNAi results are described here. We examined whether the ratio of Tau4R/Tau3R was altered when tau minigene TauEx9−11d5(N279K) was transfected into cells treated with Tra2β-specific or SRp40-specific or control RNAi oligonucleotides. 48 h after co-transfection of RNAi oligonucleotide and the tau splicing substrate, the expression of Tra2β was measured by a sensitive RT-PCR assay (Fig. 9A). Whereas the control oligonucleotide or SRp40 oligonucleotide had no effect on Tra2β expression, the Tra2β-specific oligonucleotide reduced Tra2β expression by ∼60−70% (Fig. 9A, compare lane 1 with lane 2 or 3). This reduction was Tra2β-specific, because the same oligonucleotide did not reduce γ actin (Fig. 9C) or other genes examined, including tubulin and bcl-x (data not shown). Although we were not able to completely block Tra2β expression even after repeated treatment using the oligonucleotide, a significant effect on tau exon 10 splicing was detected when Tra2β expression was decreased. Down-regulation of Tra2β reduced tau exon 10 splicing as compared with that in the control or SRp40 RNAi-treated cells (Fig. 9, C and D). The treatment by the same Tra2β oligonucleotide did not affect alternative exon inclusion in other genes examined such as caspase-2 (data not shown), suggesting relative specificity of Tra2β in regulation of tau gene alternative splicing. Consistent with the lower level of Tra2β protein associated with the wild-type tau transcript as compared with the N279K transcript (Figs. 4 and 5), the effect of Tra2β reduction on wild-type tau exon 10 splicing was detectable but less dramatic (data not shown).
Fig.9. Down-regulation of Tra2β by RNAi decreases the inclusion of exon 10 in tau minigene.
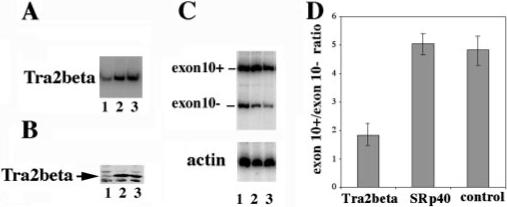
RNAi oligonucleotide specific for human Tra2β (lane 1) or SRp40 (lane 2) or control oligonucleotide (lane 3) was cotransfected with tau minigene TauEx9−11d5(N279K) into HEK293 cells. A shows the specific down-regulation of Tra2β expression in the RNAi oligonucleotide-treated cells. The total RNAs of HEK293 cells were prepared 48 h after transfection. Tra2β-specific primers were used for RT-PCR to detect the RNA level of Tra2β. B, Western blotting results using specific anti-Tra2β in the corresponding RNAi oligonucleotide-treated cells. C, tau exon 10 splicing pattern as detected by RT-PCR. RNA amounts were comparable as shown with the internal γ actin control as shown in the lower portion of C. The ratio of exon 10+/exon 10− splicing products was decreased by Tra2β-specific RNA interference. Quantification of the results derived from three independent experiments is shown in D.
To confirm that Tra2β gene is indeed expressed in the human brain, we used a previously established RT-PCR assay (17) with cDNA samples prepared from the frontal cortex, the major region affected in FTDP-17 patients. Specific Tra2β signals were detected in the cDNA samples prepared from both fetal and adult brain tissues (Fig. 10). Consistent with our observation, a recent study was published while this manuscript was in preparation, documenting the expression of Tra2β in the rat brain (80). Therefore, Tra2β gene is expressed in the brain, where the balance of different tau alternative splicing isoforms is crucial for proper neuronal function.
Fig. 10. The detection of Tra2β gene expression in the brain.
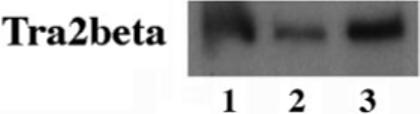
cDNA samples were prepared from the frontal cortical region of human brain tissues and used for RT-PCR as described (17). The specific Tra2β band was detected in either fetal (lane 1) or adult brains (lanes 2 and 3).
Taken together, our results establish tau pre-mRNA as one of targets for Tra2β splicing regulator and support a role of Tra2β in the regulation of alternative splicing of exon 10 in the tau gene. The in vivo significance of Tra2β in regulation of tau alternative splicing will be further investigated in our future study using approaches such as gene knock-out.
DISCUSSION
In this study, we report characterization of an exonic splicing enhancer located in exon 10 of the human tau gene with AATAAGAAG as the wild-type sequence. This AG-rich region contains the sequence in which mutations associated with FTDP-17 have been identified. Specifically, two mutations have been found inside this enhancer element, N279K (AAGAAGAAG) and Del280K (AATAAG). We made an additional mutant, dEn, lacking the entire AG-rich element. By systematically comparing the splicing efficiency, protein-RNA interaction profiles, and the splicing complex formation between the wild type and mutant transcripts, we conclude that these mutations alter the activity of this splicing enhancer by increasing (for N279K) or decreasing (for Del280K) interaction with protein factors interacting with this AG-rich element. Among the trans-acting factors interacting with this AG-rich element, we have identified human Tra2β protein. N279K mutation stimulates, whereas Del280K or dEn mutations reduce, the binding of Tra2β to the AG-rich exonic enhancer. The interaction of Tra2β protein with this exonic element correlates well with the copy number of AAG repeats inside the exonic enhancer. Furthermore, down-regulation of endogenous Tra2β expression using RNA interference led to a reduction in tau exon 10 splicing. Our study suggests that Tra2β or Tra2β-like protein(s) may play an important role in affecting the delicate balance of tau isoforms and in pathogenesis of tauopathies involving aberrant tau exon 10 alternative splicing. Further investigation is necessary to determine the spectrum and specificity of individual splicing activators such as Tra2β. Nonetheless, the down-regulation of endogenous Tra2β reduced the tau exon 10 inclusion in a relatively specific manner, and such a treatment partially reverses the exon 10 splicing effect caused by N279K mutation. Our results suggest a new direction for designing therapy for certain FTDP patients (such as those carrying the N279K mutation) based on down-regulation of certain specific splicing activators for tau exon 10 splicing.
The importance of tau exon 10 alternative splicing has been demonstrated by a number of genetic studies that link the mutations affecting exon 10 splicing in the human tau gene to the development of FTDP-17 (15, 16, 20, 23, 26-28, 37, 38, 48-50). These mutations are located either in the intron downstream of exon 10 or inside exon 10. Our previous study has demonstrated that some intronic mutations act at the level of U1 snRNP binding to the 5′ splice site of exon 10 during the earliest step of spliceosome assembly (17). Exonic mutations inside exon 10 have been studied by several groups (15, 16, 20, 25-29, 37, 50-52). Some of the exonic mutations have been shown to affect the microtubule binding properties of Tau protein(s) (14, 16, 25, 30, 37, 40), whereas other exon 10 mutations alter the Tau4R/Tau3R ratio presumably by influencing exon 10 splicing (26, 27, 37). However, the molecular mechanisms underlying the splicing mutations inside exon 10 were not understood.
Exonic splicing enhancers (ESEs) are sequence elements within exons that promote pre-mRNA splicing. A number of ESEs have been identified in tissue-specific or developmentally regulated exons, which typically have weak splice sites and require the ESE for exon inclusion. As shown by our study, the upstream 3′ splice site of exon 10 is weak. Utilization of this suboptimal 3′ splice site was only detectable in the in vitro splicing assay when additional U2AF65 protein was provided in the splicing reactions. The presence of an ESE in exon 10, together with a suboptimal 3′ splice site, provides a fine-tuning mechanism for the regulation of exon 10 inclusion in certain tissues or at certain developmental stages. It is conceivable that during fetal stages of human development, tau exon 10 inclusion is repressed because of either insufficient levels of splicing activator(s) or the presence of trans-acting factors that counteract the activity of embryonically expressed splicing activators. It is equally possible that the fetal pattern of tau exon 10 splicing (almost exclusively exon 10 skipping) is a result of the combination of the above mentioned mechanisms. We are actively investigating these possibilities using both molecular and biochemical approaches.
The sequence motif for ESEs can be divergent, although purine-rich motifs represent one of the best characterized elements. Often ESEs function by interacting with SR domain-containing splicing regulators (reviewed in Refs. 53-57). ESEs were initially identified by their ability to activate upstream 3′ splice sites (58-60). Recent studies also identified splicing enhancers that stimulate downstream 5′ splice site utilization (61-63). There have also been reports of bidirectional enhancer elements (62, 63), and these elements contain multiple domains with distinct regions functioning to facilitate splicing of either upstream or downstream exons. In our study, we have used biochemical approaches to identify a simple AG-rich element that acts bidirectionally to promote exon 10 splicing with both exon 9 and exon 11. We show that one of the important trans-acting factors involved in the function of this exonic splicing enhancer is human Tra2β protein that acts as an alternative splicing activator. Using a transient transfection assay, we were able to express Tra2β in a functionally active form at a level comparable with that detected in the rat brain tissue by Western blotting (data not shown). Similar to previous studies, our results show that Tra2β by itself is not sufficient to complement S100 cytoplasmic extracts for the splicing activity in the manner that certain essential splicing factors of the SR family do (46). It is likely that Tra2β protein acts in concert with certain basic essential splicing factors to activate tau exon 10 splicing. Furthermore, Tra2β protein interacts with tau pre-mRNA by binding to the AG-rich exonic splicing enhancer, as shown by site-specific UV cross-linking experiments. The addition of the transiently expressed Tra2β protein into the in vitro splicing reaction further stimulates exon 10 splicing, and such stimulatory effect is more pronounced in the presence of the N279K mutation. Thus, Tra2β acts as a splicing activator in tau exon 10 splicing. The bidirectional activity of tau gene exonic splicing enhancers and involvement of alternative splicing regulators, in addition to essential splicing factors, make the similarity more obvious between splicing enhancers and transcription enhancers.
SR domain-containing splicing factors play important roles in mammalian pre-mRNA splicing and alternative splicing regulation (for recent reviews, see Refs. 54, 55, 57, 64, and 65). In addition to interacting with pre-mRNAs via their RNA recognition motifs, these splicing factors have been proposed to mediate interactions both across introns and across exons (59). More recently, SR proteins have been reported to support basal AT-AC splicing and to stimulate exonic splicing enhancer important for AT-AC intron splicing (66). Tra2β is an SR domain-containing protein initially identified as one of the human homologues of Drosophila SR protein transformer 2 (Tra2) (67). Systematic biochemical experiments demonstrated that human Tra2β is a splicing activator interacting with exonic enhancer sequences rich in A and G or GAA repeats rather than a constitutive splicing factor (46). Tra2β has been reported to activate the splicing of exon 7 of survival of motor neuron genes when overexpressed by transfection (68). The mechanism by which Tra2β activates splicing was not clear. Our UV cross-linking and RNase H cleavage/U1 snRNP protection experiments show that increased exon 10 splicing in the tau N279K mutant is associated with increased Tra2β binding to the exonic splicing enhancer and that the addition of Tra2β promotes the formation of U1 snRNP-dependent complex at the 5′ splice site of exon 10. Protein-protein interaction between Tra2β and a U1 snRNP protein U1 70K has been observed,2 and the functional significance of such interaction in promoting U1 binding will be further investigated. These results suggest that one mechanism by which Tra2β acts to stimulate exon 10 splicing may be by facilitating U1 snRNP binding to the 5′ splice site of exon 10.
A previous study reported that overexpression of human Tra2β in a chimeric tau minigene system had no effect on human tau gene alternative splicing (69). It is possible that in this previous study, the failure to detect the effect of Tra2β was a result of using the artificial chimeric tau minigene rather than the native tau minigene (69). Alternatively, the presence of endogenous Tra2β was high enough to mask the effect of overexpression. Our systematic biochemical analyses of the interaction between Tra2β and the AG-rich splicing enhancer in exon 10 and results from Tra2β down-regulation experiments strongly suggest an important role of Tra2β in tau exon 10 alternative splicing regulation. A definitive in vivo role of Tra2β in this alternative splicing event awaits future exploration using strategies such as targeted gene deletion.
Acknowledgments
We thank Drs. Benoit Chabot, Marc T. McNally, Woan-Yuh Tarn, Rui-Ming Xu, and James Manley for generously providing various reagents. We are grateful to Drs. E. Johnson, C. Hicks, and J. Cote and to Angie Hantak for helpful discussions and/or critical reading of the manuscript.
* This work was supported by National Institutes of Health Grant RO1 GM53945/AG17518 and a grant from the Muscular Dystrophy Association (to J. Y. W.) and by a scholarship from the Leukemia Society of America (to J. Y. W.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: snRNP, small nuclear ribonucleoprotein; Ad, adenovirus; RT, reverse transcriptase; WT, wild type; RNAi, RNA interference; ESE, exonic splicing enhancer.
S. Stamm, unpublished results.
REFERENCES
- 1.Cleveland DW, Hwo SY, Kirschner MW. J. Mol. Biol. 1977;116:227–247. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- 2.Drubin DG, Kirschner MW. J. Cell Biol. 1986;103:2739–2746. doi: 10.1083/jcb.103.6.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanai Y, Takemura R, Oshima T, Mori H, Ihara Y, Yanagisawa M, Masaki T, Hirokawa N. J. Cell Biol. 1989;109:1173–1184. doi: 10.1083/jcb.109.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanai Y, Chen J, Hirokawa N. EMBO J. 1992;11:3953–3961. doi: 10.1002/j.1460-2075.1992.tb05489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Mol. Biol. Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 7.Brandt R, Lee G. J. Biol. Chem. 1993;268:3414–3419. [PubMed] [Google Scholar]
- 8.Goode BL, Denis PE, Panda D, Radeke MJ, Miller HP, Wilson L, Feinstein SC. Mol. Biol. Cell. 1997;8:353–365. doi: 10.1091/mbc.8.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Preuss U, Biernat J, Mandelkow EM, Mandelkow E. J. Cell Sci. 1997;110:789–800. doi: 10.1242/jcs.110.6.789. [DOI] [PubMed] [Google Scholar]
- 10.Mandelkow E. Nature. 1999;402:588–589. doi: 10.1038/45095. [DOI] [PubMed] [Google Scholar]
- 11.Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA. Brain Res. 1986;387:271–280. doi: 10.1016/0169-328x(86)90033-1. [DOI] [PubMed] [Google Scholar]
- 12.Andreadis A, Brown WM, Kosik KS. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 13.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 14.Goedert M, Spillantini MG, Davies SW. Curr. Opin. Neurobiol. 1998;8:619–632. doi: 10.1016/s0959-4388(98)80090-1. [DOI] [PubMed] [Google Scholar]
- 15.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Heutink P, et al. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 16.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 17.Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY. Mol. Cell. Biol. 2000;20:4036–4048. doi: 10.1128/mcb.20.11.4036-4048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goode BL, Chau M, Denis PE, Feinstein SC. J. Biol. Chem. 2000;275:38182–38189. doi: 10.1074/jbc.M007489200. [DOI] [PubMed] [Google Scholar]
- 19.Lee VM, Goedert M, Trojanowski JQ. Annu. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 20.Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D'Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC. Proc. Natl. Acad. Sci. U. S. A. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois B, Saugier-Veber P, Martin C, Penet C, Charbonnier F, Agid Y, Frebourg T, Brice A. Hum. Mol. Genet. 1998;7:1825–1829. doi: 10.1093/hmg/7.11.1825. [DOI] [PubMed] [Google Scholar]
- 22.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Ann. Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 23.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC. Am. J. Pathol. 1998;153:1359–1363. doi: 10.1016/S0002-9440(10)65721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasegawa M, Smith MJ, Goedert M. FEBS Lett. 1998;437:207–210. doi: 10.1016/s0014-5793(98)01217-4. [DOI] [PubMed] [Google Scholar]
- 26.Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M. FEBS Lett. 1999;443:93–96. doi: 10.1016/s0014-5793(98)01696-2. [DOI] [PubMed] [Google Scholar]
- 27.D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P. Am. J. Hum. Genet. 1999;64:414–421. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B. J. Neuropathol. Exp. Neurol. 1999;58:667–677. doi: 10.1097/00005072-199906000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Spillantini MG, Yoshida H, Rizzini C, Lantos PL, Khan N, Rossor MN, Goedert M, Brown J. Ann. Neurol. 2000;48:939–943. doi: 10.1002/1531-8249(200012)48:6<939::aid-ana17>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- 31.Tolnay M, Grazia Spillantini M, Rizzini C, Eccles D, Lowe J, Ellison D. Neuropathol. Appl. Neurobiol. 2000;26:368–378. doi: 10.1046/j.1365-2990.2000.00109.x. [DOI] [PubMed] [Google Scholar]
- 32.Yasuda M, Takamatsu J, D'Souza I, Crowther RA, Kawamata T, Hasegawa M, Hasegawa H, Spillantini MG, Tanimukai S, Poorkaj P, Varani L, Varani G, Iwatsubo T, Goedert M, Schellenberg DG, Tanaka C. Ann. Neurol. 2000;47:422–429. [PubMed] [Google Scholar]
- 33.Lippa CF, Zhukareva V, Kawarai T, Uryu K, Shafiq M, Nee LE, Grafman J, Liang Y, St. George-Hyslop PH, Trojanowski JQ, Lee VM. Ann. Neurol. 2000;48:850–858. [PubMed] [Google Scholar]
- 34.Pastor P, Pastor E, Carnero C, Vela R, Garcia T, Amer G, Tolosa E, Oliva R. Ann. Neurol. 2001;49:263–267. doi: 10.1002/1531-8249(20010201)49:2<263::aid-ana50>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 35.Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, Xuereb JH, Crowther RA, Spillantini MG. J. Neuropathol. Exp. Neurol. 2000;59:990–1001. doi: 10.1093/jnen/59.11.990. [DOI] [PubMed] [Google Scholar]
- 36.Neumann M, Schulz-Schaeffer W, Crowther RA, Smith MJ, Spillantini MG, Goedert M, Kretzschmar HA. Ann. Neurol. 2001;50:503–513. doi: 10.1002/ana.1223. [DOI] [PubMed] [Google Scholar]
- 37.Iseki E, Matsumura T, Marui W, Hino H, Odawara T, Sugiyama N, Suzuki K, Sawada H, Arai T, Kosaka K. Acta Neuropathol. 2001;102:285–292. doi: 10.1007/s004010000333. [DOI] [PubMed] [Google Scholar]
- 38.Miyamoto K, Kowalska A, Hasegawa M, Tabira T, Takahashi K, Araki W, Akiguchi I, Ikemoto A. Ann. Neurol. 2001;50:117–120. doi: 10.1002/ana.1083. [DOI] [PubMed] [Google Scholar]
- 39.Rosso SM, van Herpen E, Deelen W, Kamphorst W, Severijnen LA, Willemsen R, Ravid R, Niermeijer MF, Dooijes D, Smith MJ, Goedert M, Heutink P, van Swieten JC. Ann. Neurol. 2002;51:373–376. doi: 10.1002/ana.10140. [DOI] [PubMed] [Google Scholar]
- 40.Dayanandan R, Van Slegtenhorst M, Mack TG, Ko L, Yen SH, Leroy K, Brion JP, Anderton BH, Hutton M, Lovestone S. FEBS Lett. 1999;446:228–232. doi: 10.1016/s0014-5793(99)00222-7. [DOI] [PubMed] [Google Scholar]
- 41.DeTure M, Ko LW, Yen S, Nacharaju P, Easson C, Lewis J, van Slegtenhorst M, Hutton M, Yen SH. Brain Res. 2000;853:5–14. doi: 10.1016/s0006-8993(99)02124-1. [DOI] [PubMed] [Google Scholar]
- 42.Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M. J. Biol. Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 43.Varani L, Spillantini MG, Goedert M, Varani G. Nucleic Acids Res. 2000;28:710–719. doi: 10.1093/nar/28.3.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cote J, Dupuis S, Jiang Z, Wu JY. Proc. Natl. Acad. Sci. U. S. A. 2001;98:938–943. doi: 10.1073/pnas.031564098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tacke R, Chen Y, Manley JL. Proc. Natl. Acad. Sci. U. S. A. 1997;94:1148–1153. doi: 10.1073/pnas.94.4.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tacke R, Tohyama M, Ogawa S, Manley JL. Cell. 1998;93:139–148. doi: 10.1016/s0092-8674(00)81153-8. [DOI] [PubMed] [Google Scholar]
- 47.Seiwert SD, Steitz JA. Mol. Cell. Biol. 1993;13:3134–3145. doi: 10.1128/mcb.13.6.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4113–4118. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker M, Kwok JB, Kucera S, Crook R, Farrer M, Houlden H, Isaacs A, Lincoln S, Onstead L, Hardy J, Wittenberg L, Dodd P, Webb S, Hayward N, Tannenberg T, Andreadis A, Hallupp M, Schofield P, Dark F, Hutton M. Ann. Neurol. 1997;42:794–798. doi: 10.1002/ana.410420516. [DOI] [PubMed] [Google Scholar]
- 50.Iijima M, Tabira T, Poorkaj P, Schellenberg GD, Trojanowski JQ, Lee VM, Schmidt ML, Takahashi K, Nabika T, Matsumoto T, Yamashita Y, Yoshioka S, Ishino H. Neuroreport. 1999;10:497–501. doi: 10.1097/00001756-199902250-00010. [DOI] [PubMed] [Google Scholar]
- 51.Spillantini MG, Crowther RA, Goedert M. Acta Neuropathol. 1996;92:42–48. doi: 10.1007/s004010050487. [DOI] [PubMed] [Google Scholar]
- 52.Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, Yen SH. FEBS Lett. 1999;447:195–199. doi: 10.1016/s0014-5793(99)00294-x. [DOI] [PubMed] [Google Scholar]
- 53.Black DL. RNA. 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- 54.Fu XD. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- 55.Manley JL, Tacke R. Genes Dev. 1996;10:1569–1579. doi: 10.1101/gad.10.13.1569. [DOI] [PubMed] [Google Scholar]
- 56.Blencowe BJ. Trends Biochem. Sci. 2000;25:106–110. doi: 10.1016/s0968-0004(00)01549-8. [DOI] [PubMed] [Google Scholar]
- 57.Graveley BR. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Manley JL. RNA. 1995;1:335–346. [PMC free article] [PubMed] [Google Scholar]
- 59.Wu JY, Maniatis T. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 60.Zuo P, Manley JL. Proc. Natl. Acad. Sci. U. S. A. 1994;91:3363–3367. doi: 10.1073/pnas.91.8.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cote J, Simard MJ, Chabot B. Nucleic Acids Res. 1999;27:2529–2537. doi: 10.1093/nar/27.12.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bourgeois CF, Popielarz M, Hildwein G, Stevenin J. Mol. Cell. Biol. 1999;19:7347–7356. doi: 10.1128/mcb.19.11.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Selvakumar M, Helfman DM. RNA. 1999;5:378–394. doi: 10.1017/s1355838299981050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blencowe BJ, Bowman JA, McCracken S, Rosonina E. Biochem. Cell Biol. 1999;77:277–291. [PubMed] [Google Scholar]
- 65.Hastings ML, Krainer AR. Curr. Opin. Cell Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- 66.Hastings ML, Krainer AR. RNA. 2001;7:471–482. doi: 10.1017/s1355838201002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beil B, Screaton G, Stamm S. DNA Cell Biol. 1997;16:679–690. doi: 10.1089/dna.1997.16.679. [DOI] [PubMed] [Google Scholar]
- 68.Hoffman BE, Lis JT. Mol. Cell. Biol. 2000;20:181–186. doi: 10.1128/mcb.20.1.181-186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao QS, Memmott J, Lafyatis R, Stamm S, Screaton G, Andreadis A. J. Neurochem. 2000;74:490–500. doi: 10.1046/j.1471-4159.2000.740490.x. [DOI] [PubMed] [Google Scholar]
- 70.Jiang ZH, Zhang WJ, Rao Y, Wu JY. Proc. Natl. Acad. Sci. U. S. A. 1998;95:9155–9160. doi: 10.1073/pnas.95.16.9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dignam JD, Lebovitz RM, Roeder RG. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krainer AR, Maniatis T. Cell. 1985;42:725–736. doi: 10.1016/0092-8674(85)90269-7. [DOI] [PubMed] [Google Scholar]
- 73.Ruskin B, Krainer AR, Maniatis T, Green MR. Cell. 1984;38:317–331. doi: 10.1016/0092-8674(84)90553-1. [DOI] [PubMed] [Google Scholar]
- 74.Lapham J, Yu YT, Shu MD, Steitz JA, Crothers DM. RNA. 1997;3:950–951. [PMC free article] [PubMed] [Google Scholar]
- 75.Yu YT. Methods. 1999;18:13–21. doi: 10.1006/meth.1999.0752. [DOI] [PubMed] [Google Scholar]
- 76.Eperon IC, Ireland DC, Smith RA, Mayeda A, Krainer AR. EMBO J. 1993;12:3607–3617. doi: 10.1002/j.1460-2075.1993.tb06034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chabot B, Blanchette M, Lapierre I, La Branche H. Mol. Cell. Biol. 1997;17:1776–1786. doi: 10.1128/mcb.17.4.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zahler AM, Lane WS, Stolk JA, Roth MB. Genes Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 79.Elbashir SM, Lendeckel W, Tuschl T. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Daoud R, Mies G, Smialowska A, Olah L, Hossmann KA, Stamm S. J. Neurosci. 2002;22:5889–5899. doi: 10.1523/JNEUROSCI.22-14-05889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]