

Abstract
Homeobox genes of the Hox class are required for proper patterning of skeletal elements, but how they regulate the differentiation of specific tissues is unclear. We show here that overexpression of a Hoxc-8 transgene causes cartilage defects whose severity depends on transgene dosage. The abnormal cartilage is characterized by an accumulation of proliferating chondrocytes and reduced maturation. Since Hoxc-8 is normally expressed in chondrocytes, these results suggest that Hoxc-8 continues to regulate skeletal development well beyond pattern formation in a tissue-specific manner, presumably by controlling the progression of cells along the chondrocyte differentiation pathway. The comparison to Hoxd-4 and Isl-1 indicates that this role in chondrogenesis is specific to proteins of the Hox class. Their capacity for regulation of cartilage differentiation suggests that Hox genes could also be involved in human chondrodysplasias or other cartilage disorders.
Hox genes encode transcription factors that are important regulators of pattern formation during the development of the vertebrate skeleton (1, 2). In the mouse, the appearance of skeletal elements is determined by the expression of one or more Hox genes in a given body region (refs. 3–5, reviewed in ref. 6). On the basis of results from targeted gene disruptions in mice, it has been suggested that Hox genes control the condensation, proliferation, or differentiation of skeletogenic precursor cells (2). To investigate this hypothesis, we generated mice that express Hox transgenes specifically in skeletogenic cells.
Ectopic expression or overexpression of Hox genes in mice has previously been reported to result in defects in skeletal patterning, such as supernumerary ribs (7, 8), limb defects (9, 10), and homeotic transformations of vertebrae (7, 9, 11). However, little is known about the cellular and molecular basis for these abnormalities. A particular problem in investigating Hox-transgenic mice has been their premature lethality, which leaves only founder animals for analysis (for detailed discussions, see refs. 12 and 13). We have therefore employed a binary transgenic mouse system that provides a reproducible supply of genetically identical animals for morphological and histological analyses (14, 15).
METHOD
Transgenic Mice.
The transgenic mouse strains have been described elsewhere (14, 15). Genotyping was performed by semiquantitative PCR on DNA isolated from yolk sacs. Reaction mixtures consisted of genomic DNA at 320 ng/ml, dNTPs at 200 nM, and each primer at a final concentration of 20 μg/ml (primer sequences are described in ref. 15) and were performed in the presence of 5% dimethyl sulfoxide for up to 35 cycles under the following conditions: denaturation for 5 min at 94°C, annealing at 60°C for 1 min, extension at 72°C for 2 min, followed by denaturation for 1 min at 94°C. The intensity of ethidium bromide-stained bands on a 1% agarose gel (determined by using the histogram function in Adobe PhotoShop) was normalized to the DNA concentration for each sample and divided by the lowest value obtained. Samples from mice hemizygous for the transgene locus gave results around 1, while those from mice homozygous for a transgene locus gave results close to 2. The accuracy of these estimates was confirmed by crossing transgenic mice of various genotypes to wild-type animals and analyzing their progeny for segregation of the transgene loci (Y.G.Y. and C.K., unpublished work).
Preparation and Staining of Skeletons.
Cadavers of newborn mice were deskinned, eviscerated, and fixed in ethanol for 4 days and in acetone for 3 days. After rinsing with water, the specimens were incubated for 5 days in the staining solution: 0.0075% alizarin red/0.003% Alcian blue/5% acetic acid/82.5% ethanol (vol/vol). Skeletons were rinsed with water and transferred into 20% (vol/vol) glycerol/1% potassium hydroxide at 37°C until the tissue cleared, and then gradually into 100% ethanol for storage.
Immunohistochemistry and in Situ Hybridizations.
Immunoperoxidase staining for proliferating cell nuclear antigen (PCNA) was performed on deparaffinized, rehydrated sections of 10 μm thickness by using the antibody PC10 (Boehringer) at 50 μg/ml and the Vectastain Elite ABC and DAB substrate kits and following the manufacturer’s protocol (Vector). Sections were weakly counterstained with hematoxylin (Bio-Tek, Burlington, VT). In situ hybridizations were performed as described by Lee et al. (16).
RESULTS AND DISCUSSION
We used a binary transgenic mouse system, which enabled us (i) to achieve reproducible expression of homeobox transgenes in mice and (ii) to define transgene expression levels by virtue of combinations of transgene loci. This two-tiered system (14) (Fig. 1A) allows the activation of transgenes that are stably transmitted in silent form, thus circumventing the problem of premature lethality typical for Hox-transgenic mice. To drive the expression of VP16, we used the 5-kb upstream promoter fragment from the murine Hoxc-8 gene (17). The IE-LacZ transresponder (TR) gene indicates activity of the Hoxc-8 promoter in the growth zones of the skeleton at later stages of development (Fig. 1B) in cells of the chondrocyte lineage (Fig. 1 C–F). This pattern of expression reflects the expression of the endogenous Hoxc-8 gene in chondrocytes at 15.5 and 18.5 days of development (Fig. 1 G–J and data not shown). Some β-galactosidase activity was found more anterior to the normal domain of Hoxc-8 expression (18) in mesenchyme around the eye and the cranial cartilage. This staining may reflect a weak basal activity of the Hoxc-8 promoter in these tissues that was enhanced by the VP16 transactivator (TA) (unpublished observations). As the effector gene, we chose Hoxc-8, whose absence causes homeotic transformations (18), and which, upon ectopic expression, has been shown to alter skeletal patterning (8). The Hoxc-8 transgene was linked to a viral immediate early (IE) gene promoter. When both a TA and a TR transgene are present in the same individual, the IE-Hoxc-8 transgene will be transcriptionally activated by VP16 (15) (Fig. 1A).
Figure 1.
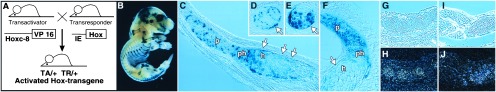
Expression of homeobox transgenes and Hoxc-8 in the developing skeleton. Our binary transgenic mouse system (A) is based on the potent viral transactivator VP16. The combination of two transgenes, namely the transactivator (TA) and a transresponder (TR) transgene, in the same individual leads to activation of the TR transgene. (B) Sites of IE-LacZ transgene activation indicated by β-galactosidase activity. VP16 expression under control of the Hoxc-8 promoter leads to TR gene expression in growth zones and cartilage of the skeleton at 17.5 days of development. (C) Section through a rib of the embryo in B showing staining for β-galactosidase in proliferating (p), prehypertrophic (ph), and some hypertrophic (h) chondrocytes. (D and E) Sections through two ribs at different planes of the respective growth zones with prehypertrophic cells (D) and hypertrophic cells (E) exhibiting β-galactosidase activity. (F) β-Galactosidase expression in proliferating cells in the prospective neural arch of a vertebra. Notably, there is no detectable transgene activation in perichondrial cells (arrows). (G–J) In situ hybridizations with a Hoxc-8 antisense probe to parasagittal sections of a mouse embryo at 15.5 days (G and H) and 18.5 days (I and J). Cells of the developing rib cartilage (bright-field exposure in G and I) express Hoxc-8, whereas the surrounding tissue is negative (H and J). Sense probe for Hoxc-8 gave no appreciable signal (not shown).
The consequences of activation of the Hoxc-8 transgene were found to increase in severity with the progressive combination of transgene loci (Table 1). Mice that inherited only TA or TR transgenes appeared normal at birth, in either hemizygous (TA/+) or homozygous (TA/TA) condition (TR/+ or TR/TR, respectively), indicating that either transgene alone did not interfere with development. Mice hemizygous for both transgenes (TA/+ TR/+) were slightly smaller and exhibited open eyes at birth, but they are viable and fertile. Crossing double transgenic mice (TA/+ TR/+ × TA/+ TR/+) produced lethality in approximately 25% of the progeny within a day after birth. The animals displayed increased flexibility and pliability, particularly in the lumbar and sacral regions, indicative of structural defects in their skeletons. Genotype analyses revealed that the lethality was associated with inheritance of both TA and TR transgene loci in excess of hemizygosity (TA/TA TR/+, or TA/+ TR/TR). Mice homozygous for both transgenes (TA/TA TR/TR) were found dead at birth. Thus, the severity of the phenotype induced by activation of Hoxc-8 was correlated with transgene dosage.
Table 1.
Phenotypes of TA and TR crosses
| Genotype | Phenotype | |
|---|---|---|
| Hoxc-8 transgene | ||
| TA/++/+ | Viable | |
| TA/TA+/+ | Viable | |
| +/+TR/+ | Viable | |
| +/+TR/TR* | Viable | |
| TA/+TR/+*† | Open eyes at birth, viable | |
| TA/+TR/TR† | Open eyes, neonatal lethal | |
| TA/TATR/+† | Open eyes, neonatal lethal | |
| TA/TATR/TR‡ | Open eyes, perinatal lethal | |
| Hoxd-4 transgene | ||
| +/+TR/+ | Viable | |
| TA/+TR/+‡ | Open eyes, neonatal lethal | |
| Isl-1 transgene | ||
| +/+TR/+ | Viable | |
| TA/+TR/+† | Posterior growth defect | |
All transgenes were on the FVB inbred background. In all TA mice used, VP16 expression is controlled by the Hoxc-8 promoter (14). The generation of TR strains was reported elsewhere (15). Results were obtained as follows: ∗, with two independent TA strains:
, with two independent TR stains; or
, with one combination of TA and TR strain.
Morphological analyses of the skeletons from newborn transgenic mice (Fig. 2) revealed the likely cause of death: Rib cartilage was soft and highly pliable (Fig. 2 B and D), suggesting that a lack of structural rigidity compromised lung inflation, resulting in pulmonary failure. Staining with Alcian blue, a marker for sulfated proteoglycans, was significantly reduced or absent in the cartilaginous parts of the ribs, the vertebrae, and in intervertebral spaces (Fig. 2 B and D). The defects in cartilage and the reduction in strength were most severe in the axial skeleton, indicating that abnormalities were predominant at sites of endochondral bone formation. The limb cartilage was defective in some animals (Fig. 2C), whereas cranial bones and the tail were always unaffected. This result could be due to regional differences in Hoxc-8 enhancer activity, and hence low levels of expression of the Hoxc-8 transgene. By virtue of β-galactosidase staining intensity, TR gene activation was found to be weaker in cranial and the most caudal cartilages (data not shown). Alternatively, the susceptibility of cells to perturbation by Hoxc-8 could vary with anatomical location. Detailed examinations of the thoracic (Fig. 2 E–H), lumbar (Fig. 2 I–L), and sacral (Fig. 2 M–P) regions revealed cartilage and bone abnormalities that correlated with transgene dosage, including the progressive loss of Alcian blue staining. Skeletons from double hemizygous neonates (TA/+ TR/+) were indistinguishable from wild-type skeletons (compare Fig. 2 A and E). In animals with higher transgene dosage, the vertebrae were abnormally ossified, and the neural arches were reduced. While the bony parts of the ribs seemed normal, the rib and axial cartilages were profoundly weakened, resulting in disassembly of the skeleton in the most affected animals (Fig. 2 H, L, and P). All mice had the expected number of skeletal elements, and Alcian blue staining revealed normal cartilaginous anlagen in transgenic embryos of all genotypes at 14.5 days of development (data not shown). Thus, although the initial formation of cartilage anlagen was normal in Hoxc-8 transgenic mice, these animals developed severe defects during cartilage differentiation at later stages of development. From these results, we concluded that increasing levels of Hoxc-8 expression affect cartilage differentiation in a dose-dependent manner.
Figure 2.

Cartilage abnormalities in Hox gene transgenic mice. Skeletons from newborn mice were stained with Alizarin red (bone) and Alcian blue (cartilage). (A–D) Cartilage defects upon overexpression of Hoxc-8 (B and C) or Hoxd-4 (D) transgenes. (A) Skeleton of a wild-type newborn FVB mouse. (B and C) Reduced Alcian blue staining in ribs and knee cartilage of a Hoxc-8 transgenic mouse that died shortly after birth. (D) Alcian blue staining was reduced in ribs and vertebral column of a Hoxd-4 transgenic animal. Note that the cartilaginous portions of the ribs were present (arrows in D, compare with B) and that tracheal cartilage was normal. (E–P) Severity of cartilage abnormalities increased with transgene dosage. The skeleton from an animal hemizygous for both TA and TR loci resembled staining of the wild-type situation (compare E to A). The thoracic (E–H), lumbar (I–L), and sacral regions (M–P) of the skeletons from animals with the genotypes TA/+ TR/+ (E, I, M), TA/+ TR/TR (F, J, N), TA/TA TR/+ (G, K, O), and TA/TA TR/TR (H, L, P) are shown. All animals with transgene loci in excess of hemizygosity died shortly after birth or were dead at birth. The distortion and flexibility of the skeleton were highest in the animal homozygous for both transgenes.
To determine if the role in cartilage differentiation was unique to Hoxc-8, we also expressed a Hoxd-4 transgene in our transgenic system. The protein sequences of Hoxc-8 and Hoxd-4 share 67% identity within the homeodomain and 50% in the hexapeptide motif but little similarity in the remaining 70% of the molecules. Hoxd-4-deficient mice develop abnormalities in cervical vertebrae C1 to C3 and the basioccipital bone (19), and ectopic expression of Hoxd-4 leads to defects in cranial bones (20), highlighting the patterning function of Hoxd-4. When we transactivated an IE-Hoxd-4 TR gene, we found abnormalities in the neonatal skeleton similar to those induced by Hoxc-8 (Fig. 2D). Thus, Hoxc-8 and Hoxd-4 appear to act on chondrocyte differentiation in a similar manner. The data could imply that both transcription factors regulate the same target gene(s) or that the forced expression of any homeodomain protein may alter chondrocyte differentiation. To control for the latter possibility, we used a homeobox-containing gene that is not known to be associated with skeletal development, the gene encoding Islet-1 (Isl-1). Isl-1 was originally cloned as an insulin-promoter-binding protein (21) and has been demonstrated to be required for the development of motor neurons and certain interneurons (22) as well as pancreatic cells (23). Isl-1 shares no significant sequence similarities with Hoxc-8 or Hoxd-4, and the presence of an additional metal-binding domain classifies Islet-1 as a structurally distinct homeodomain transcription factor. Upon activation of IE-Isl-1 TR genes with the same TA used before, we did not observe a cartilage phenotype (Table 1). Instead, Isl-1 transgenic mice exhibited abnormalities consistent with posterior growth defects (data not shown). The differences in the phenotypes generated by Isl-1, as opposed to Hoxc-8 and Hoxd-4, imply that the cartilage abnormalities were specifically induced by Hox genes.
To define the cellular basis for abnormal cartilage in Hoxc-8 transgenic mice, we histologically analyzed tissues obtained at 16.5 days of development (Fig. 3) from embryos of the genotypes TA/+ +/+ (Fig. 3 A, C, E, G, and I) and TA/TA TR/+ (Fig. 3 B, D, F, H, and J). Within the developing vertebrae of the embryo that inherited both transgenes, the hypertrophic cartilage was reduced, with prehypertrophic and hypertrophic cells being almost absent from the vertebral bodies and neural arches. Sulfated proteoglycans, a major component of cartilage, could be detected, but the staining with Alcian blue was markedly reduced in the abnormal cartilage (compare Fig. 3 E and F). In addition, there was generally less extracellular matrix, which may cause the weakness of cartilage at birth. The skeletal structures contained an increased number of cells that morphologically resembled proliferating chondrocytes. The accumulation of such cells was most profound in the developing vertebrae (Fig. 3 D, F, H, and J) and the ribs (not shown). Immunohistochemistry using antibody against proliferating cell nuclear antigen (PCNA/cyclin), a marker for proliferating chondrocytes (24) (Fig. 3 G–J), confirmed the proliferative phenotype of the accumulated cells. The increase in cell number could be accounted for almost entirely by the increase of proliferative cells (Fig. 3 K and L). Collectively, these data show that Hoxc-8 affects differentiation of cartilage in a way that leads to the accumulation of proliferating immature chondrocytes or chondrocyte precursor cells. The presence of modified cartilage and of ossified bone in the skeletons of transgenic newborns, however, indicates that Hoxc-8 does not completely block chondrogenesis. Rather, the level of Hoxc-8 expression modulates, in a dose-dependent fashion, the progression of chondrocytes along their differentiation pathway.
Figure 3.

Alterations in cartilage differentiation in Hoxc-8 transgenic mice. Embryos (genotypes TA/+ +/+ and TA/TA TR/+) were isolated at 16.5 days of development. Corresponding sections at the lumbar level were stained with hematoxylin/eosin (A–D), Alcian blue (E and F), or an antibody that detects proliferating cell nuclear antigen (PCNA; G–J). For enumeration of cells, photographs I and J were enlarged and every cell with a nucleus within the vertebral center was counted (K and L). (M and N) In situ hybridization for collagen II mRNA to sections from 17.5 day embryos (genotypes +/+ TR/+ and TA/TA TR/TR) that cut through more than one developing vertebra at the lower thoracic level. Both photographs were taken under identical exposure conditions. Sections hybridized with the sense probe were negative (not shown).
To define the specific stage at which Hoxc-8 interferes with chondrocyte differentiation, we performed in situ hybridizations using the expression of collagen II as a marker for immature chondrocytes (25), Indian hedgehog (ihh) as a marker for prehypertrophic and hypertrophic chondrocytes, and patched (ptc) as a marker for the perichondrium (26). The choice of the latter markers was motivated by a recent report implicating ihh and its receptor patched (27–30) in cartilage maturation (26). There were no significant differences between control and transgenic tissues in the expression of patched or ihh in the developing vertebrae at 17.5 days (data not shown). However, we found a marked increase in the expression of collagen II mRNA, a marker for immature chondrocytes, in terms of both cell number and signal intensity (compare Fig. 3 M and N). Taken together, our results identified the accumulated cells as proliferating, collagen II-expressing cells, hence, immature chondrocytes. Transcriptionally up-regulated expression of collagen II is associated with increased protein production (31), which in turn has been shown to disrupt collagen fibril formation (32). Thus, the cartilage defect in our transgenic mice may result from increased collagen expression in combination with an overall decrease in extracellular matrix. The accumulation of proliferating chondrocyte precursor cells suggests that Hoxc-8 affects cartilage differentiation by regulating either the maintenance of cells in the precursor state or the entry of cells into or their progression along the chondrocyte differentiation pathway.
Several lines of evidence substantiate a role for Hoxc-8 in normal cartilage differentiation in the axial skeleton: (i) The Hoxc-8 gene is normally expressed in chondrogenic cells (Fig. 1 H and J), and we observed altered proliferation of those cells in Hoxc-8 transgenic mice; (ii) the Hoxc-8 promoter is active in chondrocytes and precursor cells (Fig. 1 C and F); and (iii) a lacZ gene in place of the endogenous Hoxc-8 locus is expressed in cartilage (18, 33). Further support comes from the analysis of limb defects in compound Hox gene mutants (4, 34, 35). The gradual size reduction of skeletal elements in the limb by cumulative loss of Hox gene function implicates Hox genes in growth control (4) in prechondrogenic condensations (34) as well as in chondroblasts (35). It is conceivable that the different stages of chondrogenesis are differentially susceptible to changes in Hox gene expression levels, emphasizing the importance of temporal regulation for Hox gene function. In this regard, it is important to note that our Hoxc-8 transgenic mice do not develop homeotic transformations as observed upon ectopic Hoxc-8 expression (8) or in Hoxc-8-deficient mice (18), which exhibit supernumerary ribs on the first lumbar vertebrae. This is best explained by considering that the experimental models target different temporal stages of Hoxc-8 expression. Three phases can be distinguished: (i) early expression throughout the posterior region of the embryo (days 8–10; refs. 14 and 36); (ii) a more localized domain of expression with strict anterior boundaries in the neural tube and mesoderm (from day 10.5 on; refs. 17 and 37); and (iii) restricted expression during cartilage and tissue differentiation (this work and unpublished observations). The first phase is not affected by either gain or loss of Hoxc-8 function (8, 18). It is the second phase in which Hoxc-8 is required for patterning in Hoxc-8 mutants, but there is no specific dosage requirement as heterozygotes develop normally (18). In addition, the localization of defects to only a portion of the Hoxc-8 expression domain argues for functional complementation by other Hox genes (1, 18). The interpretation of phenotypes induced by ectopic Hoxc-8 (8) is complicated in that the use of a heterologous promoter combines ectopic presence of Hoxc-8 in cells where it is not normally expressed with altered temporal regulation in its own domain. This affects both patterning and the growth of ribs (38). In contrast, in our transgenic system, Hoxc-8 is activated under control of its own promoter in cells where the endogenous Hoxc-8 gene is also expressed. In this condition, even with overexpression of the transgene in the early and intermediate phases (14, 15), the critical stage of susceptibility to Hoxc-8 overexpression appears to be the immature proliferating chondrocyte. Since, within a given structure, there is mosaicism of Hoxc-8 nonexpressing and expressing cells (ref. 33; see also Fig. 1 C–F), the particular stage of their differentiation becomes an important factor for Hox gene function. These considerations suggest that Hoxc-8 (and possibly other Hox genes) may exert different roles at different developmental time points, affecting patterning initially, and, subsequently, cell differentiation.
Collectively, our results demonstrate that Hoxc-8 can regulate chondrocyte differentiation at the level of the proliferating chondrocyte or its immediate precursor (Fig. 4). This places Hoxc-8 upstream of the Indian hedgehog signaling pathway, which acts primarily at the level of prehypertrophic chondrocytes (26) and controls the progression to hypertrophy. It remains to be investigated whether Hoxc-8 (and Hoxd-4) actively promote proliferation of cells or if they inhibit differentiation of chondrocytes (39). In both situations, the rate of chondrocyte differentiation could be modulated by the repertoire of various Hox proteins in chondrogenic cells. A testable prediction from our transgenic model is that higher concentrations of Hox transcription factors would favor proliferation, whereas a reduction would stimulate differentiation of chondrocytes.
Figure 4.

Model for the regulation of cartilage differentiation by Hoxc-8. Hoxc-8 leads to accumulation of proliferating precursor cells, thus interfering negatively with the progression of differentiation. The signaling molecule Indian hedgehog (ihh) has been shown to negatively modulate the rate of differentiation at the level of prehypertrophic cells. Because patched, the receptor for ihh, is expressed in the adjoining perichondrium, this signalling pathway acts indirectly (broken line) on chondrocyte differentiation (26). Hoxc-8 is expressed in and directly affects chondrocytes prior to the prehypertrophic stage upstream of ihh.
In summary, our data provide evidence that Hox genes are involved in regulating the progression of cells along the chondrogenic differentiation pathway after the initial formation of the cartilage anlagen. We demonstrate that Hoxc-8, by virtue of its role in chondrocyte differentiation, is involved in tissue-specific gene regulation. While the factors that control Hox gene expression in cartilage have yet to be identified, our results establish the functional importance of Hox genes, beyond the process of pattern formation, in the regulation of chondrocyte differentiation.
Acknowledgments
We thank B. de Crombrugghe, M. Scott, C. Tabin, and A. Vortkamp for probes; A. Jennings for sectioning; T. Tinder for in situ hybridizations; M. Ruona and J. Jensen for the composition of figures; and S. Cormier-Regard, J. Loftus, C. H. Rundle, J. M. Salbaum, P. J. Yaworsky, and D. Zacharias for comments on the manuscript. This work was supported in part by a grant from the Arthritis Foundation (to C.K.) and by the Mayo Foundation for Medical Education and Research.
ABBREVIATIONS
- TA
transactivator
- TR
transresponder
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Krumlauf R. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 2.Capecchi M R. Ann NY Acad Sci. 1996;785:34–37. doi: 10.1111/j.1749-6632.1996.tb56241.x. [DOI] [PubMed] [Google Scholar]
- 3.Horan G S B, Ramirez-Solis R, Featherstone M S, Wolgemuth D J, Bradley A, Behringer R R. Genes Dev. 1995;9:1667–1677. doi: 10.1101/gad.9.13.1667. [DOI] [PubMed] [Google Scholar]
- 4.Davis A P, Capecchi M R. Development (Cambridge, UK) 1996;122:1175–1185. doi: 10.1242/dev.122.4.1175. [DOI] [PubMed] [Google Scholar]
- 5.Favier B, Rijli F M, Fromental-Ramain C, Fraulob V, Chambon P, Dolle P. Development (Cambridge, UK) 1996;122:449–460. doi: 10.1242/dev.122.2.449. [DOI] [PubMed] [Google Scholar]
- 6.Rijli F M, Chambon P. Curr Opin Genet Dev. 1997;7:481–487. doi: 10.1016/s0959-437x(97)80074-3. [DOI] [PubMed] [Google Scholar]
- 7.Jegalian B G, DeRobertis E M. Cell. 1992;71:901–910. doi: 10.1016/0092-8674(92)90387-r. [DOI] [PubMed] [Google Scholar]
- 8.Pollock R A, Jay G, Bieberich C J. Cell. 1992;71:911–923. doi: 10.1016/0092-8674(92)90388-s. [DOI] [PubMed] [Google Scholar]
- 9.Charite J, de Graaff W, Shen S B, Deschamps J. Cell. 1994;78:589–601. doi: 10.1016/0092-8674(94)90524-x. [DOI] [PubMed] [Google Scholar]
- 10.Knezevic V, DeSanto R, Schughart K, Huffstadt U, Chiang C, Mahon K A, Mackem S. Development (Cambridge, UK) 1997;124:4523–4536. doi: 10.1242/dev.124.22.4523. [DOI] [PubMed] [Google Scholar]
- 11.Kessel M, Balling R, Gruss P. Cell. 1990;61:301–308. doi: 10.1016/0092-8674(90)90810-2. [DOI] [PubMed] [Google Scholar]
- 12.Byrne G W, Ruddle F H. Proc Natl Acad Sci USA. 1989;86:5473–5477. doi: 10.1073/pnas.86.14.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrne G W, Kappen C, Schughart K, Utset M, Bogarad L, Ruddle F H. Biotechnology. 1991;16:135–152. [PubMed] [Google Scholar]
- 14.Gardner D P, Byrne G W, Ruddle F H, Kappen C. Transgenic Res. 1996;5:37–48. doi: 10.1007/BF01979920. [DOI] [PubMed] [Google Scholar]
- 15.Rundle, C. H., Macias, M. P., Gardner, D. P., Yueh, Y. G. & Kappen, C. (1998) Biochim. Biophys. Acta, in press. [DOI] [PubMed]
- 16.Lee J J, Radice G, Perkins C P, Costantini F. Development (Cambridge, UK) 1992;115:277–288. doi: 10.1242/dev.115.1.277. [DOI] [PubMed] [Google Scholar]
- 17.Bieberich C J, Utset M F, Awgulewitsch A, Ruddle F H. Proc Natl Acad Sci USA. 1990;87:8462–8466. doi: 10.1073/pnas.87.21.8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LeMouellic H, Lallemand Y, Brulet P. Cell. 1992;69:251–264. doi: 10.1016/0092-8674(92)90406-3. [DOI] [PubMed] [Google Scholar]
- 19.Horan G S B, Kovacs E N, Behringer R R, Featherstone M S. Dev Biol. 1995;169:359–372. doi: 10.1006/dbio.1995.1150. [DOI] [PubMed] [Google Scholar]
- 20.Lufkin T, Mark M, Hart C P, Dolle P, LeMeur M, Chambon P. Nature (London) 1992;359:835–841. doi: 10.1038/359835a0. [DOI] [PubMed] [Google Scholar]
- 21.Karlsson O, Thor S, Norberg T, Ohlsson H, Edlund T. Nature (London) 1990;344:879–882. doi: 10.1038/344879a0. [DOI] [PubMed] [Google Scholar]
- 22.Pfaff S L, Mendelsohn M, Stewart C L, Edlund T, Jessell T M. Cell. 1996;84:309–320. doi: 10.1016/s0092-8674(00)80985-x. [DOI] [PubMed] [Google Scholar]
- 23.Ahlgren U, Pfaff S L, Jessell T M, Edlund T, Edlund H. Nature (London) 1997;385:257–260. doi: 10.1038/385257a0. [DOI] [PubMed] [Google Scholar]
- 24.Amizuka N, Henderson J E, Hishi K, Warshawsky H, Ozawa H, Goltzman D, Karaplis A C. Endocrinology. 1996;137:5055–5067. doi: 10.1210/endo.137.11.8895380. [DOI] [PubMed] [Google Scholar]
- 25.Sandell L J, Morris N, Robbins J R, Goldring M B. J Cell Biol. 1991;114:1307–1319. doi: 10.1083/jcb.114.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vortkamp A, Lee K, Lanske B, Segre G V, Kronenberg H M, Tabin C. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, Struhl G. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- 28.Goodrich L V, Johnson R L, Milenkovic L, McMahon J A, Scott M P. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- 29.Marigo V, Davey R A, Zuo Y, Cunningham J M, Tabin C J. Nature (London) 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- 30.Stone D M, Hynes M, Armanini M, Swanson T A, Gu Q, Johnson R L, Scott M P, Pennica D, Goddard A, Phillips H, Noll M, Hooper J E, de Sauvage F, Rosenthal A. Nature (London) 1996;384:129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 31.Sandell L J. Microsc Res Tech. 1994;28:470–482. doi: 10.1002/jemt.1070280603. [DOI] [PubMed] [Google Scholar]
- 32.Garofalo S, Metsaranta M, Ellard J, Smith C, Horton W, Vuorio E, de Crombrugghe B. Proc Natl Acad Sci USA. 1993;90:3825–3829. doi: 10.1073/pnas.90.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tiret L, LeMouellic H, Lallemand Y, Maury M, Brulet P. C R Acad Sci Ser III. 1993;316:1017–1024. [PubMed] [Google Scholar]
- 34.Fromental-Ramain C, Warot X, Messadecq N, LeMeur M, Dolle P, Chambon P. Development (Cambridge, UK) 1996;122:2997–3011. doi: 10.1242/dev.122.10.2997. [DOI] [PubMed] [Google Scholar]
- 35.Zakany J, Duboule D. Nature (London) 1996;384:69–71. doi: 10.1038/384069a0. [DOI] [PubMed] [Google Scholar]
- 36.Shashikant C S, Bieberich C J, Belting H-G, Wang J C H, Borbely M A, Ruddle F H. Development (Cambridge, UK) 1995;121:4339–4347. doi: 10.1242/dev.121.12.4339. [DOI] [PubMed] [Google Scholar]
- 37.Breier G, Dressler G R, Gruss P. EMBO J. 1988;7:1329–1336. doi: 10.1002/j.1460-2075.1988.tb02948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sreenath T L, Pollock R A, Bieberich C J. Proc Natl Acad Sci USA. 1996;93:9636–9640. doi: 10.1073/pnas.93.18.9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duboule D. Curr Opin Genet Dev. 1995;5:525–528. doi: 10.1016/0959-437x(95)90058-o. [DOI] [PubMed] [Google Scholar]