

Abstract
A new method for predicting protein-coding regions in microbial genomic DNA sequences is presented. It uses an ab initio iterative Markov modeling procedure to automatically perform the partition of genomic sequences into three subsets shown to correspond to coding, coding on the opposite strand, and noncoding segments. In contrast to current methods, such as genemark [Borodovsky, M. & McIninch, J. D. (1993) Comput. Chem. 17, 123–133], no training set or prior knowledge of the statistical properties of the studied genome are required. This new method tolerates error rates of 1–2% and can process unassembled sequences. It is thus ideal for the analysis of genome survey and/or fragmented sequence data from uncharacterized microorganisms. The method was validated on 10 complete bacterial genomes (from four major phylogenetic lineages). The results show that protein-coding regions can be identified with an accuracy of up to 90% with a totally automated and objective procedure.
The complete genome sequencing of Haemophilus influenzae (1) initiated a tremendous interest in bacterial genomics. At the end of 1997, 11 complete bacterial genomes had been published (1–11). In the meantime, the first eukaryote genome had also been completed (12), allowing global genomic studies between five major phylogenetic lineages (13). Venter’s group developed the now-popular direct shotgun approach (1) for the complete sequencing of megabase-sized genomes. An essential feature of this strategy is that no prior knowledge of the genome is required. As a consequence, most of the numerous genomes (more than 40) targeted for sequencing in the near future (see The Institute for Genomic Research microbial database at: http://www.tigr.org/tdb/mdb/mdb.html) correspond to relatively uncharacterized microorganism species. In particular, the public databases do not contain any protein-coding gene sequences for most of these organisms.
In contrast, all current computer methods for locating genes require some prior knowledge of the sequence statistical properties (such as codon usage, positional preference, etc., see review in ref. 14) that have to be estimated from previously identified protein-coding genes of the microorganism. For instance, genemark (15, 16), the most popular program today, requires a sizable training set for estimating the numerous parameters of a nonhomogeneous seven-state Markov model of up to order 4. Although a general method derived from basic principles would have obvious advantages, training set- based methods presently dominate the field of gene identification (17). Those methods are inherently conservative. Once trained and optimized on a set of “typical” genes, these programs tend to be successful at only detecting more of the same. Furthermore, training set-dependent programs may also perpetuate biases unknowingly introduced in the original data set. The main goal of complete genome sequencing, i.e., an exhaustive gene annotation eventually leading to the discovery of surprising and atypical features, thus is not well served by this type of approach. In addition, the fundamental interest in trying to decipher the genomic information in an objective way is also to identify the truly universal and biologically significant signals, transcending the peculiarities of any given organism. For the same reason, it is desirable to avoid the use of too many adjustable parameters and empirical thresholds, a weakness of current gene-recognition programs.
Typical bacterial genes correspond to ORFs spanning an average of 950 nt. Because ORFs longer than 300 nt are very unlikely to occur by chance (18), most coding regions can be located within an assembled genomic sequence by simply sieving on ORF size. However, there is an increasing demand for a new method capable of identifying most coding regions in the context of inexpensive genome survey projects [1- to 2-fold redundancy shotgun sequencing (19)]. The challenge then becomes to analyze sequence data distributed between a multitude of small islands of one or two individual gel readings with a typical 1–2% error rate.
The original method presented here predicts coding regions without learning species-specific features from an arbitrary training set. Its accuracy is comparable to the popular program genemark (15). In addition, our method can work on totally unassembled sequence data and tolerate a simulated error rate of 1–2%. Finally, the method involves only two parameters (a window size, w, and a Markov chain order, k), the optimal values of which are fixed once and do not depend on the bacterial genome being analyzed. The method has been successfully tested on 10 complete genomes, including species from the four major phylogenetic lineages (Gram-negative, Gram-positive, cyanobacteria, archebacteria). Given the differences (in size, composition, G+C content, etc.) in the properties of those genomes, it is likely that the method will work equally well on any of the bacteria to be studied in the future.
METHODS
Homogeneous Markov Modeling.
The procedure starts from an initial data set consisting of all the genomic sequences available for a given organism. It could be a complete and fully assembled sequence, a number of large contigs, or the many disjoined sequence fragments resulting from a low-redundancy shotgun sequencing project. The driving principle of our algorithm is to regroup similar sequences into the same categories. However, unlike most clustering/partition procedures used in sequence analysis, our approach does not involve pairwise comparisons. Instead, the sequences will be distributed between N classes on the basis of their best match to N homogeneous Markov models. Markov chains often have been used to model DNA sequences (see, for instance, refs. 20–23). Given a word of length k (the order of the Markov chain), a Markov transition matrix consists of all the probabilities for any of the possible k words (4k of them) to be followed by one of nucleotides A, C, G, or T. The probability for a specific DNA sequence W of length L to be emitted by a given Markov transition matrix M is given by:
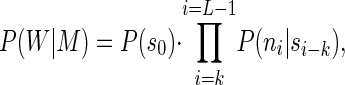 |
1 |
where si is the word of length k starting at position i in the sequence W, and ni is the nucleotide occurring at position i.
Markov models traditionally are used in sequence analysis for the purpose of mapping distinct (nonoverlapping), functional domains (coding/noncoding regions, promoters, etc.). In this context, several Markov transition matrices are used, one for each of the domains to be recognized. Among N competing Markov models, the problem is now to determine which one is the most likely to have emitted a given input sequence. This is done by inverting Eq. 1 using Bayes’ theorem:
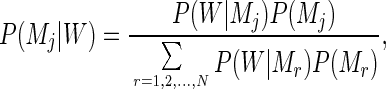 |
2 |
where P(Mj) is the probability of the matrix Mj to correspond to any sequence before input. In the following, the N classes have been taken as a priori equiprobable; that is, P(Mj) = 1/N. We can now use Eq. 2 to classify (or partition) any new input sequence into the best matching of the N functional categories. For the specific purpose of identifying protein-coding regions in bacterial genomic sequences, a natural number of nonoverlapping classes is n = 3, corresponding to sequence regions (i) coding on the direct strand, (ii) coding on the complementary strand (reverse coding), and (iii) noncoding sequences. Three distinct, homogeneous Markov models then will be used (i.e., n = 3 in Eq. 2) for which we now have to build three transition matrices. It is usually at this stage that the notion of “training sets” would come into play. Classically, sequences previously known to belong to one of the three classes would be collected in three training subsets, and the three Markov transition matrices would be computed from their respective, hopefully different, statistical properties. Here, we want to show that an alternative procedure is possible, leading to a fully objective way of analyzing bacterial genomes in the absence of any prior functional (or database similarity) information. To avoid the use of training sets, the task of annotating the sequence will be treated as a more abstract, optimal partition problem. A concept of “similarity” between objects is required to regroup them into different classes. Here, two sequence segments will be considered “similar” (denoted “≈”) and as belonging to the same j class, if they are best recognized (on average, over a minimal window length w) by the same transition matrix Mj: W1 ≈ W2 if both P(Mj|W1) and P(Mj|W2) are maximal.
The Markov model Mj of the j class then will be computed from the statistical features of the subset of j-type sequences. Indeed, this definition is self-referential and, at first, does not alleviate the need for three training sequence subsets corresponding to the j (j = 1, 2, 3) classes. However, we used a simple iterative procedure to overcome this problem. The whole data set of genomic sequence (assembled or not) first was randomly cut into nonoverlapping pieces w nucleotides in length (with w ≅ 100, see below). These pieces then are randomly distributed between three distinct subsets, from which three initial Markov transition matrices are built. The genomic sequence data then are scanned by using a sliding window of w nucleotides. Within each window, the most likely emitter (M1, M2, or M3) was determined according to Eq. 2. The window is then shifted over five positions, and the process is repeated. A decision about the classification of the current sequence segment occurs when two successive windows cease to be associated with the same Mj transition matrix. If the same Mj has been called for n (or more) successive windows (such as 5n ≥ w), the largest, locally j-consistent segment (from the middle point of the first window to the middle point of the last consistent one) is collected into the j data set. Otherwise, the current sequence segment remains unclassified. Requiring the same Markov transition matrix to be the best match over the entire window length alleviates the need for an arbitrary probability threshold. After completion of the analysis of the whole genomic sequence data, three new Markov transition matrices are built from each of the three data sets, and the procedure is reiterated. Convergence is reached when the size (and content) of each of the j data sets does not vary significantly (<0.5%) from one iteration to the next.
This simple algorithm exhibited two essential features. First, convergence was obtained quite rapidly, usually after 50 iterations or less (Fig. 1). Second, from any initial random partitioning of the data, the procedure kept converging to the same Markov transition matrices and j data set content. This suggested that the three classes defined by the algorithms could have a functional interpretation. To proceed from the abstract three-way partition performed by the iterative algorithm to the practical assignment of the three classes as “coding,” “reverse coding,” or “noncoding” requires a supplementary step establishing the correct functional correspondence. A possible approach might consist of searching for similarities between known protein sequences and the content of the three data sets. The data set corresponding to the noncoding sequences should exhibit the fewest protein similarities, whereas the orientation of the matches from the two other data sets will indicate which one corresponds to the coding (direct strand matches) or reverse-coding (opposite strand matches) regions. The next section describes a better approach, in which the functional assignment of the three classes of sequences is performed without using prior information and is combined with a significant refinement of the original partition.
Figure 1.

Convergence of the iterative homogeneous Markov modeling. The numbers of nucleotides correctly assigned as “coding” or “reverse coding” are plotted to follow the convergence of the iterative procedure. (A) Influence of the Markov chain order. (B) Influence of the window size. (C) Influence of the simulated error rate. (D) Specificity of the recognition of coding (+) and reverse coding (o) segments for 10 genomes of different G+C content. Mj, M. jannaschii; Mg, M. genitalium; Mp, M. pneumoniae; Hi, H. influenzae; Hp, H. pylori; Bs, B. subtilis; Mt, M. thermoautotrophicum; Syn, Synechocystis sp.; Af, A. fulgidus; Ec, E. coli. The discrepancies between the recognition of coding and reverse-coding regions in the Mg and Mp genomes indicate an actual strand asymmetry.
Inhomogeneous Markov Model Refinement.
The functional attributes (coding, reverse coding, and noncoding) are determined as follows. For each of the three data sets obtained after convergence of the homogeneous Markov modeling, the fraction of sequences totally open in reading frames 1, 2, 3, and −1, −2, −3 is computed. The data set for which the fraction of totally open sequences on the direct (respectively opposite) strand is maximal then is equated to the “coding” (respectively “reverse coding”) class. The remaining data set is equated to the noncoding class. The assignment of functional classes thus is done objectively, without using protein similarity. The only information needed at this point is the proper genetic code to apply.
Once the coding, reverse coding, and noncoding sequence subsets are identified, they can be optimized according to a theoretical framework well established by Borodovsky and McIninch (15) and implemented in the popular genemark program (15, 16). Briefly, seven Markov transition matrices are built, three for capturing the phase-dependent (in frame, in frame +1, in frame +2) statistical regularities of coding regions, three for capturing the phase-dependent statistical regularities of reverse coding (also referred to as “shadow”) regions, and one for modeling noncoding regions. The construction of these matrices from sets of examples has been described previously in detail (15, 16). It is referred to as “inhomogeneous Markov modeling,” to emphasize that different transition matrices M1, M2, and M3 (or M−1, M−2, or M−3) have to be used in a proper phase-dependent context to properly model the coding and reverse-coding regions.
The iterative procedure that we described above using three homogeneous Markov transitions matrices can now be extended as follows. From the data sets assigned to the coding and reverse-coding classes we extracted the sequences containing a single totally opened ORF. Alternatively, the presence of statistically significant ORFs (i.e., >300 nt) can be used. These unambiguous ORFs then were used to compute the starting set of transition matrices M1, M2, and M3. Matrices M−1, M−2, and M−3 were computed similarly from unambiguous ORFs from the reverse-coding class. The noncoding transition matrix M7 was computed from all sequences of the noncoding class. These seven matrices were then used to reanalyze the original genomic data, using a window of w nucleotides, according to the inhomogeneous (i.e., phase-dependent) Markov model procedure. The analysis was repeated after sliding the window over six positions. For a sequence segment to be classified in the j class (Mj being the most likely emitter, starting at position 1), we required the Mj Markov model to be consistently verified over n (or more) successive windows (now chosen such as 6n ≥ w, for an easy implementation of the phase-dependent Markov analysis). The locally j-consistent segment then is collected into the j data set (e.g., coding region in phase j, or noncoding region). As before, segments failing the consistency test were left unclassified. The analysis of the input sequence then resumed with a new analysis window shifted over six positions. Upon completion of the genomic data scan, the data sets corresponding to the three coding classes are used to compute the next set of M1, M2, and M3 transition matrices. The data sets corresponding to the three reverse-coding classes are used similarly to compute the M−1, M−2, and M−3 matrices. The noncoding transition matrix also is actualized. As before, the iteration is finally ended when the content of the seven data sets becomes stable. Again, the convergence was fast (less than 50 iterations) for the 10 bacterial genomes tested (Table 2).
Table 2.
Results of the refined partition procedure
| Total predicted | Coding | Reverse coding | Other |
|---|---|---|---|
| Haemophilus influenzae (1,830,140 nt) | |||
| 1,651,409 nt, 90% | 744,614 nt | 775,845 nt | 317,850 nt |
| C+ pred. (702,129 nt) | 636,824 (91%) | 1,197 | 64,108 |
| C− pred. (712,993 nt) | 1,579 | 662,504 (93%) | 48,910 |
| No pred. (236,287 nt) | 43,277 | 42,475 | 150,535 |
| Methanococcus jannaschii (1,664,977 nt) | |||
| 1,487,006 nt, 89% | 759,425 nt | 679,908 nt | 225,644 nt |
| C+ pred. (699,314 nt) | 661,740 (95%) | 2,897 | 34,677 |
| C− pred. (620,763 nt) | 6,173 | 587,600 (95%) | 26,990 |
| No pred. (166,929 nt) | 19,057 | 31,841 | 116,031 |
| Synechocystis PCC6803 (3,573,470 nt) | |||
| 3,236,025 nt, 91% | 1,621,700 nt | 1,471,880 nt | 479,890 nt |
| C+ pred. (1,324,169 nt) | 1,279,612 (97%) | 3,109 | 41,448 |
| C− pred. (1,217,840 nt) | 5,604 | 1,178,788 (97%) | 33,448 |
| No pred. (694,016 nt) | 191,933 | 175,121 | 326,962 |
| Escherichia coli (4,638,858 nt) | |||
| 4,214,577 nt, 91% | 1,994,205 nt | 2,084,634 nt | 560,019 nt |
| C+ pred. (1,610,214 nt) | 1,582,668 (98%) | 2,313 | 25,233 |
| C− pred. (1,705,110 nt) | 8,103 | 1,665,355 (98%) | 31,652 |
| No pred. (899,253 nt) | 214,115 | 238,100 | 447,038 |
| Helicobacter pylori (1,667,867 nt) | |||
| 1,543,591 nt, 93% | 722,915 nt | 780,576 nt | 164,376 nt |
| C+ pred. (655,117 nt) | 634,753 (97%) | 658 | 19,706 |
| C− pred. (690,480 nt) | 2,135 | 663,511 (96%) | 24,834 |
| No pred. (197,994 nt) | 34,574 | 57,019 | 106,401 |
| Mycoplasma pneumoniae (816,394 nt) | |||
| 754,571 nt, 92% | 299,312 nt | 414,027 nt | 103,055 nt |
| C+ pred. (313,976 nt) | 284,212 (91%) | 4,267 | 25,497 |
| C− pred. (407,335 nt) | 3,862 | 377,941 (93%) | 25,532 |
| No pred. (33,260 nt) | 1,317 | 15,027 | 16,916 |
| Mycoplasma genitalium (580,073 nt) | |||
| 557,245 nt, 96% | 285,729 nt | 226,875 nt | 73,180 nt |
| C+ pred. (284,455 nt) | 266,667 (94%) | 2,709 | 15,079 |
| C− pred. (241,256 nt) | 4,328 | 215,916 (90%) | 21,012 |
| No pred. (21,534 nt) | 2,665 | 18 | 18,851 |
| Bacillus subtilis (4,214,814 nt) | |||
| 3,683,449 nt, 87% | 1,797,237 nt | 1,877,565 nt | 540,012 nt |
| C+ pred. (1,491,612 nt) | 1,438,578 (96%) | 1,748 | 51,286 |
| C− pred. (1,495,245 nt) | 6,276 | 1,447,961 (97%) | 41,008 |
| No pred. (696,592 nt) | 127,671 | 209,008 | 359,833 |
| Archeoglobus fulgidus (2,178,400 nt) | |||
| 2,001,668 nt, 92% | 1,008,654 nt | 1,010,811 nt | 158,935 nt |
| C+ pred. (878,849 nt) | 838,321 (95%) | 5,242 | 35,286 |
| C− pred. (907,581 nt) | 14,113 | 870,252 (96%) | 23,216 |
| No pred. (215,238 nt) | 62,801 | 54,280 | 98,157 |
| Methanobacterium thermoautotrophicum (1,751,377 nt) | |||
| 1,636,136 nt, 93% | 777,122 nt | 810,068 nt | 164,187 nt |
| C+ pred. (690,437 nt) | 671,485 (97%) | 5,608 | 13,344 |
| C− pred. (697,590 nt) | 2,351 | 675,861 (97%) | 19,378 |
| No pred. (248,109 nt) | 49,280 | 84,271 | 114,558 |
Classified genome sequence segments are collected in three automatically defined data sets: C+ (predicted coding), C− (predicted reverse coding), and No (predicted noncoding). The number of nucleotides in these various data sets annotated in Genbank as “coding,” “reverse coding,” or “other” are indicated for 10 species.
RESULTS
The Three-Class Homogeneous Markov Partition Identifies Protein-Coding Regions.
Table 1 summarizes the result of the partition of the H. pylori (Gram-negative eubacteria, 1.67 Mb) and M. jannaschii (anaerobic archebacteria, 1.67 Mb) genome sequences using the ab initio homogeneous Markov modeling (k = 5, w = 100) without further refinement. Upon convergence, 82% and 84% of the total nucleotides of H. pylori and M. jannaschii are respectively distributed between three data sets (DB1, DB2, DB3) corresponding to the sequence segments with a consistent best match to the three Markov transition matrices built by the procedure. The association between a data set and a coding status is a priori unknown and varies from one experiment to the next. The correlation between the abstract partition in three classes and the coding, reverse coding, and noncoding functional classes was computed by reference to GenBank (24) annotations. In Table 1, the iteration for H. pylori mostly collected coding segments (95%) in the DB1 data sets and reverse-coding segments (94%) in DB3. For M. jannaschii, the iteration shown collected coding segments (93%) in DB3 and reverse-coding segments (93%) in DB1. The cross-contamination between coding regions in opposite orientation is very small. Starting from an initial window size of 100 nt, the final average lengths of the segments of the coding/reverse-coding classes are 328/330 for H. pylori and 410/516 for M. jannaschii. For both organisms, the third and smallest data set, DB2, collected a mixture of noncoding (i.e., unannotated), coding, and reverse-coding segments. Thus, the correspondence between this third data set and the noncoding class is weaker. Noncoding regions consist of regulatory sequences mixed with true intergenic regions and might be less statistically homogeneous. The results shown in Table 1 are typical of all microbial genomes we analyzed so far.
Table 1.
Performance of the iterative homogeneous Markov partition
| Data sets | GenBank annotation | ||
|---|---|---|---|
| Helicobacter pylori (1,667,867 nt) | |||
| (+) Coding (722,915) | (−) Coding (780,576) | Other (164,376) | |
| DB1 (565,176) | 537,242 (95%) | 8710 (1.5%) | 19,224 (3.5%) |
| DB2 (254,346) | 50,294 (21.5%) | 110,225 (43.5%) | 93,827 (37%) |
| DB3 (544,572) | 9,152 (2%) | 513,606 (94%) | 21,814 (4%) |
| Methanococcus jannaschii (1,664,977 nt) | |||
| DB1 (553,666) | 6,997 (1%) | 514,406 (93%) | 32,263 (6%) |
| DB2 (187,380) | 28,258 (15%) | 57,650 (31%) | 101,472 (54%) |
| DB3 (665,857) | 619,818 (93%) | 4,877 (1%) | 41,162 (6%) |
Upon convergence, sequence segments are collected into three automatically defined data sets: DB1, DB2, and DB3. The sizes of these various data sets annotated as “coding” (+), “reverse coding” (−), or “other” in GenBank are indicated for H. pylori (top) and M. jannaschii (bottom). The numbers corresponding to the cognate DB/annotation matches are in bold. A total of 1,364,094 nt (82%) and of 1,406,903 nt (84.5%) are classified (i.e., collected in DB 1–3) for H. pylori and M. jannaschii, respectively.
Influence of the Window Size and Markov Chain Order on the Initial Sequence Partition.
Our procedure for the spontaneous partition of the data set in coding, reverse-coding, and noncoding sequences involves two parameters: the analysis window size w, and the order (k in Eq. 1) of the three Markov models to be built. Fig. 1A shows the H. pylori convergence behavior for k = 3, k = 4, and k = 5. More coding nucleotides are classified successfully as k increases. The convergence is satisfactory for k ≥ 4, with k = 5 resulting in a faster, more symmetrical, and more accurate partition. Markov models of order 5 are built from the frequencies of hexamers. The power of hexamer statistics to discriminate between coding and noncoding regions has been recognized previously (14, 25) and is confirmed here. Larger (k ≥ 6) Markov model orders do not result in significant improvement and may even cause convergence problems by lack of sufficient data to populate all transition matrix elements. The value k = 5 was then retained throughout this work. Another important parameter is the size w of the analysis window over which a consistent best match to one of the Markov models, Mj, is required for classification in the j class. Fig. 1B shows the H. pylori convergence behavior for w = 50, 100, and 200. A window of 100 allows more coding nucleotides to be classified successfully than a window of 50. However, using a window of 200 produced an uneven recognition between the coding and reverse-coding regions (this pathological behavior originates from the over-/underprediction of a class and also is encountered when using nonoptimal Markov orders). A window size of 100 nt thus was retained throughout this work.
Influence of Sequencing Errors.
The iterative homogeneous Markov modeling leads to an accurate detection of the protein-coding regions without explicitly involving the concept of an ORF. It is thus suitable for the automated annotation of sequence data in the context of a genome survey, where each position is determined only once or twice on average (19), precluding contigs much larger than individual runs (400–500 nt) to be assembled. However, our method will be practical only if it can handle the typical sequencing error rate associated with this approach. Fig. 1C compares the H. pylori convergence behavior of the method in the presence of 0, 1, 2, and 5% simulated sequence errors (point mutations, insertions, and deletions each amounting for a third of the total rate). The method is clearly able to handle a realistic 2% error rate without degradation in performances, whereas a rate of 5% induces the asymmetrical recognition pattern, indicative of a failure of the procedure as already seen for other nonoptimal parameter choices (Fig. 1 A and B).
Influence of the G+C Composition.
Because it relies on the existence of distinct hexamer vocabularies between coding and noncoding regions, the success of our iterative homogeneous Markov modeling procedure might be expected to depend on some statistical characteristics, such as the G+C composition. Fig. 1D shows the average specificity (predicted/annotated as coding) achieved by our method for each of the 10 available microbial genomes. A specificity above 90% is achieved for largely different (32–51%) G+C genome contents. There also is no obvious correlation between the size of the genome (or its gene density) and the accuracy of the coding region prediction. The mycoplasma genomes are atypical, because they exhibit a marked difference (92 vs. 76%) in the recognition of coding regions in opposite orientations. These results suggest a strand asymmetry that is already visible in the number of coding nucleotides annotated in the database: a strand difference of 30 and 20% for M. pneumoniae and M. genitalium, respectively. These variations probably are not a result of annotation errors because the numbers of putative ORFs are also different: 274 vs. 204 and 203 vs. 152 for ORFs spanning more than 600 nt in M. pneumoniae or M. genitalium, respectively. Thus, we interpret our results as suggesting that a significant fraction of the genes encoded on the low-coding density strand have a distinct statistical make-up that is less easily captured by homogeneous Markov models (albeit no significant difference was found in codon usage). Fortunately, the problem is corrected by the inhomogeneous Markov refinement (Table 2).
Performances After Inhomogeneous Markov Modeling.
Table 2 summarizes the results obtained on 10 bacterial genomes with the ab initio partition procedure used to seed the inhomogeneous Markov model refinement described in Methods. The improvement over the simpler iterative homogeneous Markov procedure is 2-fold. First, a larger fraction of the genomic sequence data is classified (from 84 to 89% for M. jannaschii, and from 82 to 93% for H. pylori). Second, the prediction specificity also is increased (from 93 to 95% for M. jannaschii, and from 94 to 96% for H. pylori). The best specificity (98%) is found for Escherichia coli, probably the most reliably annotated genome. The discrepancies between the coding and reverse-coding regions previously noted for the mycoplasma genomes (Fig. 1D) also are largely attenuated.
CONCLUSION
A General ab Initio Method Can Perform as Well as a Species-Specific, Training Set-Dependent Method.
For 10 bacterial genomes tested, covering four major lineage (Gram-negative, Gram-positive, cyanobacteria, and archea), we have shown that a totally objective recognition of coding regions is possible. Furthermore, there is no significant difference in the performance of our new ab initio iterative procedure and the accuracy of genemark trained on a specific E. coli gene subset (15, 26). The convergence properties and the performance of our procedure do not appear linked to phylogenetic, compositional, or size characteristics of the genomes tested. It thus is likely that the method is generally applicable and will work equally well for any new bacterial genome data becoming available in the future. For the species tested so far, the coding regions are recognized with an average specificity of 95% (range: 90–98%) and an overall accuracy (the product of sensitivity by specificity) of 87% (range: 82–90%). These performances are estimated by reference to the database annotations, which probably are not entirely correct. For E. coli, the best-known and most reliably annotated genome, our procedure specificity is 98% for a sensitivity of 91%; hence, an overall false-negative rate (percentage of missed or incorrectly recognized coding nucleotides) of 10.8% (provided the database is error-free). On the other hand, the false-positive rate (percentage of nucleotides not known to be coding but predicted as “coding” or “reverse coding”) is 10%. Again, this rate assumes that all coding nucleotides are annotated correctly in the database. Some ORFs, for instance, might extend 5′ to the actual translation initiator codon. Interestingly, the agreement between our predictions and the E. coli genome annotations improved with time: between January 16, 1997 and September 12, 1997, we gained 1% of the nucleotides predicted as “coding” or “noncoding.” Finally, we noticed that most of the coding regions remaining undetected by our method are from genes with a low codon usage bias [class III genes (27)]. Fortunately, those genes represent a small proportion of the E. coli genome (about 10%).
Preliminary Results with Less Compact Genomes.
The method presented here is primarily intended for the analysis of new bacterial genomes. Its extension to the analysis of less compact genomes is being studied. We verified, for instance, that the homogeneous iterative Markov modeling converged when applied to the yeast genomic sequence. Our ab initio procedure recognizes coding regions with a specificity of about 80% and a sensitivity of about 68%. Those rates are clearly lower estimates given that GenBank annotations for the yeast genome are far from being complete and accurate. The three-class homogeneous Markov modeling also converged when applied to human genomic sequences, albeit with quite different results. In this case, two of the three self-defined classes collected the most frequent repeats: Alu and Line-1 sequences. The recognition of genes in complex genomes thus might be possible after adapting the iterative procedure for the presence of repeats that could be masked out in successive steps. Alternatively, a larger number of classes could be imposed on the iterative Markov modeling procedure. The curious convergence properties of this algorithm need to be studied in more detail. For instance, what happens when the number of classes does not correspond to a “natural” partition scheme for the input data? For bacterial sequences, the use of only two classes (instead of three) significantly decreases the specificity of the recognition process although coding/reverse coding categories still dominate the final partition. On the other hand, imposing four classes may result in two scenarios: one of the dominant classes (coding or reverse coding) becomes distributed between two data sets of roughly equal sizes, or, alternatively, one of the data sets eventually shrinks to a small number of nucleotides.
The iterative partition procedure presented here is unlike other ab initio genome analysis methods proposed so far (3, 28, 29). However, it is reminiscent of the Gibbs sampling approach (30) in the sense that it combines a mathematical model of the data with a randomized optimization procedure. The algorithm selects sequence segments according to their conditional probabilities, then uses the resulting partition to update those probabilities. In our case, the convergence is driven by the maximization of the number of sequence segments locally consistent with N coevolving Markov models. The fast and accurate convergence observed with bacterial genome sequences when using Markov models of order 5 confirms that the hexamer/dicodon bias (14, 25) is the essential characteristic exhibited by protein-coding regions.
Acknowledgments
We thank Drs. Chantal Abergel, Rob Ewing, and David Robertson for reading the manuscript, and gratefully acknowledge the financial support from Incyte Pharmaceuticals, Inc. The programs are available at http://igs-server.cnrs-mrs.fr.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, Kerlavage A R, Bult C J, Tomb J F, Dougherty B A, Merrick J M, et al. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 2.Fraser C M, Gocayne J D, White O, Adams M D, Clayton R A, Fleischmann R D, Bult C J, Kerlavage A R, Sutton G, Kelley J M, et al. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 3.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, Sutton G G, Blake J A, FitzGerald L M, Clayton R A, Gocayne J D, et al. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 4.Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S, et al. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- 5.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li B C, Herrmann R. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraser C M, Casjens S, Huang W M, Sutton G, Clayton R, Lathigra R, White O, Ketchum K A, Dodson R, Hickey E K, et al. Nature (London) 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 7.Tomb J F, White O, Kerlavage A R, Clayton R A, Sutton G G, Fleischmann R D, Ketchum K A, Klenk H P, Gill S, Dougherty B A, et al. Nature (London) 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 8.Blattner F R, Plunkett G, III, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, et al. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 9.Smith D R, Doucette-Stamm L A, Deloughery C, Lee H, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbert K, et al. J Bacteriol. 1997;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, Bertero M G, Bessieres P, Bolotin A, Borchert S, et al. Nature (London) 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 11.Klenk H P, Clayton R A, Tomb J F, White O, Nelson K E, Ketchum K A, Dodson R J, Gwinn M, Hickey E K, Peterson J D, et al. Nature (London) 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 12.Goffeau A, Barrell B G, Bussey H, Davis R W, Dujon B, Feldmann H, Galibert F, Hoheisel J D, Jacq C, Johnston M, et al. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 13.Tatusov R L, Koonin E V, Lipman D J. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 14.Fickett J W, Tung C S. Nucleic Acids Res. 1992;20:6441–6450. doi: 10.1093/nar/20.24.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borodovsky M, McIninch J D. Comput Chem. 1993;17:123–133. [Google Scholar]
- 16.Borodovsky M, McIninch J D, Koonin E V, Rudd K E, Medigue C, Danchin A. Nucleic Acids Res. 1995;23:3554–3562. doi: 10.1093/nar/23.17.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Claverie J-M. Hum Mol Genet. 1997;6:1735–1744. doi: 10.1093/hmg/6.10.1735. [DOI] [PubMed] [Google Scholar]
- 18.Claverie J-M, Poirot O, Lopez F. Comput Chem. 1997;21:203–214. doi: 10.1016/s0097-8485(96)00039-3. [DOI] [PubMed] [Google Scholar]
- 19.Claverie J-M. Genomics. 1994;23:575–581. doi: 10.1006/geno.1994.1545. [DOI] [PubMed] [Google Scholar]
- 20.Almagor H. J Theor Biol. 1983;104:633–645. doi: 10.1016/0022-5193(83)90251-5. [DOI] [PubMed] [Google Scholar]
- 21.Borodovsky M, Sprizhitsky I, Golovanov E I, Aleksandrov A A. Mol Biol (Moscow) 1986;20:1024–1033. [PubMed] [Google Scholar]
- 22.Scherer S, McPeek M S, Speed T P. Proc Natl Acad Sci USA. 1994;91:7134–7138. doi: 10.1073/pnas.91.15.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Audic S, Claverie J-M. Comput Chem. 1997;21:223–227. doi: 10.1016/s0097-8485(96)00040-x. [DOI] [PubMed] [Google Scholar]
- 24.Benson D A, Boguski M S, Lipman D J, Ostell J, Ouellette B F. Nucleic Acids Res. 1998;26:1–7. doi: 10.1093/nar/26.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Claverie J-M, Bougueleret L. Nucleic Acids Res. 1986;14:179–196. doi: 10.1093/nar/14.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleffe J, Hermann K, Borodovsky M. Comput Chem. 1996;20:123–133. doi: 10.1016/s0097-8485(96)80014-3. [DOI] [PubMed] [Google Scholar]
- 27.Medigue C, Rouxel T, Vigier P, Henault A, Danchin A. J Mol Biol. 1991;222:851–856. doi: 10.1016/0022-2836(91)90575-q. [DOI] [PubMed] [Google Scholar]
- 28.Salzberg S L, Delcher A L, Kasif S, White O. Nucleic Acids Res. 1998;26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tiwari S, Ramachandran S, Bhattacharya A, Bhattacharya S, Ramaswamy R. Comput Appl Biosci. 1997;13:263–270. doi: 10.1093/bioinformatics/13.3.263. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence C E, Altschul S F, Boguski M S, Liu J S, Neuwald A F, Wootton J C. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]