

Abstract
Zn2+ is a key structural/functional component of many proteins and is present at high concentrations in the brain and retina, where it modulates ligand-gated receptors. Therefore, a study was made of the effects of zinc on homomeric neuronal nicotinic receptors expressed in Xenopus oocytes after injection of cDNAs encoding the chicken wild or mutant α7 subunits. In oocytes expressing wild-type receptors, Zn2+ alone did not elicit appreciable membrane currents. Acetylcholine (AcCho) elicited large currents (IAcCho) that were reduced by Zn2+ in a reversible and dose-dependent manner, with an IC50 of 27 μM and a Hill coefficient of 0.4. The inhibition of IAcCho by Zn2+ was competitive and voltage-independent, a behavior incompatible with a channel blockade mechanism. In sharp contrast, in oocytes expressing a receptor mutant, with a threonine-for-leucine 247 substitution (L247Tα7), subnanomolar concentrations of Zn2+ elicited membrane currents (IZn) that were reversibly inhibited by the nicotinic receptor blockers methyllycaconitine and α-bungarotoxin. Cell-attached single-channel recordings showed that Zn2+ opened channels that had a mean open time of 5 ms and a conductance of 48 pS. At millimolar concentrations Zn2+ reduced IAcCho and the block became stronger with cell hyperpolarization. Thus, Zn2+ is a reversible blocker of wild-type α7 receptors, but becomes an agonist, as well as an antagonist, following mutation of the highly conserved leucine residue 247 located in the M2 channel domain. We conclude that Zn2+ is a modulator as well as an activator of homomeric nicotinic α7 receptors.
Zn2+ is present in several regions of the brain, stored in synaptic vesicles of nerve terminals and released upon stimulation (1, 2); and a large body of evidence indicates that Zn2 has pleiotropic functions in cell tissues. For instance, it modulates postsynaptic neurotransmitter receptors in the central nervous system (3–5) and plays a role in the modulation of transcription processes and protein activity involved in gene regulation (6).
The α7 nicotinic acetylcholine receptor (nAcChoR) is an α-bungarotoxin-sensitive ligand-gated ion channel exhibiting fast desensitization, nonlinear current– voltage (I– V) relation, and low-affinity for AcCho, and is largely expressed in the retina and hippocampus, where Zn2+ is particularly abundant (1, 2). Moreover, it is known that Zn2+ alters the function of glycine, γ-aminobutyric acid type A (GABAA), GABAρ, and glutamate receptors (2, 5, 7–9), which play key roles in the synaptic activity of the brain and retina. Therefore, we thought it would be interesting to investigate whether Zn2+ also modulates the function of the α7 nAcChoR. We report that at μM concentrations Zn2+ blocks considerably the α7 nAcChoRs expressed in Xenopus oocytes.
A threonine-for-leucine 247 substitution (L247Tα7), in the channel domain, renders the receptor I– V relation linear, increases its affinity for AcCho, gives rise to an additional channel conductance, and decreases receptor desensitization (10). Strikingly, even in the absence of AcCho, oocytes that express mutated L247Tα7 nAcChoRs exhibit a significant inward current that is blocked by nicotinic antagonists and that is attributed to spontaneous openings of the mutated α7 nAcChoR channels (11, 12). We used the wild and the mutated receptors as tools to gain some insight on the mechanisms whereby Zn2+ modulates receptor function.
MATERIALS AND METHODS
Oocyte Injection.
Full-length cDNAs encoding the chicken wild-type α7 or the mutated L247Tα7 neuronal nAcChoR subunits were kindly provided by M. Ballivet (Univ. of Geneva, Geneva, Switzerland) and were expressed as described previously (13, 14). Stage VI oocytes were injected intranuclearly with cDNA clones. Preparation of oocytes and nuclear injection procedures were as detailed elsewhere (13–15).
Electrophysiology.
Two to four days after injection, whole-cell membrane currents were recorded in voltage-clamped oocytes by using two microelectrodes filled with 3 M KCl (15). The oocytes were placed in a recording chamber (volume, 0.1 ml) and perfused continuously, 11–12 ml/min, with oocyte Ringer (82.5 mM NaCl/2.5 mM KCl/2.5 mM CaCl2/1 mM MgCl2/5 mM Hepes, adjusted to pH 7.4 with NaOH) at room temperature (20–22°C). To obtain dose/response relations AcCho was applied to the oocytes at 3-min intervals. The half-inhibitory concentration (IC50) of Zn2+, as well as the half-dissociation constant (EC50) of AcCho were estimated by fitting the data to Hill equations, using least-square routines:
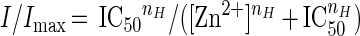 |
1 |
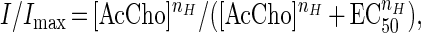 |
2 |
where [Zn2+] and [AcCho] are the doses of Zn2+ and AcCho, respectively, nH is the Hill coefficient, and Imax is the maximum current response.
Single-channel currents were recorded from the animal pole of the oocytes injected with L247Tα7 cDNA by using the patch-clamp technique in the cell-attached mode, as reported (15–17). Unless otherwise stated, the Zn2+ in the patch pipette was 10−8 M. If no events were detected within 60 s after seal formation, at 0–40 mV pipette potential, or if the frequency of openings was below 0.1 Hz, the patch was discarded. Typically, a successful patch was stable for 5–25 min and had >400 opening transitions. Current recordings were filtered at 2 kHz, sampled at 10 kHz, and analyzed by pclamp 6.0.2 routines (Axon Instruments) using a threshold-crossing criterion. Events briefer than 0.2 ms were incompletely resolved and were excluded from the open-time histograms, which, therefore, represent apparent mean open-times. Histograms of amplitudes (400–2,000 events) were fitted with a single Gaussian function, and open-times were fitted with the sum of exponentials. Burst duration was studied by grouping openings separated by a specific critical time, which was calculated for each patch from the fitted parameters of the shut-time distribution. For each patch, slope conductances were obtained by linear fitting of current–voltage relations constructed by hyperpolarizing the patch membrane potential up to 90 mV and by depolarizing the patch by up to 100 mV. For further details see ref. 17.
Zn2+ solutions made from ZnCl2 and Zn2+ acetate were purchased from Sigma (catalog numbers Z0173 and Z4875) and Fluka (catalog numbers 96458 and 96469). All four gave similar results.
RESULTS
Zn2+ Blocks IAcCho Generated by Wild-Type α7 Receptors. Oocytes expressing wild-type α7 (WTα7) receptors and held at −100 mV responded to 150 μM AcCho (18) with an inward current (IAcCho), which peaked to −460 ± 69 nA (mean ± SEM; range: −80 to −1,150 nA; 25 oocytes/5 donors) and decayed to 10% (T0.
1) in 123 ± 42 ms. Zn2+ alone (0.1 μM– 10 mM) did not elicit obvious current responses in either noninjected oocytes or oocytes expressing WTα7 nAcChoRs, and, when coapplied with AcCho (150 μM), Zn2+ failed to alter IAcCho. However, an additional pretreatment with Zn2+ for 20–30 s led to a large and reversible decrease of IAcCho peak amplitude (see Inset in Fig. 1). The inhibition of IAcCho by Zn2+ was not enhanced when the pretreatment with Zn2+ was prolonged to 10 min, but it increased as the concentration of Zn2+ was raised. The IAcCho was suppressed completely by 10 mM Zn2+ pretreated for 30 s and coapplied with AcCho (150 μM). The mean Zn2+ dose– IAcCho response relation fitted to Eq. 1 (see Methods) gave values for IC50 and nH of 27 μM and 0.4, respectively (Fig. 1). At this Zn2+ concentration the T0.1 was unchanged (105 ± 14 ms and 113 ± 13 ms in control and Zn2+-treated oocytes; 9 oocytes/3 donors). As previously reported (14, 18), the IAcCho– voltage relation for WTα7 receptors shows strong rectification at positive potentials. This pattern was not modified by Zn2+ (30 μM) (e.g., Fig. 2), indicating that the inhibitory action of Zn2+ on WTα7 nAcChoR was not changed by membrane hyperpolarization.
Figure 1.

Zn2+ concentration– AcCho current response relation in oocytes expressing WTα7 receptors. The peak currents evoked by AcCho (150 μM) coapplied with Zn2+, at the concentrations indicated in the abscissa were normalized to the response to AcCho alone. Data are mean ± SEM (10 oocytes, 3 donors). Data without bars, mean of 2–4 oocytes. (Inset) Sample currents elicited by 150 μM AcCho alone (horizontal, continuous bar) or together with Zn2+ (horizontal, dashed bar). First and last traces, control and recovery. Oocytes held at −80 mV and pretreated for 30 s with Zn2+ before applying AcCho plus Zn2+.
Figure 2.

Voltage-independent inhibition of IAcCho by Zn2+ in WTα7 oocytes. (A) The percentage block is the inhibition of IAcCho (AcCho, 150 μM) by Zn2 (20 μM) at various holding potentials. Each point is the mean ± SEM of 7 oocytes and 2 donors. Oocytes were pretreated with Zn2+ as in Fig. 1. (B) Current– voltage relation in one oocyte expressing WTα7 nAcChoRs. Peak currents evoked at various holding potentials by 150 μM AcCho (■) and by 150 μM AcCho coapplied with 30 μM Zn2+ (□). AcCho was applied at 3-min intervals and Zn2+ for 30 s before coapplication with AcCho. Solid lines represent second-order polynomial fits to the data. Note strong current rectification at positive potentials.
To see whether Zn2+ altered the binding affinity of the receptor for AcCho, the control AcCho dose– current response relation was compared with that obtained in oocytes treated with Zn2+ at about IC50 concentration. The dose– response curve was shifted to the right and the EC50 increased from 123 ± 13 μM in untreated to 166 ± 36 μM in Zn2+-treated oocytes (11/2), while the Hill coefficient remained unchanged (nH =1.2).
Zn2+ Activates L247Tα7 nAcChoRs in the Absence of AcCho.
It is known that, because of “spontaneously” active mutant AcCho receptors, the holding current required to clamp an oocyte is greater for cells expressing L247Tα7 mutant receptors than for those expressing WTα7 receptors (11, 12). Zn2+ (1 mM), applied to oocytes expressing the L247Tα7 nAcChoRs, gave rise to an outward current of 130 ± 31 nA (IZn; 7 oocytes/3 donors) followed by a large inward current after Zn2+ withdrawal (Fig. 3A and C). αBuTx (100 nM) and methyllycaconitine (MLA) (1 μM) also elicited an outward current, and both of them prevented Zn2+ from generating the outward currents as well as the Zn2+-off current (Fig. 3 B and C). It should be noted that αBuTx elicits first a small inward current, presumably because it acts as an agonist of the mutant receptor before blocking it (cf. also ref. 11). The I– V curve for the outward current showed a null potential at about −18 mV, and the outward current elicited by 10 mM Zn2+ was similar to that elicited by 1 mM Zn2+ whereas 0.5 mM Zn2+ did not elicit an appreciable outward current. In contrast, Zn2+ failed to elicit outward currents in noninjected or injected but nonexpressing oocytes, although it is known that sometimes Zn2+ triggers oscillatory currents because of activation of the phosphatidyl inositol system (19).
Figure 3.

Zn2+, AcCho, αBuTx, and MLA currents in oocytes expressing L247Tα7 receptors. (A) Currents activated by Zn2+ (1 mM) at −100 mV. Note an outward current followed by a rapid “off” inward current after Zn2+ withdrawal. (B) Outward current evoked by 1 μM MLA at −100 mV in another oocyte. Note that the Zn2+ currents (1 mM) were completely blocked. (C) AcCho, Zn2+, and αBuTx currents in one L247Tα7 oocyte. First record is control AcCho current (0.2 μM). A few minutes later, Zn2+ (1 mM) was applied, then αBuTx (100 nM), and, after 4 min, AcCho and Zn2+ were reapplied, still in the presence of αBuTx. Note that αBuTx generated first an inward current followed by an outward current. Note also that the responses to both AcCho and Zn2+ were abolished by αBuTx. In each frame the dotted lines indicates the resting baseline current.
Interestingly, at concentrations below 0.5 mM, Zn2+ evoked a short latency inward current in the oocytes expressing L247Tα7 receptors. For instance, in oocytes held at −60 mV the inward current elicited by 10 nM Zn2+ was −1.78 ± 0.28 μA (range −288 nA to −4.6 μA; 25 oocytes/4 donors). This inward current was again blocked by αBuTx and by MLA (Fig. 4). The ability of low concentrations of Zn2+ to induce inward currents may explain the “off current” elicited after withdrawal of high concentrations of Zn2+ (1 mM) (e.g., Fig. 3 A–C).
Figure 4.

Zn2+ dose–current response relationship in oocytes expressing L247Tα7 receptors. Peak inward currents evoked by Zn2+ are expressed as percentage of the response to 10 nM Zn2+. Each point represents mean ± SEM of 4–16 values (24 oocytes, 3 donors). Holding potential was −60 mV. Note the currents elicited at low concentrations possibly from, at least partially, Zn2+ contamination of the oocyte Ringer. (Inset, Left) Zn2+ current activated in one oocyte at the concentration indicated and blocked by MLA (1 μM). (Inset, Right) Zn2+ currents activated in another oocyte by Zn2 applied alone. Holding potential, −60 mV.
The Zn2+ dose– inward current response relationship over the wide range of Zn2+ concentrations tested (10 fM to 1 mM) (Fig. 4) showed a peak with 1–10 nM, suggesting a dual action of Zn2+ on the L247Tα7 receptors. At low concentrations, Zn2+ activated an inward current that increased in amplitude with Zn2+ concentrations, reached a peak at 1–10 nM, and then decreased to 0 with about 1 mM Zn2+. The current elicited by 10 nM Zn2+ was linearly related to the membrane potential, similarly to IAcCho, and inverted direction at −13 ± 7 mV (n = 4), a value that is close to the reversal potential of IAcCho (14) (Fig. 5).
Figure 5.

IZn–voltage relationship in an L247Tα7 oocyte. Peak currents evoked by Zn2+ (10 nM) at various holding potentials. Note the lack of rectification and the null potential at −18 mV. Zn2+ was applied at 3-min intervals and holding potential was −50 mV. Curve fitting was as in Fig. 2. (Inset) Sample currents elicited in another oocyte at +45 mV (Upper) and −100 mV (Lower).
Preliminary experiments substituting Co2+ for Zn2+ showed that Co2+ did not trigger a current like IZn and that IAcCho was not greatly influenced by Co2+ concentrations as high as 1 mM or as low as 10 nM, suggesting that the action of Zn2+ on L247Tα7 is very likely specific to that ion. Thus, it appeared that at low concentrations, Zn2+ was gating directly some membrane channels.
Zn2+-Gated L247Tα7 nAcChoR Channels.
In an attempt to detect the channel openings gated by Zn2+, cell-attached patch-clamp recordings were made from oocytes expressing L247Tα7 receptors. With 10 nM Zn2+ in the patch pipette, and in the absence of AcCho, analyses of unitary events revealed only one channel conductance of 47.5 ± 1.3 ps (5 oocytes/2 donors) (Fig. 6). No transitions from higher to lower amplitude channels were observed, and each oocyte exhibited a homogeneous channel population. All these observations indicate that the estimated mean channel conductance is associated to a channel population with a single conductance level. This population of channel openings showed a mean open-time (τop) of 5.3 ± 1.1 ms (mean ± SEM; 5 patches, 5 oocytes/2 donors), made up of a briefer (τ1 = 1.6 ± 0.3 ms; 60%) and a longer (τ2 = 16.6 ± 2.0 ms; 40%) exponential component at an extrapolated membrane potential of −59 ± 3 mV (Fig. 6C). Similar values were observed with 1 nM Zn2+ in the patch pipette (3 oocytes). Flickering activity was practically absent under our recording conditions, as shown by the burst mean duration (τb), which was only slightly longer (6.2 ± 1.3 ms) than τop, a behavior indicating the absence of open-channel blockage by the agonist itself (20). Channel activity (≈7 Hz at −60 mV extrapolated membrane potential) and amplitude were rather stable over time at a given patch pipette potential, with only rare overlapping events. As the patch membrane was hyperpolarized, the amount of voltage required to change the opening frequency e-fold was 18 mV, and the τop did not change with a hyperpolarization of 30 mV.
Figure 6.

Properties of channels activated by Zn2+ in oocytes injected with L247Tα7 subunit cDNA. A– D refer to a cell-attached patch of the same L247Tα7-injected oocyte, with 10 nM Zn2+ in the pipette. (A) Single-channel currents at −63 mV extrapolated membrane potential. Inward currents are represented by upward deflections. (B) Distribution of single-channel amplitudes, at the same membrane potential. (C) Histogram of open durations fitted by the sum of two exponential functions with τ1 = 2.7 ± 0.03 (84%), τ2 = 13.9 ± 0.1 (16%), and τop as indicated. (D) Mean channel current amplitudes at different potentials plotted vs. pipette potential and fitted by linear regression (solid line), yielding the slope conductance indicated.
Zn2+ Modulates IAcCho in L247Tα7 Mutant cDNA-Injected Oocytes. Oocytes expressing L247Tα7 mutant receptors responded to 0.
2 μM AcCho (≈EC50; refs. 10 and 14) with an IAcCho whose peak amplitude (at −100 mV) averaged −935 ± 180 nA (24/4, range: −230 nA to −3,480 nA) and decayed with a T0.1 >10 s. When Zn2+ (1 mM) was coapplied with AcCho, the IAcCho was reduced in amplitude (−274 ± 112 nA; 9 oocytes/2 donors), decayed with similar kinetics, and was followed by a large “Zn2+-off” current after withdrawal of AcCho and Zn2+ (Fig. 7A). Both IAcCho and the Zn2+ currents were abolished by the nicotinic receptor blockers αBuTx and MLA (1 μM) (not shown). The blockage of IAcCho by Zn2+ increased as the Zn2+ concentration was increased, and IAcCho was completely suppressed with 10 mM Zn2+ (not shown). Interestingly, Zn2+ had the same blocking effect on IAcCho if the oocytes were pretreated for 30–60 s with Zn2+, which is in contrast with the results obtained in oocytes expressing WTα7 receptors, where Zn2+ was able to block IAcCho only after a brief Zn2+ pretreatment. Furthermore, the blockage of IAcCho by Zn2+ in L247Tα7 oocytes was voltage-dependent with a drastic increase at hyperpolarized potentials, as illustrated in Fig. 7 B and C.
Figure 7.

Zn2+ modulation of IAcCho in L247Tα7 mutant cDNA-injected oocytes. (A) Examples of inward current activated in the same oocyte by AcCho alone or by AcCho plus Zn2+. Note the large inward current after Zn2+ withdrawal. Holding potential, −100 mV. (B) Voltage-dependent inhibition of IAcCho by Zn2+. Inhibition of IAcCho (AcCho, 0.2 μM) by Zn2+, measured as the percentage of IAcCho alone to that in the presence of Zn2+ (10 mM). Data represent mean from 6 oocytes and 2 donors. (C) IAcCho– voltage relationship in an oocyte held at −50 mV. Peak currents evoked at various test potentials by 0.2 μM AcCho (•) or by 0.2 μM AcCho plus 1 mM Zn2+ (○). AcCho was applied at 3-min intervals. The solid lines are three-order polynomial fits to the data. Note that IAcCho was reduced considerably at hyperpolarized potentials. (D) Potentiation of IAcCho by Zn2+ at lower doses.
In contrast to the inhibition of IAcCho by high concentrations of Zn2+, low concentrations (Zn2+ 1 nM– 1 μM) increased IAcCho (AcCho, 0.1–1 μM; Fig. 7D). This Zn2+-induced potentiation was not observed when AcCho was 100 μM, a nAcChoR-saturating dose (14). This suggests that the potentiation is a result of additional channel openings gated by Zn2+.
The AcCho dose– IAcCho response relationship was shifted toward the right in the presence of Zn2+ (1 mM), and the EC50 increased from 0.34 to 1.22 μM, while the nH remained at 1.0 (not shown), suggesting that Zn2+ may act on, or near, the nAcChoR-binding site.
DISCUSSION
During neurotransmission, nerve terminals can release, together with the neurotransmitter, a variety of molecules including peptides, nucleotides, and ions, which act on the postsynaptic cells and serve multiple functions, such as cell development and survival and modulation of postsynaptic receptors (2, 21, 22). In particular, in the central nervous system zinc ions are released with neurotransmitters and may reach μM concentrations (1, 2). Furthermore, a large body of evidence indicates that Zn2+ plays a key role in transmitter– receptor binding and in the opening of ligand-gated channels as, for example, GABAρ, GABAA, glutamate, and glycine receptors.
α7 nAcChoRs are largely expressed in both the central and peripheral nervous systems, and it is believed that their dysfunction is involved in various neurological disorders including epileptic seizures and schizophrenia (23, 24). In here, we have shown that Zn2+ reversibly blocks, in a dose-dependent manner, the IAcCho elicited by activation of WTα7 nAcChoRs expressed in Xenopus oocytes. Because the inhibition of IAcCho by Zn2+ was not voltage-dependent and the receptor-binding affinity for the transmitter was reduced by Zn2+, it is likely that the action of Zn2+ is from a competition and/or an allosteric inhibition, rather than from an open channel blockage. Furthermore, given that a pretreatment with Zn2+ was necessary to inhibit the WTα7 receptor, it seems that activation of those receptors by AcCho is a faster process then their blockage by Zn2+.
It is known that Zn2+ modulates various ligand-gated channels, namely glutamate, GABAA, GABAρ, glycine, and purinergic receptors (5, 7–9, 25, 26). Our findings indicate that Zn2+ acts differently on α7 nAcChoRs. For instance, similar to its action on GABAρ1 and purinergic receptors (2, 4, 7, 25, 26), but unlike that on N-methyl-d-aspartate receptors in cortical neurons (27) and on GABAA receptors in dentate gyrus basket cells (9), the inhibition of α7 nAcChoRs by Zn2+ is competitive and voltage-independent. That Zn2+ changes the apparent affinity of the WTα7 receptors for AcCho suggests an interaction of this metal ion at or near the agonist-binding sites, as reported previously for glycine receptors (5).
In contrast, the action of ZnCl2 on the mutant receptors is more complicated and appears to be bimodal, at least. On the one hand, at mM concentrations, Zn2+ reversibly blocks the action of AcCho on the L247Tα7 receptors without the need of a Zn2+ pretreatment, and the block is voltage-dependent. This resembles the effects of fluoxetine, which is a competitive inhibitor of WTα7 receptors, but acts as a channel blocker on the mutant α7 receptors (12). On the other hand, and very strikingly, Zn2+ appears to act as a very potent agonist on the mutant α7 receptors and is able to generate currents even at picomolar concentrations.
Considering that Zn2+ blocks Cl− channels (19) and also some types of GABA receptors (2, 7, 9), the fact that Zn2+ blocks the WTα7 receptors is not entirely unexpected. The question is, how does Zn2+ generate a current in oocytes expressing the mutant α7 receptors? Because Zn2+ evoked no current in oocytes expressing WTα7 receptors, or in noninjected oocytes, it seems very likely that Zn2+ is acting directly on the mutant receptors. The mechanism whereby Zn2+ elicits the currents is still unknown, but it is appropriate to consider here a few possibilities. For example, it is known that in oocytes expressing mutant receptors, and in the absence of AcCho, there is a membrane current that is abolished by αBuTx and MLA, two specific nicotinic receptor blockers. Until now, this current has been attributed to spontaneous openings of the mutant α7 receptors (11). Therefore, one possibility is that, as with Sr2+ and La3+ (28, 29), Zn2+ increased the lifetime of the AcChoR channels. This possibility is not likely because in the L247Tα7 oocytes the lifetime of the channels opened by Zn2+ was actually about one-half of that of the channels gated by AcCho (17). For both Zn2+ and AcCho there were two lifetimes: one short and one long. For AcCho the corresponding open times were 2.6 ms and 35 ms (17) compared with 1.6 ms and 16.6 ms for Zn2+. Furthermore, the conductance of the channels gated by Zn2+ was similar to that of AcCho-gated channels (47 ps and 44 ps, respectively).
Thus, it appears that Zn2+ is not modulating “spontaneously” active L247Tα7 receptors, but seems to be actually acting as an agonist. A very potent agonist indeed, because it generates currents at picomolar concentrations. Such an action provides a different explanation for the resting current seen in oocytes expressing L247Tα7 receptors; namely, that the mutant receptors are not spontaneously active but that they are being gated by a small Zn2+ contamination of one, or more, of the salts that make the oocyte Ringer solution. Such a contamination may account for the “shoulder” seen in the Zn2+ dose– current response curve (Fig. 4). It is, of course, possible that another contaminant may be responsible for the Zn2+ current but, if that were the case, it would need to be present in the three different types of ZnCl2 and one Zn2+ acetate tested; and the contaminant would have to exist at a high concentration to withstand the very large dilutions used.
In conclusion, it seems that Zn2+ acts mainly as a competitive antagonist on WTα7 receptors, but has a dual action on the mutant receptors. So far, the simplest explanation of our results is that, similar to its action on the WTα7 receptors, Zn2+ binds at, or near, the AcCho-binding site of mutant receptors. The unexpected finding was that the mutation converted Zn2+ into a very potent agonist, in addition to its antagonistic action. It is tempting to speculate that similar effects may play a role in some neurodiseases in which an L247T or equifunctional mutation might exist.
Acknowledgments
We are very grateful to Dr. Francesca Grassi and Professor Enzo Wanke for the critical reading of the manuscript. This work was supported by the Ministero Universitá Ricerca Scientifica Tecnologica (to F.E.), Telethon fellowship 222/bi (to E.P.), and the National Science Foundation (to R.M.).
ABBREVIATIONS
- IZn
Zn2+-activated current
- AcCho
acetylcholine
- IAcCho
AcCho-activated current
- MLA
methyllycaconitine
- αBuTx
α-bungarotoxin
- nAcChoR
nicotinic AcCho receptor
- L247Tα7
threonine-for-leucine 247 α7-subunit mutant
- nH
Hill coefficient
References
- 1.Assaf S Y, Chung S H. Nature (London) 1984;308:734–738. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- 2.Smart T G, Xie X, Krishek B J. Prog Neurobiol. 1994;42:393–341. doi: 10.1016/0301-0082(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 3.Draguhn A, Verdoorn T A, Ewert M, Seeburg P H, Sakmann B. Neuron. 1990;5:781–788. doi: 10.1016/0896-6273(90)90337-f. [DOI] [PubMed] [Google Scholar]
- 4.Smart T G, Moss S J, Xie X, Huganir R L. Brit J Pharmacol. 1991;103:1837–1839. doi: 10.1111/j.1476-5381.1991.tb12337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laube B, Kuhse J, Rundstrom N, Kirsch J, Schmieden V, Betz H. J Physiol (London) 1995;483:613–619. doi: 10.1113/jphysiol.1995.sp020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg J M, Shi Y. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 7.Calvo D J, Vasquez D E, Miledi R. Proc Natl Acad Sci USA. 1994;91:12725–12729. doi: 10.1073/pnas.91.26.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paoletti P, Ascher P, Neyton J. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berger T, Schwarz C, Kraushaar U, Monyer H. J Neurosci. 1998;18:2437–2448. doi: 10.1523/JNEUROSCI.18-07-02437.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Revah F, Bertrand D, Galzi J L, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux J P. Nature (London) 1991;352:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 11.Bertrand S, Devillers-Thiery A, Palma E, Buisson B, Edelstein S J, Corringer P J, Changeux J P, Bertrand D. NeuroReport. 1997;8:3591–3596. doi: 10.1097/00001756-199711100-00034. [DOI] [PubMed] [Google Scholar]
- 12.Maggi, L., Palma, E., Miledi, R. & Eusebi, F. (1998) Mol. Psychiatry, in press. [DOI] [PubMed]
- 13.Palma E, Bertrand S, Binzoni T, Bertrand D. J Physiol (London) 1996;491:151–161. doi: 10.1113/jphysiol.1996.sp021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palma E, Mileo A M, Eusebi F, Miledi R. Proc Natl Acad Sci USA. 1996b;93:11231–11235. doi: 10.1073/pnas.93.20.11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miledi R, Parker I, Sumikawa K. Proc R Soc London Ser B. 1983;218:481–484. doi: 10.1098/rspb.1983.0053. [DOI] [PubMed] [Google Scholar]
- 16.Methfessel C, Witzemann V, Takahashi T, Mishina M, Numa S, Sakmann B. Pflügers Arch Eur J Physiol. 1986;407:577–588. doi: 10.1007/BF00582635. [DOI] [PubMed] [Google Scholar]
- 17.Palma E, Maggi L, Eusebi F, Miledi R. Proc Natl Acad Sci USA. 1997;94:9915–9919. doi: 10.1073/pnas.94.18.9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couturier S, Bertrand D, Matter J M, Hernandez M C, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 19.Miledi R, Parker I, Woodward R. J Physiol (London) 1989;417:173–195. doi: 10.1113/jphysiol.1989.sp017796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogden D C, Colquhoun D. Proc R Soc London Ser B. 1985;225:329–355. doi: 10.1098/rspb.1985.0065. [DOI] [PubMed] [Google Scholar]
- 21.Westbrook G L, Mayer M L. Nature (London) 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 22.Sensi S L, Canzoniero L M T, Yu S P, Ying H S, Koh J Y, Kerchner G A, Choi D W. J Neurosci. 1997;17:9554–9564. doi: 10.1523/JNEUROSCI.17-24-09554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elmslie F V, Rees M, Williamson M P, Kerr M, Kjeldsen M, Pang K A, Sundqvist A, Friis M L, Chadwock D, Richens A, et al. Hum Mol Genet. 1997;6:1329–1334. doi: 10.1093/hmg/6.8.1329. [DOI] [PubMed] [Google Scholar]
- 24.Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Odincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, et al. Proc Natl Acad Sci USA. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang T L, Hackman A, Guggino W B, Cutting G R. J Neurosci. 1995;15:7684–7691. doi: 10.1523/JNEUROSCI.15-11-07684.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C, Peoples R W, Weight F F. J Physiol (London) 1997;505:641–653. doi: 10.1111/j.1469-7793.1997.641ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christine C W, Choi D W. J Neurosci. 1990;10:108–116. doi: 10.1523/JNEUROSCI.10-01-00108.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heuser J, Miledi R. Proc R Soc London Ser B. 1971;179:247–260. doi: 10.1098/rspb.1971.0096. [DOI] [PubMed] [Google Scholar]
- 29.Miledi R, Parker I. J Physiol (London) 1980;306:567–577. doi: 10.1113/jphysiol.1980.sp013415. [DOI] [PMC free article] [PubMed] [Google Scholar]