

Abstract
Dystrophin is a multidomain protein that links the actin cytoskeleton to laminin in the extracellular matrix through the dystrophin associated protein (DAP) complex. The COOH-terminal domain of dystrophin binds to two components of the DAP complex, syntrophin and dystrobrevin. To understand the role of syntrophin and dystrobrevin, we previously generated a series of transgenic mouse lines expressing dystrophins with deletions throughout the COOH-terminal domain. Each of these mice had normal muscle function and displayed normal localization of syntrophin and dystrobrevin. Since syntrophin and dystrobrevin bind to each other as well as to dystrophin, we have now generated a transgenic mouse deleted for the entire dystrophin COOH-terminal domain. Unexpectedly, this truncated dystrophin supported normal muscle function and assembly of the DAP complex. These results demonstrate that syntrophin and dystrobrevin functionally associate with the DAP complex in the absence of a direct link to dystrophin. We also observed that the DAP complexes in these different transgenic mouse strains were not identical. Instead, the DAP complexes contained varying ratios of syntrophin and dystrobrevin isoforms. These results suggest that alternative splicing of the dystrophin gene, which naturally generates COOH-terminal deletions in dystrophin, may function to regulate the isoform composition of the DAP complex.
Keywords: dystrophin, muscular dystrophy, syntrophin, dystrobrevin, mdx mice
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive disease caused by defects in the dystrophin gene (Koenig et al. 1987; Emory, 1993). Although the exact function of dystrophin is unclear, it is postulated to play both structural and signaling roles in protecting muscle fibers from contraction-induced injury (Zubrzycka-Gaarn et al. 1988; Ervasti and Campbell 1991; Cox et al. 1993; Petrof et al. 1993; Grady et al. 1999). Dystrophin is a 427-kD multidomain protein that has an NH2-terminal actin binding motif resembling those in α-actinin and β-spectrin (for review see Amalfitano et al. 1997). The majority of the dystrophin molecule is a rod-like domain composed of 24 spectrin-like repeats and 4 hinge regions. Towards the COOH terminus, dystrophin contains multiple domains that interact with both peripheral and integral membrane proteins known as the dystrophin associated protein (DAP) complex (Ervasti and Campbell 1991). A WW domain at the beginning of this region binds to β-dystroglycan and this interaction is stabilized by the adjacent cysteine-rich domain (Jung et al. 1995). β-dystroglycan binds to α-dystroglycan, which connects to laminin, linking the DAP complex to the actin cytoskeleton and the extracellular matrix (Ibraghimov-Beskrovnaya et al. 1992; Ervasti and Campbell 1993). The sarcoglycan complex appears to stabilize the link between α-dystroglycan and β-dystroglycan (Araishi et al. 1999).
The link between β-dystroglycan and dystrophin is critical for the function of dystrophin, as deletions in the cysteine-rich domain of dystrophin eliminate binding to β-dystroglycan and prevent assembly of the sarcoglycan complex, leading to a severe dystrophy (Suzuki et al. 1992; Jung et al. 1995; Rafael et al. 1996). The dystrophin COOH-terminal domain is located adjacent to the cysteine-rich domain, and contains an alternatively spliced region and two coiled-coil motifs (Feener et al. 1989; Bies et al. 1992; Blake et al. 1995). The alternatively spliced region binds three isoforms of syntrophin in muscle, while the coiled-coil motifs bind numerous members of the dystrobrevin family (Ahn and Kunkel 1995; Dwyer and Froehner 1995; Suzuki et al. 1995; Yang et al. 1995; Sadoulet-Puccio et al. 1997). The dystrobrevins display significant homology with the COOH-terminal region of dystrophin, and the larger dystrobrevin isoforms also bind to the syntrophins (Butler et al. 1992; Wagner et al. 1993; Yoshida et al. 1995). The importance and functional significance of syntrophin and dystrobrevin remains largely unknown, although they may be involved in cell signaling pathways (Bredt 1999; Grady et al. 1999).
Three isoforms of syntrophin (α1, β1, and β2), which are encoded by separate genes, bind dystrophin in skeletal muscle (Adams et al. 1995; Ahn et al. 1996; Peters et al. 1997a). The syntrophins contain a PDZ domain that binds multiple proteins including neuronal nitric oxide synthase (nNOS), sodium channels, stress-activated protein kinase-3, and a microtubule-associated serine/threonine kinase (Brenman et al. 1996; Gee et al. 1998; Schultz et al. 1998; Hasegawa et al. 1999; Lumeng et al. 1999a). However, these interactions may not be critical for muscle fiber stability, since α1-syntrophin knockout mice have no overt signs of dystrophy (Kameya et al. 1999). While α1- and β1-syntrophin are localized along the sarcolemma, β2-syntrophin is normally localized at the troughs of the neuromuscular junction (Kramarcy and Sealock 2000).
The dystrobrevin family is encoded by at least two genes, α and β, although only the α-dystrobrevin gene is expressed at significant levels in muscle (Wagner et al. 1993; Peters et al. 1997b; Blake et al. 1998; Puca et al. 1998). Several isoforms of α-dystrobrevin are expressed in muscle due to alternative splicing of the primary transcript (Blake et al. 1996; Sadoulet-Puccio et al. 1996; Peters et al. 1998). α-dystrobrevin-1 resides primarily, but not exclusively, at the neuromuscular junction (NMJ) and contains an extended COOH terminus that is tyrosine phosphorylated. α-dystrobrevin-2 has a shorter COOH terminus, and is found along the entire sarcolemma (Balasubramanian et al. 1998; Nawrotzki et al. 1998; Peters et al. 1998). The shortest isoform, α-dystrobrevin-3, lacks both the syntrophin and dystrophin binding sites, but little is known of the expression of this isoform in muscle. As with all components of the DAP complex, syntrophin and dystrobrevin are largely absent from the sarcolemma of dystrophin-deficient mdx mice and Duchenne muscular dystrophy patients (Ohlendieck and Campbell 1991). However, mice deficient for dystrobrevin display a moderately severe muscular dystrophy with a variety of features different from those of the mdx mouse. With the exception of nNOS, all known components of the DAP complex are present on the sarcolemma of dystrobrevin-deficient mice. These mice display no signs of damage to the sarcolemma, implicating a distinct mechanism of dystrophy independent of the role of dystrophin (Grady et al. 1999). Dystrobrevin, dystrophin, and utrophin also appear to be required for postnatal maturation of neuromuscular junctions (Grady et al. 2000; Rafael et al. 2000).
To determine the importance of syntrophin and dystrobrevin binding to dystrophin, we previously generated transgenic mdx mouse strains expressing dystrophins deleted for either the syntrophin or the dystrobrevin binding domain (Rafael et al. 1994, Rafael et al. 1996). These mice displayed normal muscle function and essentially normal localization of syntrophin, dystrobrevin, and nNOS (Rafael et al. 1994, Rafael et al. 1996; Straub et al. 1997; Crosbie et al. 1998). Thus, while dystrobrevin appears to protect muscle from damage (Grady et al. 1999), removal of the dystrobrevin binding site from dystrophin does not result in a dystrophy. Subsequent studies revealed that syntrophin and dystrobrevin bind each other in addition to dystrophin, so that removal of only one of the two binding sites on dystrophin might not sever the link between dystrophin, syntrophin and dystrobrevin. To explore the nature of the interaction between dystrophin and these DAP components, we generated new transgenic mdx mouse lines that express a dystrophin lacking both the syntrophin and dystrobrevin binding domains. Surprisingly, these transgenic mice displayed normal muscle function. Our results also show that this truncated dystrophin stabilized assembly of the entire DAP complex. These results demonstrate that syntrophin and dystrobrevin can bind to the DAP complex independently from dystrophin and that this association is sufficient to prevent dystrophy.
Materials and Methods
Dystrophin Transgenic mdx Mice
The bases encoding amino acids 3402–3675 (corresponding to exons 71–78) were deleted from the full length murine dystrophin cDNA (sequence data available from EMBL/GenBank/DDBJ under accession No. M68859) by recombinant PCR, leaving the last three amino acids (exon 79) of the dystrophin protein unaltered. This dystrophin Δ71–78 cDNA was cloned into an expression vector containing bases −2139 to +239 of the human α-skeletal actin (HSA) promoter (Brennan and Hardeman 1993). A splice acceptor from the SV40 VP1 intron (isolated as a 400 bp HindIII/XbaI fragment from pSVL; Amersham Pharmacia Biotech) was inserted immediately 3′ of the HSA fragment, and the SV40 polyadenylation signal (isolated as a BamHI fragment from pCMVβ; MacGregor and Caskey 1989) was inserted 3′ of the dystrophin cDNA. The excised dystrophin Δ71–78 expression cassette was injected into wild-type C57Bl/10 × SJL/J F2 hybrid embryos, and F0 mice were screened by PCR. Five positive F0's were backcrossed onto the C57Bl/10mdx background, and most further studies focused on the line with the most uniform expression levels. Some studies used previously described transgenic mdx mice that express dystrophin constructs deleted approximately for exons 71–74 (Δ71–74) or exons 75–78 (Δ75–78), which remove amino acids 3402–3511 and 3528–3675, respectively (Rafael et al. 1996). Transgenic mdx line Dp71 expresses the Dp71 isoform of dystrophin in striated muscle (Cox et al. 1994).
Morphology
Quadriceps, soleus, extensor digitorum longus (EDL), tibialis anterior, and diaphragm muscles were removed from mice, frozen in liquid nitrogen cooled O.C.T. embedding medium (Tissue-Tek), and cut into 7-μm sections. After fixing in 3.7% formaldehyde, sections were stained in hematoxylin and eosin–phloxine. Stained sections were imaged with a Nikon E1000 microscope connected to a Spot-2 CCD camera. To determine the percentage of fibers containing central nuclei, the number of muscle fibers with centrally-located nuclei was divided by the total number of muscle fibers.
Evans Blue Assays
4-mo-old control and Δ71–78 mice were analyzed after injection with Evans blue, as described previously (Straub et al. 1997). In brief, mice were tail vein–injected with 150 μl of a solution containing 10 mg/ml Evans blue dye in PBS (150 mM NaCl, 50 mM Tris, pH 7.4). After 3 h, the animals were euthanized and mouse tissues were either fixed in 3.7% formaldehyde/0.5% glutaraldehyde to observe gross dye uptake, or frozen unfixed in O.C.T. embedding medium. To examine Evans blue uptake by individual fibers, 7-μm-thick frozen sections were fixed in cold acetone and analyzed by fluorescence microscopy.
Immunofluorescence
Quadriceps and diaphragm muscles from C57Bl/10, mdx, and Δ71–78 mice were removed, frozen in O.C.T. embedding medium, and cut into 7-μm sections. Immunofluorescence was performed with previously described antibodies against dystrophin (NH2 terminus; Rafael et al. 1996), α1-syntrophin (SYN17; Peters et al. 1997a), β1-syntrophin (Peters et al. 1997a), α-dystrobrevin-1 (αDB670), α-dystrobrevin-2 (αDB2; Peters et al. 1998), and utrophin (Lumeng et al. 1999c). After incubation with primary antibodies, cryosections were incubated with an FITC-conjugated goat anti–rabbit secondary antibody and fluorescent images were viewed on a Nikon E1000 microscope. Antibodies to α-sarcoglycan (Rabbit 98), β-sarcoglycan (Goat 26), γ-sarcoglycan (Rabbit 245), δ-sarcoglycan (Rabbit 215), sarcospan (Rabbit 235), α-dystroglycan (Goat 20), β-dystroglycan (AP 83), or nNOS (Rabbit 200) have been described previously (Duclos et al. 1998). Cy3-conjugated secondary antibodies were used and images were viewed on a Bio-Rad MRC-600 laser scanning confocal microscope. All digitized images were captured under identical conditions.
Measurements of Contractile Properties
Contractile properties of muscles from 6-mo-old Δ71–78 transgenic mice were compared with those of C57Bl/10 wild-type and mdx mice using methods described previously (Lynch et al. 1997). The samples included eight muscles each from the EDL, soleus, and diaphragm. Mice were deeply anesthetized with avertin and each muscle was isolated and dissected free from the mouse. After removal of the limb muscles, the mice were euthanized with the removal of the diaphragm muscle. The muscles were immersed in a bath filled with oxygenated buffered mammalian Ringer's solution (137 mM NaCl, 24 mM NaHCO3, 11 mM glucose, 5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 1 mM NaH2PO4, and 0.025 mM tubocurarine chloride, pH 7.4). For each muscle, one tendon was tied to a servomotor and the other tendon to a force transducer. Muscles were stretched from slack length to the optimal length for force development and then stimulated at a frequency that produced absolute isometric tetanic force (mN). After the measurements of the contractile properties, the muscles were removed from the bath, blotted and weighed to determine muscle mass. Specific force (kN/m2) was calculated by dividing absolute force by total fiber cross sectional area.
Muscle Membrane Isolation
Muscle microsomes from 12–14-mo-old C57Bl/10, mdx, Δ71–78, Δ71–74, Δ75–78, and Dp71 mice were prepared as described previously (Ohlendieck et al. 1991a). In brief, skeletal muscle was homogenized in 7.5-vol homogenization buffer plus protease inhibitor Complete (Boehringer). The homogenate was centrifuged at 14,000 g for 15 min to remove cellular debris. The supernatant was filtered through cheesecloth and spun at 142,000 g for 37 min to collect microsomes. The microsome pellet was resuspended in KCl wash buffer (0.6 M KCl, 0.3 M sucrose, 50 mM Tris-HCl, pH 7.4) plus protease inhibitors and recentrifuged at 142,000 g for 37 min to obtain KCl-washed microsomes. The final pellet was resuspended in 0.3 M sucrose and 20 mM Tris-maleate, pH 7.0. Samples were quantified by the Coomassie Plus Protein Assay Reagent (Pierce Chemical Co.) and equivalent protein loading was verified by SDS-PAGE. KCl-washed microsomes were analyzed by Western blot using antibodies against β2-syntrophin (Peters et al. 1994), pan syntrophin (Froehner et al. 1987), nNOS (Transduction Laboratories), β-dystroglycan, α-sarcoglycan (Novocastra Laboratories), and other proteins described above.
Results
Generation of Dystrophin Δ71–78 Transgenic Mice
To test the function of a dystrophin protein lacking both the syntrophin and dystrobrevin binding sites, we prepared a cDNA expression vector deleted for the COOH-terminal domain (corresponding to exons 71–78; Fig. 1 A). The structure of several dystrophin transgenic constructs we had previously tested are also shown for comparison. Mice expressing the dystrophin Δ71–78 transgene were crossed onto the mdx background and dystrophin levels were analyzed by Western blotting. The expression of the dystrophin Δ71–78 transgene in skeletal muscle was ∼10-fold higher than endogenous dystrophin (Fig. 1 B). Immunofluorescent staining of quadriceps muscle using an antibody against the NH2-terminus of dystrophin revealed that the Δ71–78 protein was localized to the sarcolemma, similar to wild-type dystrophin (Fig. 1 C). Dystrophin Δ71–78 expression was also found to be uniform in the diaphragm, EDL, and soleus muscles, but the tibialis anterior muscle displayed a mosaic expression pattern and was not further analyzed (data not shown). The human skeletal muscle α-actin promoter used in this study was not expressed in either smooth or cardiac muscle (data not shown).
Figure 1.

Generation of dystrophin Δ71–78 transgenic mdx mouse. (A) Shown are the dystrophin transgenic expression vectors that contain Dp71 and full-length cDNAs deleted for the indicated exons. The binding sites for β-dystroglycan (β-DG), syntrophin (Syn), and dystrobrevin (Db) are delineated relative to the exon boundaries of the dystrophin gene. Exon 79 encodes only three amino acids and therefore is not shown. Dystrophin structural motifs are also indicated that include the WW, EF1, EF2, ZZ, and coiled coil (CC) 1 and 2 domains. The first half of the WW domain is encoded on exon 62. All constructs contain either a mouse muscle creatine kinase [MCK] or the HSA promoter, the SV40 VP1 intron (intron), and the SV40 poly-adenylation site (“A”). (B) Western blot analysis of total skeletal muscle proteins from wild-type, mdx, and dystrophin Δ71–78 mice using an NH2-terminal specific dystrophin antibody. (C) Immunofluorescent staining for dystrophin in muscle quadriceps reveals uniform expression in wild-type and dystrophin Δ71–78 mice. Scale bar, 50 μm.
Morphology of Dystrophin Δ71–78 Mice Appears Normal
We initially analyzed transgenic mdx mouse muscle tissues for morphological signs of dystrophy. Hematoxylin and eosin–stained limb and diaphragm skeletal muscle sections of dystrophin Δ71–78 mice revealed none of the signs of fibrosis, necrotic fibers, or mononuclear cell infiltration that were apparent in age-matched mdx controls (Fig. 2A and Fig. B, and data not shown). NMJs of transgenic mice stained with rhodamine-labeled α-bungarotoxin consistently appeared normal in contrast to the varying degrees of postsynaptic folding observed in mdx NMJs (Fig. 2 C). Mdx muscle fibers have previously been shown to be highly permeable to the vital dye Evans blue in vivo, reflecting damage to the dystrophic fiber sarcolemma (Matsuda et al. 1995). Skeletal muscle fibers from dystrophin Δ71–78 mice, like wild-type animals, were not permeable to Evans blue dye (Fig. 2D and Fig. E).
Figure 2.
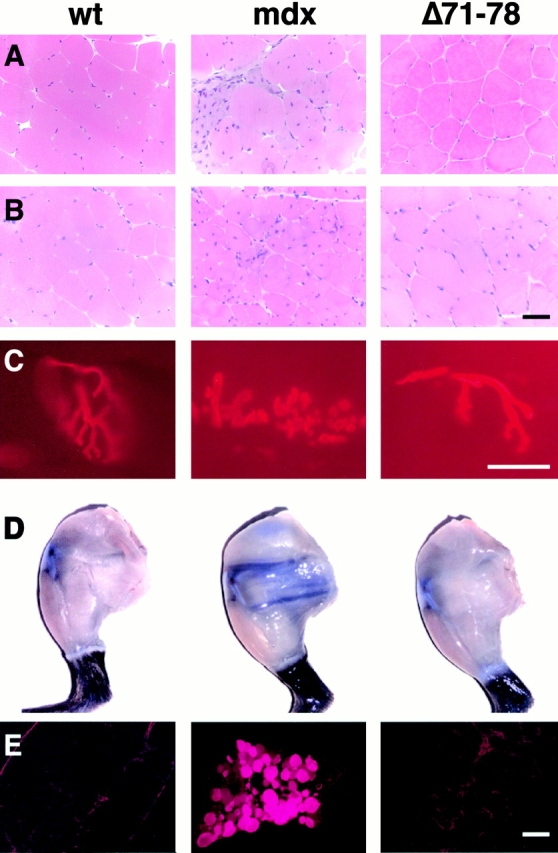
Dystrophin Δ71–78 skeletal muscles are morphologically similar to age-matched wild-type muscles. Hematoxylin and eosin staining of quadriceps shows no signs of necrosis or fibrosis in Δ71–78 mice that is apparent in mdx mice (A, 4-mo-old mice; B, 1-yr-old mice). (B) Occasional fibers in 1-yr-old Δ71–78 mice contain central nuclei. (C) Labeling of NMJs reveals no abnormalities in Δ71–78 transgenic muscles in comparison to wild-type muscles. (D) Evans blue dye is excluded from hind limb skeletal muscle fibers in wild-type and Δ71–78, but not mdx mice. (E) Cryosections from diaphragm muscle show Evans blue dye positive fibers only in mdx mice. Scale bars, 50 μm.
Analysis of Centrally Nucleated Muscle Fibers
Another hallmark of dystrophy in mdx mice is the presence of large numbers of centrally-nucleated muscle fibers, reflecting cycles of fiber degeneration and regeneration (Torres and Duchen 1987; Bockhold et al. 1998). To estimate the degree of myofiber regeneration occurring in Δ71–78 transgenic mice, we counted centrally nucleated fibers from a variety of muscle groups in age-matched wild-type, mdx, and Δ71–78 mice (Table ). By 4 mo of age, 71% of muscle fibers in mdx quadriceps muscles contained central nuclei, whereas wild-type muscles had <1%. Interestingly, 4-mo-old dystrophin Δ71–78 quadriceps muscles displayed ∼1% central nuclei, indicating that very little, if any, regeneration was occurring. However, when 1-yr-old mice were compared, an increase in centrally nucleated fibers became apparent. Quadriceps muscles from Δ71–78 mice contained 10% centrally nucleated fibers, although diaphragm muscles still displayed <1%. EDL and soleus muscles displayed 5 and 8% centrally nucleated fibers, respectively. For comparison, 1-yr-old wild-type mice had <1% centrally nucleated fibers in both limb and diaphragm muscles. Furthermore, 1-yr-old mdx limb muscles had 60% centrally nucleated fibers, whereas the diaphragm had 35%.
Table 1.
Percentage of Centrally Nucleated Fibers in Mouse Skeletal Muscles
| Line | Age | Quad | Dia | TA | EDL | Soleus |
|---|---|---|---|---|---|---|
| mo | % | |||||
| C57/Bl10 | 4 | <1 | <1 | ND | ND | ND |
| mdx | 4 | 71 | 58 | ND | ND | ND |
| 71–78 | 4 | 1 | <1 | ND | ND | ND |
| C57/Bl10 | 12 | <1 | <1 | <1 | <1 | <1 |
| mdx | 12 | 65 | 35 | 58 | 50 | 61 |
| 71–78 | 12 | 10 | <1 | ND | 5 | 8 |
| 71–74 | 15 | 5 | <1 | <1 | <1 | ND |
| 75–78 | 15 | 8 | <1 | 4 | 2 | 7 |
Quad, quadriceps; Dia, diaphragm; TA, tibialis anterior.
Our previous studies of transgenic mice expressing dystrophins deleted for exons 71–74 (Δ71–74) or exons 75–78 (Δ75–78) revealed no increase in the numbers of centrally nucleated fibers by 4 mo of age (Rafael et al. 1996). To contrast these mice with the Δ71–78 transgenics, central nuclei counts were performed on 15-mo-old Δ71–74 and Δ75–78 mice. We found that these animals had central nuclei counts in between those of wild-type and Δ71–78 mice. The Δ71–74 and Δ75–78 mice had 5 and 8% centrally nucleated fibers in quadriceps, respectively (Table ).
Contractile Properties
Compared with muscles of wild-type mice, those from mdx mice displayed a significant amount of necrosis, fibrosis, and infiltrating mononuclear cells. mdx skeletal muscles also displayed a loss of specific force–generating capacities when muscles were stimulated to contract in vitro, providing an extremely sensitive and quantitative measurement of the dystrophic process (Fig. 3 A). In contrast, dystrophin Δ71–78 mice had no major abnormalities when subjected to the same analysis (Fig. 3 B). Muscle mass for both EDL and diaphragm were not significantly different between dystrophin Δ71–78 and wild-type mice, whereas dystrophin Δ71–78 soleus muscles were slightly hypertrophied. When stimulated to contract, all three muscle groups displayed specific forces not significantly different from wild-type (P < 0.05). These results demonstrate that the dystrophin Δ71–78 protein has essentially the same functional capacity as the full-length protein.
Figure 3.

Contractile properties of EDL, soleus, and diaphragm muscles in wild-type, mdx, and dystrophin Δ71–78 mice. Muscle mass and specific force for mdx (A) and Δ71–78 (B) muscles were charted as a percentage of wild-type values. Significant differences (P < 0.05) are marked with an asterisk (*).
Localization of the DAP Complex in Δ71–78 Mice
Immunofluorescent analysis of the peripheral DAP complex revealed α1-syntrophin, β1-syntrophin, α-dystrobrevin-1, and α-dystrobrevin-2 to be localized at the sarcolemma with dystrophin, despite the lack of syntrophin and dystrobrevin binding sites in the transgene-encoded dystrophin (Fig. 4). α1-syntrophin levels were similar between wild-type and Δ71–78 mice. However, the levels of β1-syntrophin were elevated at the membrane in Δ71–78 mice, particularly in those fibers that normally express significant levels of this isoform. α-dystrobrevin-1 was primarily located at the NMJ in wild-type mice, and was exclusively located at the NMJs in mdx mice. Surprisingly, in dystrophin Δ71–78 mice, higher levels of α-dystrobrevin-1 were observed at the sarcolemma than in wild-type mice. The Δ71–78 mice also displayed a slight increase in utrophin localization along the sarcolemma, but this increase was less than the increase in mdx fibers. Immunofluorescent localization of the sarcoglycans, α- and β-dystroglycan, sarcospan, and nNOS in Δ71–78 mice revealed no differences in the expression of these proteins when compared with wild-type mice (Fig. 5). The proper localization of these proteins to the sarcolemma indicated that membrane targeting of the DAP complex components can proceed in the absence of the COOH-terminal domain of dystrophin.
Figure 4.
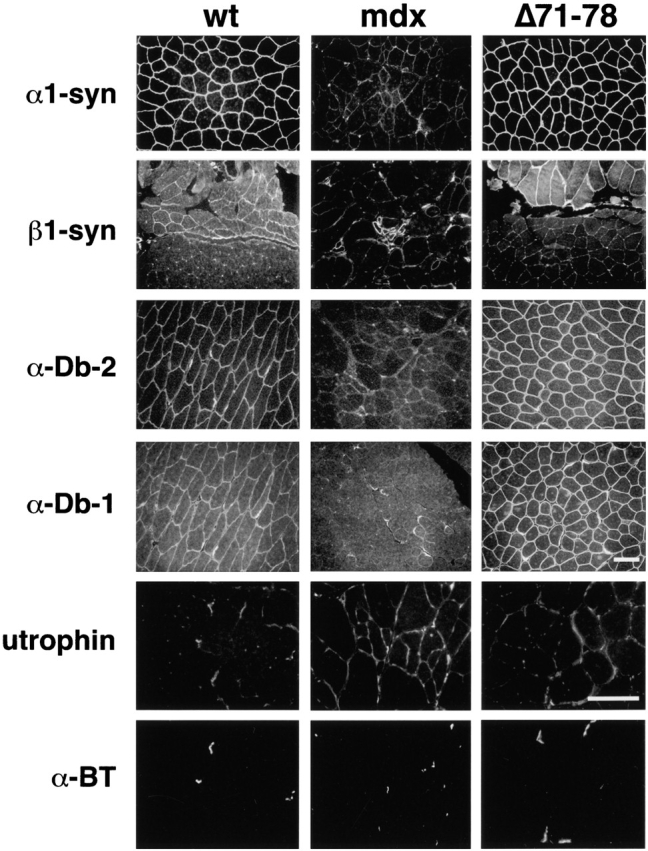
Peripheral DAP complex members are localized to the sarcolemma in quadriceps muscle of wild-type (wt) and Δ71–78 mice. Immunofluorescent staining was performed using antibodies against α1-syntrophin (α1-syn), β1-syntrophin (β1-syn), α-dystrobrevin-2 (α-Db-2), α-dystrobrevin-1 (α-Db-1), and utrophin. α-dystrobrevin-2 is primarily found at the sarcolemma in wild-type muscles. mdx muscle sections have no α1-syntrophin or α-dystrobrevin-2 staining at the sarcolemma, but α-dystrobrevin-1 remains at the NMJ. Elevated levels of β1-syntrophin were observed in regenerating fibers of mdx mice. Utrophin sections were double labeled with α-bungarotoxin (α-BT) to identify NMJs. Utrophin was expressed at the sarcolemma and in capillaries surrounding muscle fibers. Scale bars, 50 μm.
Figure 5.

Immunofluores-cent localization of the sarcoglycan complex (α-, β-, γ-, and δ-SG), dystroglycan complex (α- and β-DG), sarcospan (SSPN) and nNOS in wild-type and Δ71–78 skeletal muscle. All members of the complex are localized uniformly at the sarcolemma. Scale bar, 50 μm.
DAP Complex Protein Levels
To examine the levels of the DAP complex members that associate with dystrophin, muscle microsomes were prepared from wild-type and dystrophin Δ71–78 mice and analyzed by Western blotting (Fig. 6). This approach can provide data on the relative abundance of individual DAP complex members in muscles of separate lines of mice. Slightly elevated levels of β-dystroglycan were detected in dystrophin Δ71–78 mice, which we have previously observed whenever dystrophin is overexpressed (Cox et al. 1993; Rafael et al. 1994). Isoforms of syntrophin and dystrobrevin were present at slightly different levels when the dystrophin Δ71–78 membranes were compared with those from wild-type mice. α1-syntrophin and β2-syntrophin levels were lower than in wild-type mice, whereas the level of β1-syntrophin was elevated. Although there was approximately the same amount of α-dystrobrevin-2, there were elevated levels of α-dystrobrevin-1 in Δ71–78 microsomes. A reduction in nNOS was observed in dystrophin Δ71–78 muscle, indicating that nNOS binds weakly to the DAP complex in Δ71–78 mice. Levels of α-sarcoglycan were similar in all lines tested, and provided an internal control for protein loading.
Figure 6.
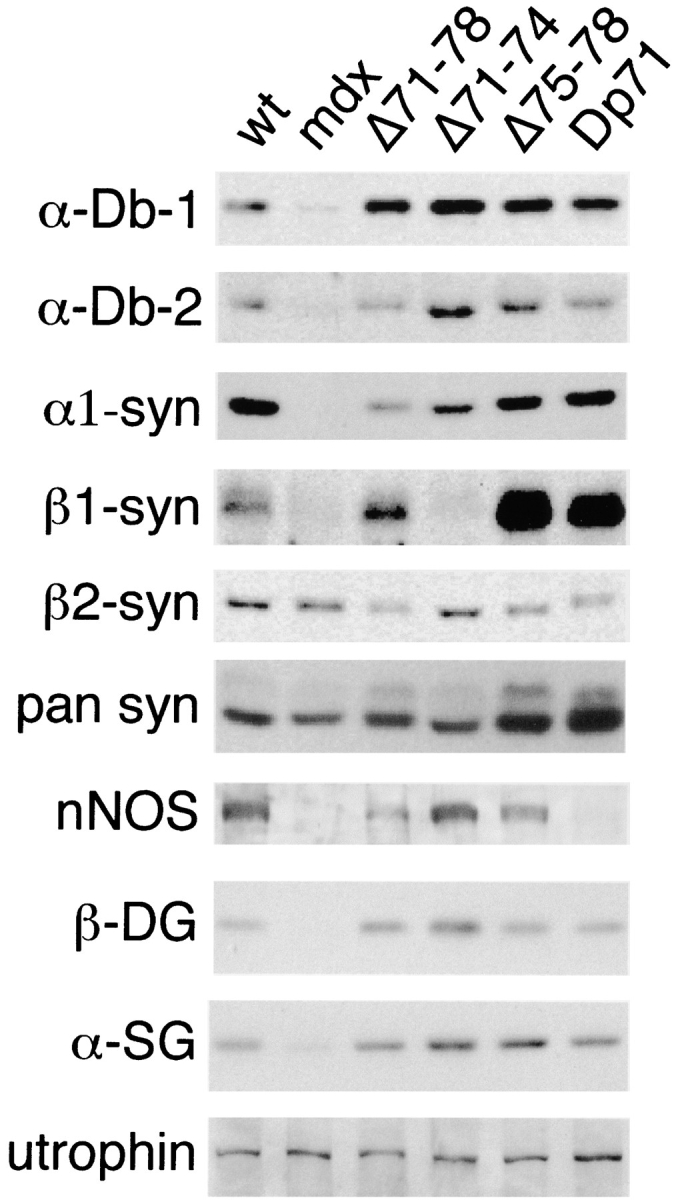
Western blot analysis of KCl-washed skeletal muscle microsome preparations from wild-type, mdx, Δ71–78, Δ71–74, Δ75–78, and Dp71 mice. 50 μg of microsome protein was analyzed by Western blot with antibodies against α-dystrobrevin-1 (α-Db-1), α-dystrobrevin-2 (α-Db-2), α1-syntrophin, β1-syntrophin, β2-syntrophin, pan syntrophins, nNOS, β-dystroglycan (β-DG), α-sarcoglycan (α-SG), and utrophin.
Since some DAP complex members exhibited isoform changes in Δ71–78 mice, we were prompted to look at purified microsomes from dystrophin Δ71–74 and Δ75–78 mice. Transgenic mdx mice that express the dystrophin isoform Dp71 in muscle were also included in this study since these dystrophic mice have the DAP complex present at the sarcolemma (Cox et al. 1994; Straub et al. 1997; Crosbie et al. 1998). α1-syntrophin levels were lower in all four transgenic lines compared with wild-type mice. Surprisingly, β1-syntrophin was absent in Δ71–74 microsomes but was highly overexpressed in Δ75–78 and Dp71 microsomes. The Δ71–74 microsomes had equivalent β2-syntrophin levels when compared with wild-type microsomes, but this isoform of syntrophin was reduced in both Δ75–78 and Dp71 microsomes. A pan syntrophin antibody, which detects all three isoforms of syntrophin, confirmed the upregulation of syntrophin in Δ75–78 and Dp71 microsomes. Similar to Δ71–78, α-dystrobrevin-1 was elevated in all dystrophin transgenic microsome preparations. However, in comparison with wild-type, α-dystrobrevin-2 was higher in Δ71–74 and Δ75–78, but equal in Dp71 microsomes. Contrary to the Δ71–78 mice, deleting either exons 71–74 or 75–78 restored nNOS to wild-type levels. However, Dp71 mice, which lack the NH2-terminal and rod domains of dystrophin, did not retain nNOS in the microsome fractions. Previous studies have also shown that utrophin is upregulated in mdx and Dp71 mice (Ohlendieck et al. 1991b; Tanaka et al. 1991; Cox et al. 1994). Therefore, we have compared utrophin levels in all transgenic lines and have found that Δ71–78, Δ71–74, and Δ75–78 mice do not have the elevated levels seen in mdx and Dp71 mice.
Discussion
The dystrophin-associated protein complex plays a critical role in protecting muscle cells from damage, but the function of each component of the DAP complex remains unclear. Integral membrane DAPs, such as β-dystroglycan and the sarcoglycans, appear to play primarily a structural role in linking the muscle cytoskeleton, via dystrophin, to the extracellular matrix. This structural link is critical for protecting muscle fibers from contraction-induced injuries and sarcolemmal damage (Petrof et al. 1993; Faulkner et al. 1997; Lynch et al. 2000). The importance of this link is illustrated by the observation that in-frame deletions within the dystroglycan binding domain of dystrophin (Δcys) inactivate the protective functions of the DAP complex and result in a dystrophy equivalent to if not worse than that resulting from the absence of dystrophin (Rafael et al. 1996). Similarly, mutations in sarcoglycan genes that destabilize the entire sarcoglycan complex typically result in a pathology resembling that resulting from dystrophin deficiency (Bönnemann et al. 1995; Noguchi et al. 1995; Nigro et al. 1996; Duclos et al. 1998).
In contrast, little is known about the peripheral dystrophin complex, and it is unclear whether it plays a structural and/or signaling role. Only one patient has been described with a deletion limited to the syntrophin and dystrobrevin binding domains of dystrophin. This patient (CM) had a deletion of exons 72–79 and displayed a mild Becker muscular dystrophy phenotype (McCabe et al. 1989; Chamberlain, J.S., unpublished observations). However, no data is available on the expression of dystrophin or other DAPs in this patient. We have studied the interaction between dystrophin, the syntrophins, and the dystrobrevins by disrupting the peripheral complex binding sites on dystrophin in vivo in mdx mice.
We found that the dystrophin Δ71–78 protein supported normal muscle function in mdx mice up to at least 6 mo of age, although very mild signs of muscle regeneration were observed in a subset of muscle groups in animals older than 1 yr. 6-mo-old Δ71–78 mice displayed normal muscle mass, morphology, NMJ structure, membrane integrity, and specific force generating capacities in at least three different muscle groups, including the diaphragm (Fig. 2 and Fig. 3; and Table ). Despite deletion of the binding sites on dystrophin for syntrophin and dystrobrevin, muscles from the Δ71–78 mice displayed an overtly normal DAP complex that included family members of both of these peripheral complex proteins, as well as nNOS (Fig. 4 and Fig. 5).
Because the lack of dystrophin in mdx mice destabilizes the entire DAP complex, it is clear that components of the DAP complex are required to localize syntrophin and dystrobrevin at the sarcolemma (Ohlendieck and Campbell 1991). Our observation that syntrophin and dystrobrevin assemble into a functional DAP complex in the absence of the dystrophin COOH-terminal domain provides several important insights into the structure and function of the DAP complex. First, this study indicates that dystrobrevin and/or syntrophin bind to another component of the DAP complex besides dystrophin. Second, the normal phenotype in the Δ71–78 mice demonstrates that a link between dystrophin and syntrophin or dystrobrevin is not critical for the function of the DAP complex.
Although young adult Δ71–78 mice displayed a normal phenotype, old transgenic mice displayed a slight increase in the presence of centrally nucleated fibers, suggesting a low level of limb muscle degeneration and regeneration. However, there were no signs of increased fibrosis or mononuclear cell infiltration, nor was there an increase in basophilic or hypercontracted fibers (Fig. 2 B, and data not shown). The number of centrally nucleated (and thus regenerated) muscle fibers was also slightly higher in 1-yr-old Δ71–78 mice than in either the Δ71–75 or Δ75–78 mice, indicating that removal of both the syntrophin and the dystrobrevin binding sites is slightly more detrimental than removal of individual sites. Interestingly, this low-level muscle regeneration was only observed in limb muscles of older transgenic mdx mice (Table ). Diaphragm muscle of 1-yr-old Δ71–78 transgenic mdx mice displayed no pathology, even though the diaphragm is the most severely affected muscle group in mdx mice (Stedman et al. 1991; Cox et al. 1993; Lynch et al. 1997). These results suggest that the peripheral DAPs play slightly different roles in different muscle types, and that these roles may require unique combinations of syntrophin and/or dystrobrevin isoforms.
A striking observation from the various lines of transgenic mice was that although all of the COOH-terminal deletions supported assembly of an overtly normal DAP complex, shifts were detected in the relative abundance of the separate dystrobrevin and syntrophin isoforms. Each of the transgenic mouse strains analyzed in Fig. 6 displayed an upregulation of α-dystrobrevin-1. In contrast, levels of α-dystrobrevin-2 displayed more modest changes by western analysis, with the least variation from wild-type found in the Δ71–78 muscles. Since α-dystrobrevin-1 is upregulated and redistributed in muscles expressing the truncated dystrophins (Fig. 4 and Fig. 6), we suggest that the alternate (non COOH-terminal dystrophin) binding site within the DAP complex may have a higher affinity for this isoform. Similarly, the COOH-terminal domain of dystrophin may have a higher affinity for α-dystrobrevin-2. Disruption of the interaction between the dystrophin COOH-terminal domain and α-dystrobrevin-2 may also make the alternate binding site more accessible to binding by α-dystrobrevin-1. This latter isoform has a tyrosine phosphorylated and extended COOH-terminal domain which might otherwise hinder access to the alternate binding site (Wagner et al. 1993; Blake et al. 1996; Balasubramanian et al. 1998; Peters et al. 1998).
The reduced level of α1-syntrophin in membrane preparations from Δ71–78 mice suggests that efficient binding of α1-syntrophin to the DAP complex requires an intact and functional dystrophin COOH terminus. Reductions of α1-syntrophin were associated with an upregulation of β1-syntrophin in each transgenic line studied, except for the line expressing a dystrophin deleted for the syntrophin binding domain (71–74). In this latter case, reduced levels of α1-syntrophin were associated with a marked upregulation of β2-syntrophin (Fig. 6). Pan-syntrophin antibodies revealed a relatively constant level of syntrophin expression in all mice studied, except for a peculiar upregulation in the Δ75–78 and Dp71 mice that was due to a large increase in β1-syntrophin expression coupled with a slight decrease in β2-syntrophin. The reduction of nNOS in microsome preparations from Δ71–78 mice could be a secondary result of weak α1-syntrophin interactions with the DAP complex. The loss of nNOS in Dp71 mice, which have normal levels of α1-syntrophin, may be a secondary consequence of the extensive pathology in these animals. Also, although Dp71 mice display elevated levels of utrophin, a significant increase was not observed in the Δ71–78 mice, indicating that ectopic utrophin expression does not account for syntrophin and dystrobrevin isoform changes. The highest utrophin levels were observed in mdx mice, which express very low levels of all DAP complex members (Fig. 6).
The Δ71–74 and Δ71–78 deletions we studied are expressed in humans and mice as naturally occurring isoforms of dystrophin (Feener et al. 1989; Bies et al. 1992; Tinsley et al. 1993). Dp71 is also a natural isoform expressed in non-muscle tissues including many cell types in the brain (Lederfein et al. 1993; Lumeng et al. 1999b; Sarig et al. 1999). Our results suggest that alternative splicing of the dystrophin COOH-terminal domains and expression of the smaller dystrophin transcripts may be important for regulating which isoforms of syntrophin and dystrobrevin assemble with the DAP complex in muscle or nonmuscle tissues.
Our results provide additional clues as to the potential signaling role of the peripheral DAP complex. The data in Fig. 6 suggest that a reduced association of nNOS with the DAP complex does not by itself contribute significantly to the dystrophic process. Mice deficient in nNOS have no abnormal muscle phenotype, and a complete lack of nNOS does not overtly exacerbate the mdx phenotype (Chao et al. 1998; Crosbie et al. 1998). These data indicate that nNOS is involved in functions separate from stabilizing the sarcolemma. Previous studies have suggested that nNOS plays a role in preventing ischemic damage in exercising muscles (Thomas et al. 1998). Mice with mutations in either dystrobrevin or syntrophin express a DAP complex that lacks nNOS, but only the dystrobrevin-deficient mice display a dystrophic phenotype (Grady et al. 1999; Kameya et al. 1999). In these dystrobrevin mutants, the remaining DAP complex is intact, indicating that the mode of dystrophy is likely different from that in mice lacking dystrophin or a sarcoglycan (Matsumura et al. 1993; Duclos et al. 1998; Hack et al. 1998; Straub et al. 1998; Araishi et al. 1999).
The ability to sever the connection between dystrophin and dystrobrevin without causing dystrophy demonstrates that the function of dystrobrevin does not depend on a tight connection to dystrophin. Such a tight connection would be expected for a protein playing a mechanical role facilitating force transduction between the muscle cytoskeleton and the extracellular matrix. Our results are therefore compatible with the suggestion by Grady et al. 1999 that dystrobrevin plays a signaling role in muscle. These observations are also in marked contrast to the severe muscle pathology present in mice with dystrophin deletions that remove the binding site for β-dystroglycan (Rafael et al. 1996).
Our results suggest that models for the organization of the DAP complex need to reflect an interaction between syntrophin and/or dystrobrevin and an additional protein besides dystrophin (Fig. 7). Utrophin is not a likely candidate, as we did not observe a correlation between utrophin levels and the expression of dystrobrevin and syntrophin isoforms (Fig. 4 and Fig. 6). We also cannot formally rule out the possibility of another binding site on dystrophin, although to date none have been detected. Yoshida et al. 2000 detected subcomplexes between dystrobrevin and the sarcoglycans when purified DAP complexes were fractionated after incubation in various conditions of ionic strength. Dystrobrevin might therefore attach to the DAP complex in the Δ71–78 mice via an interaction with the sarcoglycan complex, although the binding of either syntrophin or dystrobrevin to β-dystroglycan cannot be excluded. Another potential candidate to bind dystrobrevin or syntrophin is aquaporin-4, which was recently shown to be absent from the sarcolemma of α1-syntrophin knockout mice (Yokota et al. 2000). The identification of additional binding sites for syntrophin and dystrobrevin in the DAP complex should lead to a greater understanding of the function of the complex. Unraveling the complexities of the DAP complex will be critical to understanding how muscular dystrophies progress, and to the eventual development of therapies.
Figure 7.

Schematic diagrams of the dystrophin-associated protein complex. (A) Structure of the DAP complex in normal mouse skeletal muscle. This model reflects the presence of an interaction between dystrobrevin and the sarcoglycans and/or β-dystroglycan, in addition to dystrophin, as discussed in the text. Note that alternate binding sites to as yet unidentified DAP components are possible. (B) Proposed structure of the DAP complex in dystrophin Δ71–78 transgenic mdx mice. An interaction between dystrobrevin and the integral membrane components of the DAP complex is sufficient to localize syntrophin and dystrobrevin to the membrane in the absence of the dystrophin COOH-terminal domain.
Acknowledgments
We thank Cheryl Hassett and Paul Suding for assistance with contractile property studies; Rob Crawford and Sarah Wean for technical assistance; Sally Camper, Thom Saunders and the University of Michigan Transgenic Animal Core Facility for generation of mouse lines; and Edna Hardeman for the HSA promoter.
This study was supported by grants from the Muscular Dystrophy Association and by National Institutes of Health grants AR40864 and AG15434. Kevin P. Campbell is an investigator of the Howard Hughes Medical Institute.
Footnotes
Rachelle Crosbie's current address is Department of Physiological Science, UCLA, College of Life Sciences, Los Angeles, CA 90095.
Abbreviations used in this paper: DAP, dystrophin-associated protein; EDL, extensor digitorum longus; HSA, human α-skeletal actin; NMJ, neuromuscular junction; nNOS, neuronal nitric oxide synthase.
References
- Adams M.E., Dwyer T.M., Dowler L.L., White R.A., Froehner S.C. Mouse α1- and β2-syntrophin gene structure, chromosome localization, and homology with a discs large domain. J. Biol. Chem. 1995;270:25859–25865. doi: 10.1074/jbc.270.43.25859. [DOI] [PubMed] [Google Scholar]
- Ahn A.H., Kunkel L.M. Syntrophin binds to an alternatively spliced exon of dystrophin. J. Cell Biol. 1995;128:363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn A.H., Freener C.A., Gussoni E., Yoshida M., Ozawa E., Kunkel L.M. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J. Biol. Chem. 1996;271:2724–2730. doi: 10.1074/jbc.271.5.2724. [DOI] [PubMed] [Google Scholar]
- Amalfitano A., Rafael J.A., Chamberlain J.S. Structure and mutation of the dystrophin gene. In: Lucy J.A., Brown S.C., editors. DystrophinGene, Protein and Cell Biology. Cambridge University Press; Cambridge: 1997. pp. 1–26. [Google Scholar]
- Araishi K., Sasaoka T., Imamura M., Noguchi S., Hama H., Wakabayashi E., Yoshida M., Hori T., Ozawa E. Loss of the sarcoglycan complex and sarcospan leads to muscular dystrophy in beta-sarcoglycan-deficient mice. Hum. Mol. Genet. 1999;8:1589–1598. doi: 10.1093/hmg/8.9.1589. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S., Fung E.T., Huganir R.L. Characterization of the tyrosine phosphorylation and distribution of dystrobrevin isoforms. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1998;432:133–140. doi: 10.1016/s0014-5793(98)00804-7. [DOI] [PubMed] [Google Scholar]
- Bies R.D., Phelps S.F., Cortez M.D., Roberts R., Caskey C.T., Chamberlain J.S. Human and murine dystrophin mRNA transcripts are differentially expressed during skeletal muscle, heart, and brain development. Nucleic Acids Res. 1992;20:1725–1731. doi: 10.1093/nar/20.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake D.J., Tinsley J.M., Davies K.E., Knight A.E., Winder S.J., Kendrick-Jones J. Coiled-coil regions in the carboxy-terminal domains of dystrophin and related proteinspotentials for protein-protein interactions. Trends Biochem. Sci. 1995;20:133–135. doi: 10.1016/s0968-0004(00)88986-0. [DOI] [PubMed] [Google Scholar]
- Blake D.J., Nawrotzki R., Peters M.F., Froehner S.C., Davies K.E. Isoform diversity of dystrobrevin, the murine 87-kDa postsynaptic protein. J. Biol. Chem. 1996;271:7802–7810. doi: 10.1074/jbc.271.13.7802. [DOI] [PubMed] [Google Scholar]
- Blake D.J., Nawrotzki R., Loh N.Y., Gorecki D.C., Davies K.E. Beta-dystrobrevin, a member of the dystrophin-related protein family. Proc. Natl. Acad. Sci. USA. 1998;95:241–246. doi: 10.1073/pnas.95.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockhold K.J., Rosenblatt J.D., Partridge T.A. Aging normal and dystrophic mouse muscleanalysis of myogenicity in cultures of living single fibers. Muscle Nerve. 1998;21:173–183. doi: 10.1002/(sici)1097-4598(199802)21:2<173::aid-mus4>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Bönnemann C.G., Modi R., Noguchi S., Mizuno Y., Yoshida M., Gussoni E., McNally E.M., Duggan D.J., Angelini C., Hoffman E.P. β-Sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat. Genet. 1995;11:266–273. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- Bredt D.S. Knocking signalling out of the dystrophin complex. Nat. Cell Biol. 1999;1:E89–E91. doi: 10.1038/12085. [DOI] [PubMed] [Google Scholar]
- Brenman J.E., Chao D.S., Gee S.H., McGee A.W., Craven S.E., Santillano D.R., Wu Z., Huang F., Xia H., Peters M.F. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-Syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Brennan K.J., Hardeman E.C. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J. Biol. Chem. 1993;268:719–725. [PubMed] [Google Scholar]
- Butler M.H., Douville K., Murnane A.A., Kramarcy N.R., Cohen J.B., Sealock R., Froehner S.C. Association of the Mr 58,000 postsynaptic protein of electric tissue with Torpedo dystrophin and the Mr 87,000 postsynaptic protein. J. Biol. Chem. 1992;267:6213–6218. [PubMed] [Google Scholar]
- Chao D.S., Silvagno F., Bredt D.S. Muscular dystrophy in mdx mice despite lack of neuronal nitric oxide synthase. J. Neurochem. 1998;71:784–789. doi: 10.1046/j.1471-4159.1998.71020784.x. [DOI] [PubMed] [Google Scholar]
- Cox G.A., Cole N.M., Matsumura K., Phelps S.F., Hauschka S.D., Campbell K.P., Faulkner J.A., Chamberlain J.S. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature. 1993;364:725–729. doi: 10.1038/364725a0. [DOI] [PubMed] [Google Scholar]
- Cox G.A., Sunada Y., Campbell K.P., Chamberlain J.S. Dp71 can restore the dystrophin-associated glycoprotein complex in muscle but fails to prevent dystrophy. Nat. Genet. 1994;8:333–339. doi: 10.1038/ng1294-333. [DOI] [PubMed] [Google Scholar]
- Crosbie R.H., Straub V., Yun H.Y., Lee J.C., Rafael J.A., Chamberlain J.S., Dawson V.L., Dawson T.M., Campbell K.P. mdx muscle pathology is independent of nNOS perturbation. Hum. Mol. Genet. 1998;7:823–829. doi: 10.1093/hmg/7.5.823. [DOI] [PubMed] [Google Scholar]
- Duclos F., Straub V., Moore S.A., Venzke D.P., Hrstka R.F., Crosbie R.H., Durbeej M., Lebakken C.S., Ettinger A.J., van der Meulen J. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J. Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer T.M., Froehner S.C. Direct binding of Torpedo syntrophin to dystrophin and the 87 kDa dystrophin homologue. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1995;375:91–94. doi: 10.1016/0014-5793(95)01176-f. [DOI] [PubMed] [Google Scholar]
- Emery A.E.H. Duchenne Muscular Dystrophy 1993. Oxford Medical Publications; Oxford: pp. 392 [Google Scholar]
- Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Ervasti J.M., Campbell K.P. A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner J.A., Brooks S.V., Dennis R.G., Lynch G.S. The functional status of dystrophic muscles and functional recovery by skeletal muscles following myoblast transfer. Basic Appl. Myol. 1997;7:257–264. [Google Scholar]
- Feener C.A., Koenig M., Kunkel L.M. Alternative splicing of human dystrophin mRNA generates isoforms at the carboxy terminus. Nature. 1989;338:509–511. doi: 10.1038/338509a0. [DOI] [PubMed] [Google Scholar]
- Froehner S.C., Murnane A.A., Tobler M., Peng H.B., Sealock R. A postsynaptic Mr 58,000 (58K) protein concentrated at acetylcholine receptor-rich sites in Torpedo electroplaques and skeletal muscle. J. Cell Biol. 1987;104:1633–1646. doi: 10.1083/jcb.104.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee S.H., Madhavan R., Levinson S.R., Caldwell J.H., Sealock R., Froehner S.C. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J. Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady R.M., Grange R.W., Lau K.S., Maimone M.M., Nichol M.C., Stull J.T., Sanes J.R. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat. Cell Biol. 1999;1:215–220. doi: 10.1038/12034. [DOI] [PubMed] [Google Scholar]
- Grady R.M., Zhou H., Cunningham J.M., Henry M.D., Campbell K.P., Sanes J.R. Maturation and maintenance of the neuromuscular synapsegenetic evidence for roles of the dystrophin-glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/s0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- Hack A.A., Ly C.T., Jiang F., Clendenin C.J., Sigrist K.S., Wollmann R.L., McNally E.M. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J. Cell Biol. 1998;142:1279–1287. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M., Cuenda A., Spillantini M.G., Thomas G.M., Buee-Scherrer V., Cohen P., Goedert M. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha 1-syntrophina mechanism for specific substrate recognition. J. Biol. Chem. 1999;274:12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., Slaughter C.A., Sernett S.W., Campbell K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Jung D., Yang B., Meyer J., Chamberlain J.S., Campbell K.P. Identification and characterization of the dystrophin anchoring site on β-dystroglycan. J. Biol. Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- Kameya S., Miyagoe Y., Nonaka I., Ikemoto T., Endo M., Hanaoka K., Nabeshima Y., Takeda S. alpha1-syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J. Biol. Chem. 1999;274:2193–2200. doi: 10.1074/jbc.274.4.2193. [DOI] [PubMed] [Google Scholar]
- Koenig M., Hoffman E.P., Bertelson C.J., Monaco A.P., Feener C., Kunkel L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Kramarcy N.R., Sealock R. Syntrophin isoforms at the neuromuscular junctiondevelopmental time course and differential localization. Mol. Cell. Neurosci. 2000;15:262–274. doi: 10.1006/mcne.1999.0823. [DOI] [PubMed] [Google Scholar]
- Lederfein D., Yaffe D., Nudel U. A housekeeping type promoter, located in the 3′ region of the Duchenne muscular dystrophy gene, controls the expression of Dp71, a major product of the gene. Hum. Mol. Genet. 1993;2:1883–1888. doi: 10.1093/hmg/2.11.1883. [DOI] [PubMed] [Google Scholar]
- Lumeng C., Phelps S., Crawford G.E., Walden P.D., Barald K., Chamberlain J.S. Interactions between beta 2-syntrophin and a family of microtubule-associated serine/threonine kinases Nat. Neurosci 2 1999. 611 617a [DOI] [PubMed] [Google Scholar]
- Lumeng C.N., Hauser M., Brown V., Chamberlain J.S. Expression of the 71 kDa dystrophin isoform (Dp71) evaluated by gene targeting Brain Res 830 1999. 174 178b [DOI] [PubMed] [Google Scholar]
- Lumeng C.N., Phelps S.F., Rafael J.A., Cox G.A., Hutchinson T.L., Begy C.R., Adkins E., Wiltshire R., Chamberlain J.S. Characterization of dystrophin and utrophin diversity in the mouse Hum. Mol. Genet. 8 1999. 593 599c [DOI] [PubMed] [Google Scholar]
- Lynch G.S., Rafael J.A., Hinkle R.T., Cole N.M., Chamberlain J.S., Faulkner J.A. Contractile properties of diaphragm muscle segments from old mdx and old transgenic mdx mice. Am. J. Physiol. 1997;272:C2063–C2068. doi: 10.1152/ajpcell.1997.272.6.C2063. [DOI] [PubMed] [Google Scholar]
- Lynch G.S., Rafael J.A., Chamberlain J.S., Faulkner J.A. Contraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control mice. Am. J. Physiol. 2000;In press doi: 10.1152/ajpcell.2000.279.4.C1290. [DOI] [PubMed] [Google Scholar]
- MacGregor G.M., Caskey C.T. Construction of plasmids that express E Coli β-galactosidasein mammalian cells. Nucleic Acids Res. 171989. 2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda R., Nishikawa A., Tanaka H. Visualization of dystrophic muscle fibers in Mdx mouse by vital staining with evans blueevidence of apoptosis in dystrophin-deficient muscle. J. Biochem. (Tokyo) 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- Matsumura K., Lee C.C., Caskey C.T., Campbell K.P. Restoration of dystrophin-associated proteins in skeletal muscle of mdx mice transgenic for dystrophin gene. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1993;320:276–280. doi: 10.1016/0014-5793(93)80602-q. [DOI] [PubMed] [Google Scholar]
- McCabe E.R., Towbin J., Chamberlain J., Baumbach L., Witkowski J., van Ommen G.J., Koenig M., Kunkel L.M., Seltzer W.K. Complementary DNA probes for the Duchenne muscular dystrophy locus demonstrate a previously undetectable deletion in a patient with dystrophic myopathy, glycerol kinase deficiency, and congenital adrenal hypoplasia. J. Clin. Invest. 1989;83:95–99. doi: 10.1172/JCI113890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrotzki R., Loh N.Y., Ruegg M.A., Davies K.E., Blake D.J. Characterisation of alpha-dystrobrevin in muscle. J. Cell Sci. 1998;111:2595–2605. doi: 10.1242/jcs.111.17.2595. [DOI] [PubMed] [Google Scholar]
- Nigro V., de Sa Moreira E., Piluso G., Vainzof M., Belsito A., Politano L., Puca A.A., Passos-Bueno M.R., Zatz M. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the δ sarcoglycan gene. Nat. Genet. 1996;14:195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- Noguchi S., McNally E.M., Ben Othmane K., Hagiwara Y., Mizuno Y., Yoshida M., Yamamoto H., Bönnemann C.G., Gussoni E., Denton P.H. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819–822. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K., Campbell K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K., Ervasti J.M., Snook J.B., Campbell K.P. Dystrophin–glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma J. Cell Biol. 112 1991. 135 148a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K., Ervasti J.M., Matumura K., Kahl S.D., Leveille C.J., Campbell K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle Neuron 7 1991. 499 508b [DOI] [PubMed] [Google Scholar]
- Peters M.F., Kramarcy N.R., Sealock R., Froehner S.C. β2-syntrophinlocalization at the neuromuscular junction in skeletal muscle. NeuroReport. 1994;5:1577–1580. [PubMed] [Google Scholar]
- Peters M.F., Adams M.E., Froehner S.C. Differential association of syntrophin pairs with the dystrophin complex J. Cell Biol. 138 1997. 81 93a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M.F., O'Brien K.F., Sadoulet-Puccio H.M., Kunkel L.M., Adams M.E., Froehner S.C. β-dystrobrevin, a new member of the dystrophin familyidentification, cloning, and protein associations J. Biol. Chem. 272 1997. 31561 31569b [DOI] [PubMed] [Google Scholar]
- Peters M.F., Sadoulet-Puccio H.M., Grady M.R., Kramarcy N.R., Kunkel L.M., Sanes J.R., Sealock R., Froehner S.C. Differential membrane localization and intermolecular associations of alpha-dystrobrevin isoforms in skeletal muscle. J. Cell Biol. 1998;142:1269–1278. doi: 10.1083/jcb.142.5.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puca A.A., Nigro V., Piluso G., Belsito A., Sampaolo S., Quaderi N., Rossi E., Di Iorio G., Ballabio A., Franco B. Identification and characterization of a novel member of the dystrobrevin gene family. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1998;425:7–13. doi: 10.1016/s0014-5793(98)00097-0. [DOI] [PubMed] [Google Scholar]
- Rafael J.A., Sunada Y., Cole N.M., Campbell K.P., Faulkner J.A., Chamberlain J.S. Prevention of dystrophic pathology in mdx mice by a truncated dystrophin isoform. Hum. Mol. Genet. 1994;3:1725–1733. doi: 10.1093/hmg/3.10.1725. [DOI] [PubMed] [Google Scholar]
- Rafael J.A., Cox G.A., Corrado K., Jung D., Campbell K.P., Chamberlain J.S. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J. Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafael J.A., Townsend E.R., Squire S.E., Potter A.C., Chamberlain J.S., Davies K.E. Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum. Mol. Genet. 2000;9:1357–1367. doi: 10.1093/hmg/9.9.1357. [DOI] [PubMed] [Google Scholar]
- Sadoulet-Puccio H.M., Khurana T.S., Cohen J.B., Kunkel L.M. Cloning and characterization of the human homologue of a dystrophin related phosphoprotein found at the Torpedo electric organ post-synaptic membrane. Hum. Mol. Genet. 1996;5:489–496. doi: 10.1093/hmg/5.4.489. [DOI] [PubMed] [Google Scholar]
- Sadoulet-Puccio H.M., Rajala M., Kunkel L.M. Dystrobrevin and dystrophinan interaction through coiled-coil motifs. Proc. Natl. Acad. Sci. USA. 1997;94:12413–12418. doi: 10.1073/pnas.94.23.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarig R., Mezger-Lallemand V., Gitelman I., Davis C., Fuchs O., Yaffe D., Nudel U. Targeted inactivation of Dp71, the major non-muscle product of the DMD genedifferential activity of the Dp71 promoter during development. Hum. Mol. Genet. 1999;8:1–10. doi: 10.1093/hmg/8.1.1. [DOI] [PubMed] [Google Scholar]
- Schultz J., Hoffmüller U., Krause G., Ashurst J., Macias M.J., Schmieder P., Schneider-Mergener J., Oschkinat H. Specific interactions between the syntrophin PDZ domain and voltage-gated sodium channels. Nat. Struct. Biol. 1998;5:19–24. doi: 10.1038/nsb0198-19. [DOI] [PubMed] [Google Scholar]
- Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- Straub V., Rafael J.A., Chamberlain J.S., Campbell K.P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V., Duclos F., Venzke D.P., Lee J.C., Cutshall S., Leveille C.J., Campbell K.P. Molecular pathogenesis of muscle degeneration in the delta-sarcoglycan-deficient hamster. Am. J. Pathol. 1998;153:1623–1630. doi: 10.1016/s0002-9440(10)65751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Yoshida M., Yamamoto H., Ozawa E. Glycoprotein-binding site of dystrophin is confined to the cysteine-rich domain and the first half of the carboxy-terminal domain. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1992;308:154–160. doi: 10.1016/0014-5793(92)81265-n. [DOI] [PubMed] [Google Scholar]
- Suzuki A., Yoshida M., Ozawa E. Mammalian α1- and β1-syntrophin bind to the alternative splice-prone region of the dystrophin COOH terminus. J. Cell Biol. 1995;128:373–381. doi: 10.1083/jcb.128.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H., Ishiguro T., Eguchi C., Saito K., Ozawa E. Expression of a dystrophin-related protein associated with the skeletal muscle cell membrane. Histochemistry. 1991;96:1–5. doi: 10.1007/BF00266753. [DOI] [PubMed] [Google Scholar]
- Thomas G.D., Sander M., Lau K.S., Huang P.L., Stull J.T., Victor R.G. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. USA. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley J.M., Blake D.J., Davies K.E. Apo-dystrophin-3A 2.2kb transcript from the DMD locus encoding the dystrophin glycoprotein binding site. Hum. Mol. Genet. 1993;2:521–524. doi: 10.1093/hmg/2.5.521. [DOI] [PubMed] [Google Scholar]
- Torres L.F., Duchen L.W. The mutant mdxinherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110:269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- Wagner K.R., Cohen J.B., Huganir R.L. The 87K postsynaptic membrane protein from torpedo is a protein-tyrosine kinase substrate homologous to dystrophin. Neuron. 1993;10:511–522. doi: 10.1016/0896-6273(93)90338-r. [DOI] [PubMed] [Google Scholar]
- Yang B., Jung D., Rafael J.A., Chamberlain J.S., Campbell K.P. Identification of α-syntrophin binding to syntrophin triplet, dystrophin, and utrophin. J. Biol. Chem. 1995;270:4975–4978. doi: 10.1074/jbc.270.10.4975. [DOI] [PubMed] [Google Scholar]
- Yokota T., Miyagoe Y., Hosaka Y., Tsukita K., Kameya S., Shibuya S., Matsuda R., Wakayama Y., Takeda S. Aquaporin-4 is absent at the sarcolemma and at perivascular astrocyte endfeet in alpha 1-syntrophin knockout mice. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2000;76:22–27. [Google Scholar]
- Yoshida M., Hama H., Ishikawa-Sakurai M., Imamura M., Mizuno Y., Araishi K., Wakabayashi-Takai E., Noguchi S., Sasaoka T., Ozawa E. Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Hum. Mol. Genet. 2000;9:1033–1040. doi: 10.1093/hmg/9.7.1033. [DOI] [PubMed] [Google Scholar]
- Yoshida M., Yamamoto H., Noguchi S., Mizuno Y., Hagiwara Y., Ozawa E. Dystrophin-associated protein A0 is a homologue of the Torpedo 87K protein. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1995;367:311–314. doi: 10.1016/0014-5793(95)00574-s. [DOI] [PubMed] [Google Scholar]
- Zubrzycka-Gaarn E.E., Bulman D.E., Karpati G., Burghes A.H.M., Belfall B., Klamut H.J., Talbot J., Hodges R.S., Ray P.N., Worton R.G. The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature. 1988;333:466–469. doi: 10.1038/333466a0. [DOI] [PubMed] [Google Scholar]