

Abstract
Semliki Forest virus (SFV) is an efficient vector for cardiac gene delivery. The relatively short transgene expression induced by SFV seems appropriate for angiogenic gene therapy. We tested the effects of SFV expressing vascular endothelial growth factor (VEGF) on cardiac angiogenesis and heart failure in the mRen2 transgenic rat.
Six-week-old mRen2 rats received SFV-VEGF or control virus (n=7 each) administered intracoronarily. Twelve days after transfection, cardiac capillary density and function were assessed. Capillary density in cardiac regions where SFV expression was highest had decreased by 20% in the SFV-VEGF-treated group. The decrease in capillary density was accompanied by impaired systolic function as illustrated by increased endsystolic volumes and a 34% decrease in cardiac output.
We conclude that the time frame of SFV expression is sufficient to induce structural alterations, but that VEGF in mRen2 transgenic rats did not elicit the expected angiogenic effect. Rather, capillary density was decreased and subsequently cardiac function was impaired. This paradoxical finding is possibly related to the pathophysiology associated with this model and warrants caution if one is to pursue VEGF-mediated, angiogenic therapy before proceeding to a clinical setting. (Neth Heart J 2007;15:335-41.)
Keywords: heart failure, hypertension, Semliki Forest virus, vascular endothelial growth factor, angiogenesis
We have shown Semliki Forest virus (SFV) to be an efficient and selective vector for cardiac gene delivery with expression of a transgene lasting for at least a week.1 This time frame could be quite suitable to induce structural alterations that provide a lasting benefit to the heart. One such structural alteration is enhancement of cardiac perfusion through angiogenesis.
Hypertrophy is the common response of the heart to conditions such as hypertension or loss of viable myocardium due to myocardial infarction. The hypertrophy, typically, is not matched by sufficient angiogenesis, resulting in a decreased number of capillaries per tissue volume.2,3 The reduced perfusion capacity resulting from the decreased capillary density in combination with an increase in energy demand by the hypertrophied heart results in turn in a chronic (sub)- ischaemic state that hampers cardiomyocyte performance and facilitates the progression towards heart failure.4-6 Many experimental strategies have aimed at improving cardiac perfusion by inducing angiogenesis with various vascular growth factors, including vascular endothelial growth factor (VEGF).7,8 As methods are being sought for efficient and selective cardiac delivery of VEGF, the profile of SFV-delivered transgene expression may be suitable for this purpose.
We used the homozygous mRen2 transgenic rat to study the effect of intracoronarily delivered SFVVEGF on the cardiac microvasculature. In this animal model, the high levels of renin cause an elevated blood pressure leading to cardiac failure.9 We evaluated the cardiac capillary density in these rats 12 days after delivery of SFV-VEGF.
Materials and Methods
SFV-VEGF production
The plasmid carrying the human VEGF165 gene (GenBank accession no. AB021221) linked to a secretion signal, which is transcriptionally regulated by the cytomegalovirus promoter/enhancer, was manufactured under Good Manufacturing Practice guidelines according to Isner et al. (1996) and Sarkar et al. (2001).10,11 The plasmid was a gift from J.M. Isner, and was the same as has been used by his group. Human VEGF165 cDNA was amplified with primers containing BamHI restriction sites. The PCR product was purified and cloned into the pSFV1 plasmid. Correct orientation was verified by restriction analysis and sequence analysis. Similarly, pSFV1 encoding a non-relevant peptide (pSFV-ctrl) was constructed. SFV-VEGF, SFV-ctrl and SFV3-lacZ particles were produced in BHK cells with the pSFV3-Helper2 plasmid to obtain conditionally infectious, replication-deficient particles.12 Virus titres were determined by infection of BHK cells with serial dilutions of the SFVlacZ stock, subsequent histochemical staining for expression of β-galactosidase, and further calculation as described previously.12,13 Virus titres were found to be 5x108 IU/ml.
In vitro VEGF production
H9c2 cells were cultured in Dulbecco’s Modified Eagles Medium (DMEM; Invitrogen, Gibco, Breda, the Netherlands) with 10% foetal calf serum and 2% penicillin/streptomycin. Cells were seeded in 24-well clusters. When the cells were confluent, 0.003 μl, 0.01 μl, 0.03 μl, 0.1 μl and 0.3 μl SFV-VEGF stock (n=3 each) was added to the wells. To control wells, 0.1 μl SFV-ctrl (n=3) or no virus (n=2) was added. After 48 hours, the medium was collected and stored on ice. VEGF concentrations in the culture medium were measured by incubating 200 μl of the medium in an ELISA kit for human VEGF (R&D Systems, Abingdon, UK).
Animal experiments
Localisation of SFV transgene expression in noninfarcted myocardium was studied in seven male Hsd:SD rats (Harlan, Zeist, the Netherlands) that received SFV-lacZ. For evaluation of SFV-VEGF effects, homozygous mRen2 transgenic rats aged 6 weeks were obtained from the breeding colony at the Forschungseinrichtungen für Experimentelle Medizin (FEM), Berlin, Germany, and housed group-wise with free access to food and drinking water. Experiments were carried out at the FEM according to the United States National Institutes of Health (NIH) guidelines for the care and use of laboratory animals and were approved by the local authorities. SFV-VEGF and SFVctrl were delivered intracoronarily in seven rats each as described previously.1
Rats were anaesthetised with isoflurane (2% in O2), intubated and mechanically ventilated. During surgery animals were kept on a homeothermic blanket. A leftsided thoracotomy was made through the fifth intercostal space, and a loop was placed around the aorta and arteria pulmonalis. While the loop was tightened, 200 μl viral solution was injected into the left ventricular lumen using a 29 G needle. Upon injection, the entire cardiac wall showed a clear discoloration as the injected solution was pumped into the coronary circulation. Within 30 seconds the needle was removed and the loop loosened. During clamping of the main artery, the heart rate slowed considerably, but restored rapidly after removal of the loop. Subsequently, the thorax was closed, and when spontaneous respiration was sufficiently restored the rats were extubated and allowed to recover on a homeothermic blanket.
SFV-lacZ treated rats were anaesthetised with isoflurane 48 hours after virus delivery. The heart was excised, perfused with ice-cold saline via the aortic root, and sliced into transversal sections of ±3 mm. Sections were embedded in tissue tek OCT compound (Sakura Finetek Europe B.V., Zoeterwoude, the Netherlands) and frozen in liquid nitrogen for cryosectioning.
Haemodynamic measurements
Twelve days after virus delivery, the mRen2 rats were anaesthetised with chloral hydrate, intubated and mechanically ventilated. A 2 F microconductance pressure catheter (ARIA SPR-783; Millar-Instruments, Inc., Texas, USA) was positioned in the left ventricle (LV) for continuous registration of LV pressure volume (PV) loops in a closed-chest model, as described previously.14 Calibration of the volume signal was obtained by the hypertonic saline (10%) wash-in technique.15 Indices of cardiac function were derived from PV data obtained both at steady state and during transient preload reduction by occlusion of the abdominal vena cava. Systolic function was quantified by maximal LV systolic pressure (LVPmax), by dP/dt max as an index of LV contractility, ejection fraction (EF), end-systolic volume (ESV), and end-systolic pressurevolume relationship (ESPVR), an indicator of cardiac contractility. Furthermore, diastolic function was measured by LV end-diastolic pressure (LVEDP), dP/dt min, Tau, and the end-diastolic pressure-volume relationship (EDPVR), an indicator of LV stiffness that was determined from an exponential fit to the enddiastolic pressure-volume points. Global cardiac function was analysed by the parameters stroke volume (SV), cardiac output (CO), and heart rate (HR).
Histology
From SFV-lacZ-treated hearts, 4 μm cryosections were cut, mounted on glass slides, fixated with 1.25% glutaric aldehyde, rinsed with phosphate buffer, stained for two hours in X-gal substrate solution (1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl2, and 10% N,N-dimethylformamide in phosphate buffer) and counterstained with eosin.
Paraffin-embedded mid-ventricular samples of mRen2 rats were cut into 4 μm sections. For assessment of capillary density, sections were stained with biotin-labelled lectin (Sigma-Aldrich, Zwijndrecht, the Netherlands). To determine myocyte dimensions, sections were stained with Gomori staining (chemical purchased from Merck, Darmstadt, Germany) to visualise the circumference of the cardiomyocytes. To measure fibrosis, sections were stained with Sirius red (Klinipath, Duiven, the Netherlands) collagen staining combined with fast green (Klinipath) counterstaining of cytoplasma. Subepicardial and subendocardial transversal fields in the free LV wall were photographed at 400x magnification and the number of capillaries per ventricular area was measured (lectin-stained sections) as well as myocyte areas (Gomori-stained sections). Fibrosis was assessed globally in the left ventricle from photos taken at a magnification of 200x and counting of the total number of red pixels representing Sirius red-stained connective tissue. All measurements were performed with ImagePro software (Media Cybernetics Inc. Maryland, USA).
Statistics
Reported values are the mean ± the standard error of the mean (SEM). The Student’s t-test was used to evaluate the differences between the groups. A p value ≤0.05 was considered statistically significant.
Results
In vitro VEGF production
The cardiomyocyte cell line, H9c2, secreted increasingly high concentrations of VEGF into the culture medium after transfection with increasingly low amounts of SFVVEGF. Addition of SFV-VEGF amounts higher than indicated in figure 1 led to VEGF levels above the maximum concentration that can be detected and are therefore omitted from the figure. In contrast, VEGF levels were not elevated in the medium of VEGF-ctrltransfected cells compared with basal levels (figure 1).
Figure 1.
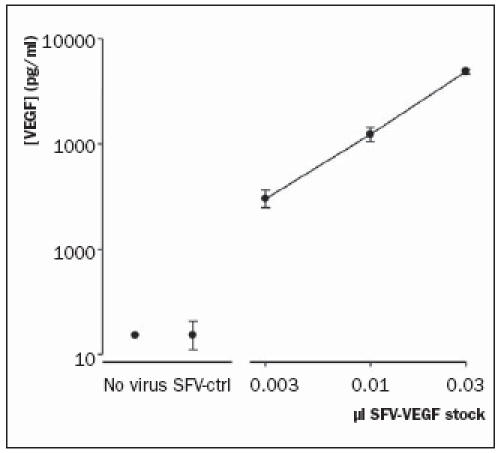
VEGF production in the medium collected from H9c2 cells 48 hours after infection with various amounts of SFV-VEGF. N=3 per data point.
Localisation of SFV-delivered transgene In X-gal stained sections of SFV-lacZ treated hearts, blue cells were visible all over the left and right ventricle, but expression was most prominent in the flow area of the left anterior descending coronary artery, i.e. in the free left ventricular wall, more specifically in the epicardial region as was also observed in MI hearts in a previous study (figure 2).1
Figure 2.
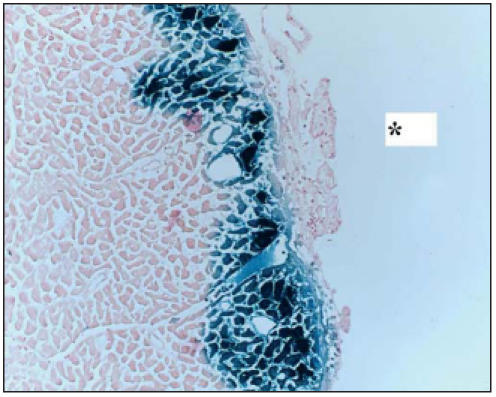
LacZ expression in rat left ventricle. Rat hearts were transfected with 108 IU SFV-LacZ. After 48 hours, the hearts were stained for β-galactosidase activity. In non-infarcted hearts, transfection efficiency was highest in the free wall of the left ventricle, particularly in epicardial regions, as was also observed in sections of infarcted hearts from a previous study.1 * left ventricular pericardial side.
Cardiac SFV-VEGF delivery in mRen2 rats
Twelve days after virus delivery, all rats were alive. General characteristics such as body weight and heart weight were similar between the SFV-ctrl and SFVVEGF groups (table 1).
Table 1.
General characteristics of mREN2 transgenic rats after treatment.
| SFV-ctrl | SFV-VEGF | P | |
|---|---|---|---|
| N | 7 | 7 | |
| Body weight (g) | 182.6±9.9 | 167.9±12.5 | 0.374 |
| Heart weight (g) | 1.12±0.11 | 0.97±0.11 | 0.349 |
| HW/BW | 6.05±0.29 | 5.71±0.32 | 0.453 |
Data are the mean ± SEM. HW/BW=heart to body weight ratio (mg/g).
Capillary density and further histological changes after SFV-VEGF delivery
Both macroscopically and histologically, SFV-VEGF treated hearts of mRen2 rats did not display obvious structural abnormalities. However, the density of lectin-reactive capillaries was markedly and significantly lower in epicardial left ventricular myocardium from SFV-VEGF-treated mRen2 rats compared with SFVctrl- treated rats. Capillary density in endocardial areas was similar between the groups (figure 3A). As capillary density might show an inverse correlation with myocyte hypertrophy, left ventricular myocyte dimensions were measured. No differences in myocyte area were observed either between treatment groups or epicardial and endocardial regions (figure 3B).
Figure 3.
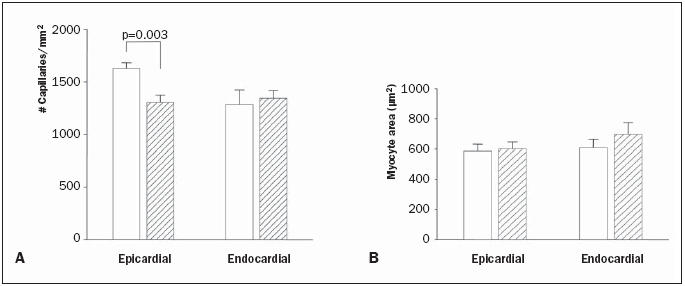
Capillary density (A) and mean myocyte size (B) in the left ventricular wall in SFV-ctrl (open bars) and SFV-VEGF (hatched bars) treated mRen2 transgenic rats, 12 days after coronary virus delivery.
Cardiac fibrosis was scored in sections stained for connective tissue using Sirius red and counterstaining of cytoplasm with fast green. There was no difference between SFV-VEGF- and SFV-ctrl-treated hearts (2179±889 vs. 2005±758 pixels per high power field, respectively).
Cardiac function
LVP was similar in both groups revealing arterial hypertension. Although left ventricular end-systolic and end-diastolic pressures were similar between the two groups, a differential effect of SFV-VEGF was observed on diastolic function and cardiac contractility. Diastolic heart function remained relatively intact showing only a difference in dP/dt min but not in LVEDP, τ, or EDPVR (table 2). Cardiac contractility was impaired in the SFV-VEGF-treated group as illustrated by a non-significant reduction in the load-dependent parameter dP/dt max, but a significant reduction of the load-independent parameter ESPVR (figure 4). In addition, pressure-volume loops in VEGF-treated rats were smaller and showed a significantly increased endsystolic volume. As a consequence, ejection fraction, stroke volume, and cardiac output were decreased in the VEGF group by approximately 35% (table 2).
Table 2.
Haemodynamic parameters.
| SFV-ctrl | SFV-VEGF | P | |
|---|---|---|---|
| HR | 347±14 | 345±19 | 0.926 |
| LVPmax | 152±6 | 155±5 | 0.787 |
| LVEDP | 21±2 | 20±2 | 0.846 |
| dPdtmax | 5479±546 | 4262±572 | 0.163 |
| dPdtmin | -5800±496 | -3932±438 | 0.022 |
| T | 11.6±0.5 | 11.8±0.2 | 0.844 |
| PHT | 8.55±0.49 | 9.12±0.50 | 0.440 |
| VES | 99.4±5.8 | 147.9±14.8 | 0.016 |
| VED | 257.5±28.5 | 240.7±9.1 | 0.590 |
| SV | 169.3±25.9 | 109.3±5.4 | 0.053 |
| CO | 58±8 | 38±3 | 0.049 |
| EF | 59±6 | 39±5 | 0.037 |
| ESPVR | 1.67±0.26 | 1.18±0.14 | 0.042 |
| EDPVR | 0.0110±0.0028 | 0.0089±0.0026 | 0.590 |
| Ea | 1.08±0.14 | 1.42±0.05 | 0.017 |
Haemodynamic parameters in mREN2 rats 12 days after intracoronary delivery of SFV-ctrl or SFV-VEGF. Data represent the mean±SEM. HR=heart rate (bpm), LVPmax=maximal LV pressure (mmHg), LVEDP=LV end-diastolic pressure (mmHg), dPdtmax/dPdtmin=maximal rate of pressure increase/decrease (mmHg/s), T=time constant of relaxation (ms), PHT=pressure half-time (ms), VES=end-systolic volume (μl), VED=end-diastolic volume (μl), SV=stroke volume (μl), CO=cardiac output (ml/min), EF=ejection fraction (%), ESPVR=slope of the end-systolic pressure volume relationship (mmHg/μl), EDPVR=end-diastolic pressure-volume relationship (ml-1), Ea=arterial elastance (mmHg/ml).
Figure 4.
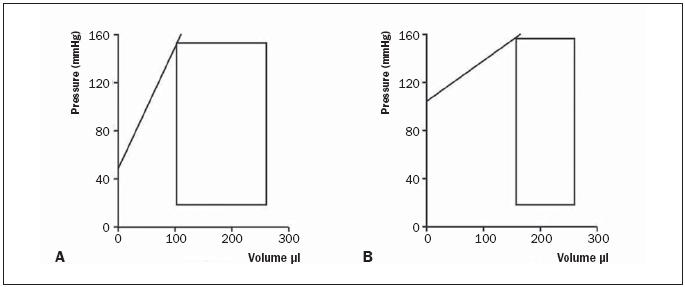
Schematic pressure volumes in (A) SFV-ctrl and (B) SFV-VEGF. Black line indicating slope of the end-systolic pressure-volumerelationship, a parameter for systolic cardiac contractility.
Discussion
Angiogenic (gene) therapy with vascular growth factors has been studied extensively, but a fail-safe protocol has not yet been defined. In a previous study we described that intracoronarily delivered SFV caused cardiospecific transgene expression for seven days and that the expression was strictly confined to the heart; results that seemed promising for successful angiogenic gene therapy.1 Therefore, we constructed a pSFV1 plasmid containing human VEGF165 cDNA to produce SFV-VEGF particles. The myocardial cell line H9c2, when transfected with this recombinant virus, secreted vast amounts of VEGF, indicating that the virus particles were functional. Subsequently, the effect of SFV-VEGF on cardiac function and histology was tested in the mRen2 transgenic rat model for hypertensioninduced heart failure.
Homozygous mRen2 transgenic rats display profound cardiac hypertrophy in combination with decreased capillary density.15 We therefore considered this rat model appropriate to study angiogenic gene therapy. Moreover, the consequences of VEGF overexpression have never been tested in a pressure-induced model for cardiac hypertrophy and dysfunction. In rats treated with control virus, epicardial capillary density was 17% higher in epicardial regions compared with endocardial regions. Similar regional differences in capillary density have been reported both in normal and in pathologically hypertrophied hearts.16-18 Unexpectedly, intracoronarily delivered SFV-VEGF caused a marked decrease in capillary density, when compared with control virus. This decrease was only observed in epicardial regions of the left ventricular wall, coinciding with the areas that displayed the highest level of transgene expression in lacZ transfected hearts. This suggests that the decrease is actually mediated by SFVmediated VEGF expression.
Gene therapy with VEGF is generally accepted as an efficient means to induce angiogenesis. VEGF165 gene therapy was shown to induce angiogenesis in normal rat heart,8 and in mouse19 and pig20,21 hearts that were made ischaemic by mechanical obstruction. To our knowledge, we are the first to study VEGF effects in pressure overload-induced heart failure. The only study that conceptually resembles the present study is a recent paper in which overexpression of VEGF decoy receptors was tested in the mouse aortic banding model for pressure-induced heart failure.22 In this study it was shown that pressure overload leads to increased VEGF levels. Without VEGF stimulus, angiogenesis could not take place in pressure-overloaded hearts, leading to impaired compensatory hypertrophy. The present study shows that further stimulation of VEGF after pressure overload leads to adverse effects through yet unknown mechanisms. Our present findings demonstrating reduced capillary density in the epicardial region of pressure overloaded hearts are in obvious contrast with the earlier reports in other types of models. The reasons for this disparity are not obvious. The level or duration of VEGF expression could have been insufficient to induce angiogenesis, but that would have resulted in the absence of any effect, rather than a decrease in vessel density. Too high local VEGF concentration in combination with acute ischaemia, on the other hand, has been described to cause angioma formation,23 which was not found in our study. An intriguing alternative to explain the observed effects is that the underlying pathophysiology in the mRen2 rats influences their response to VEGF. In contrast to the aforementioned animal models, the mRen2 rats have documented endothelial dysfunction,9 indicating that the NO availability in these rats is impaired. NO is crucial for proper VEGF signalling,24 and endothelial dysfunction impedes cardiac angiogenesis.25-27 Whether VEGF application in the absence of appropriate NO signalling can cause degradation of capillaries has not been described. Independently of this question, the reduction in capillary density induced by SFV-VEGF delivery caused functional changes in cardiac performance in the mRen2 rat.
The mRens rat model used in the current study is characterised by increased LV systolic pressure that is dependent on increased blood pressure. This leads to cardiac hypertrophy and consequently to diastolic dysfunction, indicated by increased LV end-diastolic pressure in both groups. The relevance of the observed reduction in capillary density is underlined by the decrease in cardiac systolic function. It is intriguing to speculate that this decrease in capillary density leads to cardiac ischaemia in the VEGF group. Chronic ischaemia may result in impaired cardiac systolic function directly due to impaired oxygen supply and/or indirectly due to cardiac remodelling. In our experiments a reduced systolic function is apparent. The decrease in the load-independent parameter for cardiac contractility, ESPVR, is a clear indicator for impaired cardiac systolic function. Because of the impaired cardiac contractility VEGF-treated animals were no longer able to cope with the increased blood pressure. This results in pressure-volume loops with decreased stroke volumes followed by decreased ejection fraction and cardiac output as hallmarks for early cardiac decompensation, further indicated by increased arterial elastance.
In conclusion, we show that intracoronarily delivered SFV-VEGF decreases epicardial capillary density in mRen2 transgenic rats leading to impaired cardiac performance. The cause of this paradoxical effect is unclear and will need further study, but is possibly related to the pathophysiology of the mRen2 rat. Nevertheless, the present study warrants further examination of the application of VEGF in other models of pressure overload-induced cardiac hypertrophy whilst preparing to make the step to a clinical setting.
Acknowledgements
The authors are indebted to Dr J.M. Smit for facilitating the virus production, to Dr H. Fechner for facilitating our animal research, to A. Dikkers for assisting in the in vitro experiments, and to B.J. Meijering for performing the virus delivery.
References
- 1.Loot AE, Henning RH, Deelman LE, et al. Semliki Forest virus is an efficient and selective vector for gene delivery in infarcted rat heart. J Mol Cell Cardiol 2004;37:137-42. [DOI] [PubMed] [Google Scholar]
- 2.Anversa P, Ricci R, Olivetti G. Coronary capillaries during normal and pathological growth. Can J Cardiol 1986;2:104-13. [PubMed] [Google Scholar]
- 3.Anversa P, Capasso JM, Sonnenblick EH, et al. Mechanisms of myocyte and capillary growth in the infarcted heart. Eur Heart J 1990;11(Suppl B):123-32. [DOI] [PubMed] [Google Scholar]
- 4.Lei L, Zhou R, Zheng W, et al. Bradycardia induces angiogenesis, increases coronary reserve, and preserves function of the postinfarcted heart. Circulation 2004;110:796-802. [DOI] [PubMed] [Google Scholar]
- 5.Friehs I, del Nido PJ. Increased susceptibility of hypertrophied hearts to ischemic injury. Ann Thorac Surg 2003;75:S678-84. [DOI] [PubMed] [Google Scholar]
- 6.Ashruf JF, Ince C, Bruining HA. Regional ischemia in hypertrophic Langendorff-perfused rat hearts. Am J Physiol 1999;277: H1532-9. [DOI] [PubMed] [Google Scholar]
- 7.Siddiqui AJ, Blomberg P, Wardell E, et al. Combination of angiopoietin-1 and vascular endothelial growth factor gene therapy enhances arteriogenesis in the ischemic myocardium. Biochem Biophys Res Commun 2003;310:1002-9. [DOI] [PubMed] [Google Scholar]
- 8.Sylven C, Sarkar N, Wardell E, et al. Protein and angiogenic doseresponse expression of phVEGF-A(165) gene in rat myocardium. J Thromb Thrombolysis 2001;12:151-6. [DOI] [PubMed] [Google Scholar]
- 9.Pinto YM, Buikema H, van Gilst WH, et al. Cardiovascular endorgan damage in Ren-2 transgenic rats compared to spontaneously hypertensive rats. J Mol Med 1997;75:371-7. [DOI] [PubMed] [Google Scholar]
- 10.Isner JM, Pieczek A, Schainfeld R, Blair R, Haley, L, Asahara, T, et al. Clinical evidence of angiogenesis after arterial gene transfer of phVEGF165 in patient with ischaemic limb. Lancet 1996; 348:370-4. [DOI] [PubMed] [Google Scholar]
- 11.Sarkar N, Rück A, Källner G, Y-Hassan S, Blomberg P, Islam KB, et al. Effects of intramyocardial injection of phVEGF-A165 as sole therapy in patients with refractory coronary artery disease 12-month follow-up: Angiogenic gene therapy. J Intern Med 2001;250:373-81. [DOI] [PubMed] [Google Scholar]
- 12.Berglund P, Sjoberg M, Garoff H, et al. Semliki Forest virus expression system: production of conditionally infectious recombinant particles. Biotechnology 1993;11:916-20. [DOI] [PubMed] [Google Scholar]
- 13.Daemen T, Pries F, Bungener L, Kraak M, Regts J, Wilschut J. Genetic immunization against cervical carcinoma: induction of cytotoxic T lymphocyte activity with a recombinant alphavirus vector expressing human papillomavirus type 16 E6 and E7. Gene Therapy 2000;7:1859-66. [DOI] [PubMed] [Google Scholar]
- 14.Walther T, Steendijk P, Westermann D, et al. Angiotensin deficiency in mice leads to dilated cardiomyopathy. Eur J Pharmacol 2004;493:161-5. [DOI] [PubMed] [Google Scholar]
- 15.Bachmann S, Peters J, Engler E, et al. Transgenic rats carrying the mouse renin gene—morphological characterization of a low-renin hypertension model. Kidney Int 1992;41:24-36. [DOI] [PubMed] [Google Scholar]
- 16.White FC, Witzel G, Breisch EA, et al. Regional capillary and myocyte distribution in normal and exercise trained male and female rat hearts. Am J Cardiovasc Pathol 1988;2:247-53. [PubMed] [Google Scholar]
- 17.Stoker ME, Gerdes AM, May JF. Regional differences in capillary density and myocyte size in the normal human heart. Anat Rec 1982;202:187-91. [DOI] [PubMed] [Google Scholar]
- 18.Gerdes AM, Callas G, Kasten FH. Differences in regional capillary distribution and myocyte sizes in normal and hypertrophic rat hearts. Am J Anat 1979;156:523-31. [DOI] [PubMed] [Google Scholar]
- 19.Su H, Joho S, Huang Y, et al. Adeno-associated viral vector delivers cardiac-specific and hypoxia-inducible VEGF expression in ischemic mouse hearts. Proc Natl Acad Sci USA 2004;101: 16280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rutanen J, Rissanen TT, Markkanen JE, et al. Adenoviral cathetermediated intramyocardial gene transfer using the mature form of vascular endothelial growth factor-D induces transmural angiogenesis in porcine heart. Circulation 2004;109:1029-35. [DOI] [PubMed] [Google Scholar]
- 21.Tio RA, Tkebuchava T, Scheuermann TH, et al. Intramyocardial gene therapy with naked DNA encoding vascular endothelial growth factor improves collateral flow to ischemic myocardium. Hum Gene Ther 1999;10:2953-60. [DOI] [PubMed] [Google Scholar]
- 22.Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension 2006;47:887-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarz ER, Speakman MT, Patterson M, et al. Evaluation of the effects of intramyocardial injection of DNA expressing vascular endothelial growth factor (VEGF) in a myocardial infarction model in the rat—angiogenesis and angioma formation. J Am Coll Cardiol 2000;35:1323-30. [DOI] [PubMed] [Google Scholar]
- 24.Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res 2001;49:568-81. [DOI] [PubMed] [Google Scholar]
- 25.Voisine P, Bianchi C, Ruel M, et al. Inhibition of the cardiac angiogenic response to exogenous vascular endothelial growth factor. Surgery 2004;136:407-15. [DOI] [PubMed] [Google Scholar]
- 26.Ruel M, Wu GF, Khan TA, et al. Inhibition of the cardiac angiogenic response to surgical FGF-2 therapy in a Swine endothelial dysfunction model. Circulation 2003;108(Suppl 1):II335-40. [DOI] [PubMed] [Google Scholar]
- 27.Voisine P, Bianchi C, Khan TA, et al. Normalization of coronary microvascular reactivity and improvement in myocardial perfusion by surgical vascular endothelial growth factor therapy combined with oral supplementation of l-arginine in a porcine model of endothelial dysfunction. J Thorac Cardiovasc Surg 2005;129:1414-20. [DOI] [PubMed] [Google Scholar]