

Abstract
Muscle damage has been shown to enhance the contribution of bone marrow–derived cells (BMDCs) to regenerating skeletal muscle. One responsible cell type involved in this process is a hematopoietic stem cell derivative, the myelomonocytic precursor (MMC). However, the molecular components responsible for this injury-related response remain largely unknown. In this paper, we show that delivery of insulin-like growth factor I (IGF-I) to adult skeletal muscle by three different methods—plasmid electroporation, injection of genetically engineered myoblasts, and recombinant protein injection—increases the integration of BMDCs up to fourfold. To investigate the underlying mechanism, we developed an in vitro fusion assay in which co-cultures of MMCs and myotubes were exposed to IGF-I. The number of fusion events was substantially augmented by IGF-I, independent of its effect on cell survival. These results provide novel evidence that a single factor, IGF-I, is sufficient to enhance the fusion of bone marrow derivatives with adult skeletal muscle.
Introduction
There is now extensive documentation that bone marrow–derived cells (BMDCs) can contribute to diverse nonhematopoietic tissues such as skeletal muscle, liver, brain, and epithelia after a bone marrow transplant (BMT) of genetically marked cells (GFP+, LacZ+, or Y chromosome+; Ferrari et al., 1998; Bittner et al., 1999; Gussoni et al., 1999; Lagasse et al., 2000; Mezey et al., 2000; Krause et al., 2001; LaBarge and Blau, 2002; Brazelton et al., 2003; Weimann et al., 2003; Brazelton and Blau, 2005). The physiological relevance of these events has been called into question because of their relatively low frequency (Wagers et al., 2002; Massengale et al., 2005). Nonetheless, several published studies have demonstrated that the contribution of BMDCs to adult skeletal muscle is an inherent property of these cells, which increases markedly in response to tissue stress or injury. First, diverse muscles exhibit an intrinsic 1,000-fold difference (from 0.005 to 5.0%) in their ability to incorporate BMDCs (Brazelton et al., 2003). Second, when muscle damage is inflicted by toxin injection, muscle exercise, or muscle overload, a substantial increase in skeletal muscle fibers that incorporate BMDCs is observed (Fukada et al., 2002; LaBarge and Blau, 2002; Camargo et al., 2003; Dreyfus et al., 2004; Palermo et al., 2005). Third, in parabiosis experiments in which two animals are surgically joined so that they share chimeric blood, BMDCs transit via the circulation to damaged muscle and fuse with skeletal myofibers in the absence of BMT-associated variables such as lethal irradiation and dysregulation of cytokines (Sherwood et al., 2004a; Palermo et al., 2005).
BMDCs may contribute to muscle by diverse mechanisms. We and others previously proposed that BMDCs en route to muscle can follow a biological progression. Initially, they give rise to cells with many properties typical of muscle-specific stem cells (satellite cells) and then fuse to mature myofibers under damage conditions, such as toxin-induced injury or exercise on a running wheel (Fukada et al., 2002; LaBarge and Blau, 2002; Dreyfus et al., 2004). Recently, another study showed that BMDCs that incorporate into muscle after BMT do not exhibit full myogenicity on par with endogenous cells; however, the finding that they do not fuse with each other but do fuse with muscle cells both in vitro and in vivo suggests that they share several features with myogenic cells (Sherwood et al., 2004b). It is quite possible that a proportion of BMDCs fuse directly with myofibers without passage through the satellite cell compartment. Thus, these mechanisms of BMDC contribution to skeletal muscle may coexist.
Several studies have addressed the nature of the BMDCs capable of contributing to adult skeletal muscle. When the blood was reconstituted after transplantation of a single hematopoietic stem cell (HSC), labeled myofibers containing BMDCs were readily detected in skeletal muscle, showing that these cells have the capacity to generate derivatives that fuse with muscle (Camargo et al., 2003; Corbel et al., 2003). The relevant HSC derivative that participates in muscle regeneration has been identified as the same as that suggested for liver (Willenbring et al., 2004), the myelomonocytic precursor (MMC; Lin−ckit+Sca1−; Camargo et al., 2003; Doyonnas et al., 2004; Ojima et al., 2004). After injection of notexin or cardiotoxin into the muscle, incorporation of MMCs into regenerating muscle fibers was observed. Thus, myeloid HSC derivatives have been shown to be a source of cells that can contribute to muscle fibers.
To date, knowledge of the molecular mechanisms and components responsible for BMDC contribution to muscle has been lacking. An identification of soluble factors involved in this “damage response” is of fundamental interest and is crucial if we are to increase this relatively rare event in a therapeutic context. One of the many potentially relevant soluble factors is insulin-like growth factor I (IGF-I), a secreted growth factor that regulates several diverse cellular pathways, including survival, proliferation, and differentiation (Stewart and Rotwein, 1996). Two major classes of IGF-I isoforms that result from alternative splicing have been described previously (Goldspink and Yang, 2004; Shavlakadze et al., 2005). IGF-I class 2 proteins are primarily produced in the liver and secreted into the circulation and are the major endocrine effectors of growth hormone (Yakar et al., 1999). Accordingly, conditional knockout mice in which IGF-I expression was abrogated only in the liver exhibited postnatal growth retardation, despite normal levels of growth hormone (Sjogren et al., 2002). By contrast, IGF-I class 1 isoforms are widely expressed in most tissues and remain localized within the tissue in which they are expressed. In adult skeletal muscle, two class 1 isoforms of IGF-I have been identified, IGF-IEa and mechano growth factor (MGF; Yang et al., 1996; McKoy et al., 1999), both of which are secreted but retained within muscle tissue (LeRoith and Roberts, 1991) and are up-regulated upon muscle damage (Hill and Goldspink, 2003; Hill et al., 2003). MGF has been shown in vitro to stimulate myoblast proliferation, but studies in vivo are lacking (Yang and Goldspink, 2002). IGF-IEa induces muscle hypertrophy, blocks muscle atrophy, and has been shown to be critical to satellite cell activation and regeneration of skeletal muscle (Sommerland et al., 1989; Barton-Davis et al., 1998; Musaro et al., 2001; Hill and Goldspink, 2003; Stitt et al., 2004; Schakman et al., 2005). IGF-I–null mice die immediately after birth and exhibit multiple tissue defects, including signs of skeletal muscle dystrophy (Liu et al., 1993; Powell-Braxton et al., 1993). Mice transgenic for IGF-IEa under the control of a muscle-specific promoter display marked muscle hypertrophy after birth (Musaro et al., 2001). A recent study using mice transgenic for IGF-IEa suggested that this factor could play a role in the contribution of BMDCs to skeletal muscle (Musaro et al., 2004) because after muscle damage an increased incidence of bone marrow–derived ckit+ cells present in skeletal muscle was observed relative to wild-type mice. However, interpretation of these experiments is complicated by the facts that IGF-I was present from conception onward and that multiple other damage-associated factors were up-regulated by extensive tissue injury.
In this paper, we investigated whether in the presence of mild damage, IGF-I alone could enhance the contribution of BMDCs to mature muscle fibers of adult wild-type mice. Three modes of IGF-I delivery yielded similar results, showing that the effects were not the result of the experimental paradigm used. IGF-I increased the number of GFP+ myofibers in wild-type mice that had received a BMT from GFP+ transgenic donors up to fourfold relative to untreated controls. To understand the underlying mechanism of IGF-I action, hematopoietic subfractions were co-cultured with differentiating myotubes in the presence or absence of IGF-I. We observed an increase in fusion between MMCs and myotubes that could be dissociated from the well-documented effect of IGF-I in increasing cell survival (Minshall et al., 1997; Burgess et al., 2003). Fusion was dependent on the culture media and stage of differentiation of the cells. These results are novel in that they demonstrate that a single factor, IGF-I, is sufficient to promote the formation of heterokaryons between myelomonocytic BMDC derivatives and differentiated muscle cells and can enhance their contribution to adult skeletal muscle in vivo.
Results
DNA electrotransfer of IGF-I increases contribution of BMDCs to adult skeletal muscle
Electrotransfer of DNA has been shown to be an efficient strategy for introducing genes into skeletal myofibers (Mir et al., 1999; Vilquin et al., 2001; Gollins et al., 2003; Schakman et al., 2005). The exposure of tissues to electric pulses produces a transient and reversible permeabilization of cell membranes, allowing DNA to enter target cells. The efficiency of uptake of electroporated naked DNA has been reported to be as high as 40% (Bertrand et al., 2003; Dona et al., 2003) of total myofibers, considerably higher than the 1% uptake reported for injected naked DNA (Wolff et al., 1990).
The two muscle-specific IGF-I isoforms IGF-IEa and MGF were selected for study. Both isoforms were previously reported to be up-regulated upon muscle damage (Hill and Goldspink, 2003). To confirm that this was the case, mouse skeletal muscle tissues were injected with notexin, a potent inducer of muscle damage (Harris et al., 1974; Pluskal et al., 1978; Klein-Ogus and Harris, 1983). RT-PCR analysis of total RNA from muscle revealed an increase in IGF-IEa and MGF transcripts 8 d after injury (Fig. 1 a).
Figure 1.
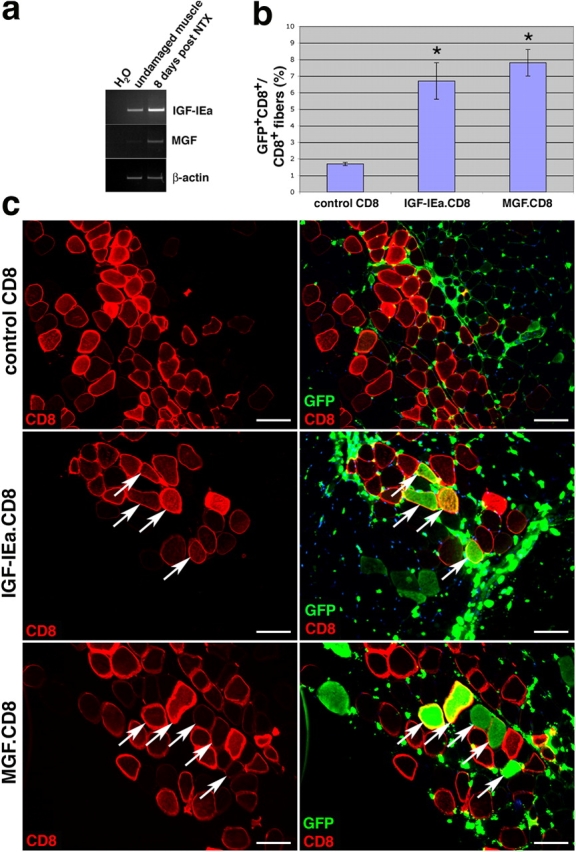
DNA electrotransfer of IGF-I increases the contribution of BMDCs to skeletal muscle. (a) The two muscle-specific isoforms, IGF-IEa and MGF, are shown to be up-regulated in the muscle after notexin damage as shown by RT-PCR analysis. (b) Plasmids encoding IGF-IEa.CD8, MGF.CD8, or CD8 only (control) were electroporated into the TA muscles of mice that had received a BMT 8 wk earlier, and muscles were harvested 4 wk after DNA delivery. The percentage of GFP+CD8+ myofibers relative to total CD8+ myofibers is shown (± SEM). P value was determined with a t test. *, P < 0.02. (c) Immunofluorescence images of transverse sections of muscle stained for CD8 only (left) or for GFP and CD8 (right). GFP+ cells <10 μm are blood cells. Arrows indicate examples of CD8+ myofibers that were also GFP+. Bars, 50 μm.
To determine whether IGF-IEa or MGF could substitute for extensive muscle damage as a signal to enhance BMDC contribution to muscle, they were each delivered to skeletal muscle by DNA electrotransfer. cDNAs encoding the two isoforms were obtained by RT-PCR of total RNA from damaged muscle and cloned into the pVIVO vector (see Materials and methods). The expression of IGF-I was linked to that of CD8 by an internal ribosome entry site sequence (IGF-IEa.CD8 and MGF.CD8), so that IGF-I–expressing electroporated myofibers could be identified by expression of CD8 on their membranes.
Tibialis anterior (TA) muscles of GFP-BMT wild-type mice 8 wk after transplantation were electroporated with plasmids encoding one of the two isoforms of IGF-I and CD8, or only CD8. Muscles were harvested from five mice in each of the three groups 4 wk later and analyzed for the presence of GFP+ myofibers (Fig. 1, b and c). Myofibers that were transduced by electrotransfer were identified by the expression of CD8 on their membranes (Fig. 1 c, left). Coincident immunofluorescent staining for CD8 and GFP allowed a determination of the frequency of myofibers that had incorporated BMDCs from the circulation (GFP+) relative to those expressing the IGF-I isoforms (CD8+; Fig. 1 c, right, arrows). GFP+ interstitial cells (<10 μm) visible in muscle transverse sections are donor-derived blood cells. The expression of IGF-I isoforms by myofibers led to a marked increase in the contribution of cells from the circulation, as demonstrated by the increase in GFP+ myofibers among the total CD8+ myofibers, relative to controls (P < 0.02; Fig. 1 b).
The effects of IGF-I supersede the effects of damage because of the mode of delivery. Electroporation appeared to result in mild damage, as the frequency of BMDC incorporation observed in the control (CD8 only; Fig. 1 b) was somewhat greater than that which we typically observed in undamaged muscle (∼0.05%; LaBarge and Blau, 2002; Brazelton et al., 2003). To establish the extent of damage, the percentage of centrally nucleated myofibers (a standard method to measure skeletal muscle injury) was assessed and determined to be 5.4 ± 0.3% in the three experimental groups tested (control, Igf-IEa, and MGF). By contrast, muscles damaged by injection of notexin, a standard means of experimentally inducing tissue damage, exhibited 73.4 ± 8.2% centrally nucleated myofibers. Thus, the damage caused by electroporation is relatively mild. To ensure that the increase in contribution of BMDCs to skeletal muscle was not attributable to the mode of IGF-I delivery but rather to the growth factor itself, two other modes of delivery were tested.
Myoblast-mediated IGF-I delivery increases contribution of BMDCs to adult skeletal muscle
Genetically engineered myoblasts provide a potent means for delivering proteins to skeletal muscle tissues. After injection into adult skeletal muscle, these cells fuse with the preexisting muscle fibers of the host. Myoblast-mediated gene delivery to skeletal or heart muscle was first shown to lead to physiological levels of secreted proteins in the circulation (e.g., human growth hormone; Barr and Leiden, 1991; Dhawan et al., 1991). Subsequently, localized secretion of proteins such as VEGF-A within heart and skeletal muscle tissues has been achieved by myoblast-mediated delivery, leading to novel insights into its mechanism of action in angiogenesis (Springer et al., 1998; Lee et al., 2000; Ozawa et al., 2004). Primary myoblasts previously engineered to express β-galactosidase (β-gal; Rando and Blau, 1994) were transduced with a retroviral vector encoding IGF-IEa.CD8, MGF.CD8, or an empty control vector (CD8 cassette only; Springer and Blau, 1997; Springer et al., 1998). All transduced cells were isolated by flow cytometry (FACS) using an antibody to CD8. IGF-I expression in these myoblast populations was confirmed by Western blot (Fig. 2 a).
Figure 2.

Myoblast-mediated delivery of IGF-I increases the contribution of BMDCs to skeletal muscle. (a) Western blot analysis of IGF-I expression by retrovirally transduced myoblasts isolated based on CD8 expression using FACS. IGF-IEa migrates as a 14-kD protein and MGF as a 12-kD protein. (b) 5 ×105 myoblasts were injected into the TA muscles of mice that had received a BMT 8 wk earlier, and muscles were harvested 4 wk after cell injection. The percentage of GFP+β-gal+ myofibers relative to total β-gal+ myofibers is shown (± SEM). P value was determined with a t test. *, P < 0.02. (c) Muscle sections were analyzed for β-gal expression (top), and serial sections were analyzed by immunofluorescence staining for GFP and laminin (bottom). Arrows indicate examples of β-gal+ myofibers that were also GFP+. Bars, 15 μm.
Wild-type mice that had been lethally irradiated and had their blood reconstituted by transplantation of GFP-expressing whole bone marrow were injected 8 wk later in the TA muscles with 5 × 105 myoblasts. These myoblasts expressed one of the two isoforms of IGF-I as well as CD8 or only CD8 (control). Muscles were harvested 4 wk later from five mice in each of the three groups and analyzed for the presence of GFP+ myofibers (Fig. 2, b and c). β-gal expression indicates those myofibers that incorporated myoblasts expressing IGF-IEa, MGF, or CD8 only, whereas GFP expression indicates those myofibers that incorporated BMDCs from the circulation. Consistent with the results obtained by electrotransfer, the expression of IGF-I isoforms by myofibers led to a marked increase in the contribution of cells from the circulation as demonstrated by the increase in GFP+ myofibers among the total β-gal+ myofibers (P < 0.02; Fig. 2 b). These results suggest that IGF-I, which is up-regulated together with many other factors in the course of muscle damage, can elicit a similar response on its own. In the myoblast-mediated delivery paradigm the muscle damage assessed as the percentage of centrally nucleated myofibers was 1.1 ± 0.4%. For comparison and to rule out the possibility that this effect was the result of myoblast injection and a consequent increase in the fusogenic environment, a third mode of IGF-I delivery was tested.
Injection of recombinant IGF-I protein increases the contribution of BMDCs to adult skeletal muscle
To prove definitively that IGF-I protein increases the contribution of BMDCs to muscle, the recombinant IGF-I protein was delivered by intramuscular injection. The commercially available recombinant protein is a purified peptide of 70 amino acids corresponding to exons 3 and 4 of the IGF-I gene, which constitutes the mature processed peptide with domains B, C, A, and D (Bell et al., 1986; Shimatsu and Rotwein, 1987). This delivery system is substantially different from the two methods described in Figs. 1 and 2 because it is transient. Different amounts of IGF-I (1, 5, and 10 μg) were injected into the TA muscles of wild-type mice that had received a BMT with GFP-labeled bone marrow 8 wk earlier, and the muscles of three mice in each group were analyzed 4 wk later. A dose-dependent increase in the number of total GFP+ myofibers per transverse section was observed in IGF-I–injected relative to the saline-injected muscles (P < 0.01; Fig. 3). By contrast, injection of a cytokine, IL-4, recently reported to play a role in the fusion of myoblasts with myocytes to form multinucleated myotubes (Horsley et al., 2003), did not increase the frequency of GFP+ myofibers (unpublished data). The extent of muscle damage assessed as the percentage of centrally nucleated myofibers was 3.3 ± 0.8%, using this mode of IGF-I delivery.
Figure 3.
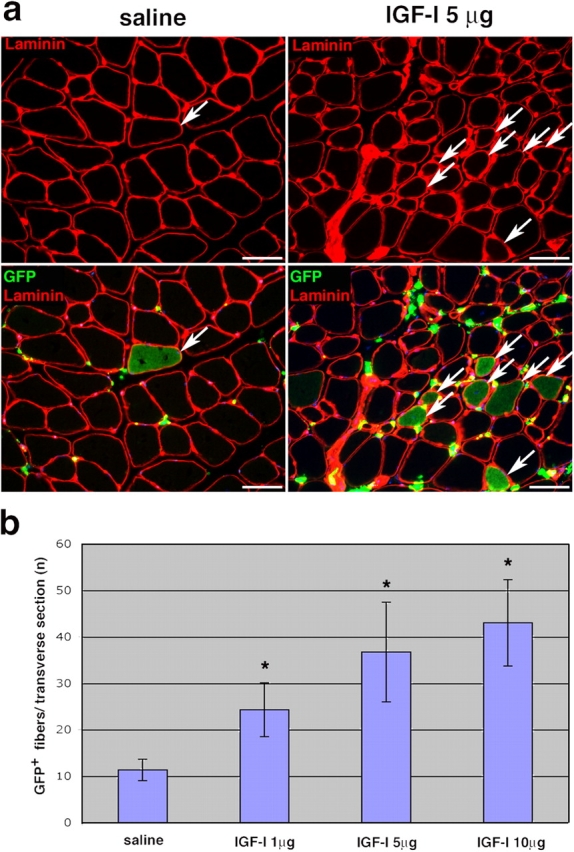
Recombinant IGF-I protein injection increases the contribution of BMDCs to skeletal muscle. (a) Saline (control) or recombinant IGF-I protein (1, 5, and 10 μg) was injected into the TA muscles of mice that had received a BMT 8 wk earlier, and muscles were harvested 4 wk after protein injection. Immunofluorescence images of transverse sections of muscles injected with saline or 5 μg IGF-I and stained for laminin only (top) or for GFP and laminin (bottom) are shown. Arrows indicate GFP+ myofibers. (b) The total numbers of GFP+ myofibers per transverse section are shown (± SEM). Asterisk indicates values that were significantly different from the control (P < 0.05). P value was determined with a t test. Significance of the values from the dose–response trendline was calculated by using the Pearson correlation coefficient (P < 0.01). Bars, 50 μm.
Because all three modes of IGF-I delivery resulted in a substantial fourfold increase in BMDC contribution to adult skeletal muscle, these effects can be attributed to IGF-I, not the delivery system used. The damage induced in all cases was minor (1–5% centrally nucleated myofibers). Yet, the frequency of GFP+ myofibers observed with all three modes of IGF-I delivery was similar to that previously reported after notexin tissue injury (∼75% centrally nucleated myofibers; LaBarge and Blau, 2002; Palermo et al., 2005), suggesting that IGF-I leads to a contribution of BMDCs to muscle on par with extensive muscle damage.
MMCs fuse spontaneously with differentiated myotubes in vitro
To elucidate the mechanism underlying the IGF-I–enhanced BMDC contribution to skeletal muscle in vivo, BMDCs were co-cultured with myogenic cells in vitro. Because we and others previously showed that the bone marrow derivatives of HSCs capable of fusing with myofibers in vivo are the MMCs (Camargo et al., 2003; Doyonnas et al., 2004; Ojima et al., 2004), experiments were designed to assess whether MMCs in co-culture with muscle cells were able to fuse with myotubes. Various experimental conditions were tested (Fig. 4 a). When freshly isolated MMCs were immediately co-cultured with myoblasts in low-serum myoblast differentiation medium (DM), fusion events were never observed, even after 6 d (Fig. 4 a, 1). By contrast, when MMCs were pretreated with high-serum myoblast growth medium (GM), fusion was observed. For instance, when MMCs and myoblasts were each cultured in GM for 3 d and then co-cultured in DM, GFP+ myotubes containing myogenin-expressing nuclei were detected as early as 4 d (Fig. 4, a [2] and b–b”). Notably, when MMCs were cultured in GM for 3 d and then plated together with 3-d-old myotubes in DM, GFP+ myotubes containing myogenin-expressing nuclei were visible as early as 12 h after co-culture (Fig. 4 a, 3), and >90% of GFP+ myotubes contained three or more nuclei (not depicted). Together, these results suggest that exposure to GM is a prerequisite for MMCs to fuse. In addition, these “activated” MMCs can fuse with well-differentiated multinucleated myotubes. In none of these conditions in eight independent experiments were unfused GFP+ mononucleated bone marrow cells that expressed myogenin ever detected (Fig. 4, b–b”, arrowheads). The frequency of these fusion events is low (∼0.013%) but comparable with that previously reported by others for adherent bone marrow mesenchymal cells that are stromal derivatives (Shi et al., 2004; Lee et al., 2005). Thus, nonadherent HSC derivatives are as capable of fusing with muscle as adherent mesenchymal cells.
Figure 4.
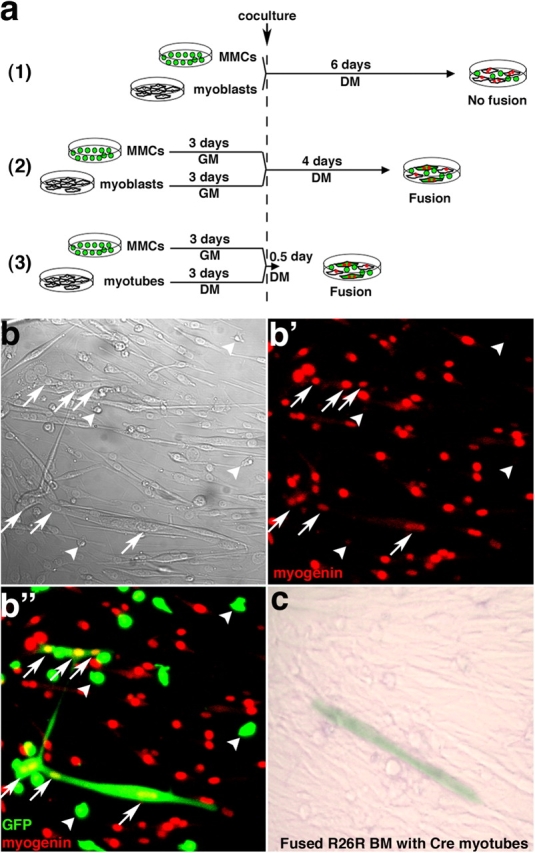
MMCs can fuse spontaneously in vitro with differentiated myotubes. (a) Schematic of the co-culture experiments. (b and c) MMCs were isolated by FACS from fresh bone marrow from GFP transgenic mice, maintained in myoblast GM for 3 d, and co-cultured with wild-type primary myoblasts and differentiation induced (DM) for 4 d, according to scheme a (2). Phase-contrast (b) and laser-scanning confocal images of immunofluorescence for myogenin only (b') or for GFP and myogenin (b”) are shown. Arrows indicate an example of GFP+myogenin+ multinucleated myotube. Arrowheads indicate GFP+ MMCs that do not express myogenin. (c) MMCs were isolated by FACS from fresh bone marrow from R26R Cre-reporter transgenic mice and co-cultured with Cre-expressing myoblasts, according to scheme a (2). A β-gal+ multinucleated myotube provides evidence of fusion between MMCs and Cre myotubes.
To determine definitively that fusion was the basis for the appearance of GFP+ myotubes rather than transdifferentiation of MMCs to myogenic cells tested under our culture conditions, an additional set of experiments was performed. MMCs were isolated from a Cre-reporter mouse (R26R; Soriano, 1999), stimulated by GM for 3 d, and co-cultured with Cre-expressing myoblasts in DM for 4 d (Fig. 4 a, 2). After fusion between MMCs and myotubes, DNA recombination mediated by the Cre recombinase resulted in β-gal expression, as shown in Fig. 4 c. This experiment provides conclusive evidence that hematopoietic MMCs can fuse with muscle cells.
Moreover, to determine whether MMCs fuse with either mononucleated myoblasts or with differentiated multinucleated myotubes, we performed time-lapse confocal microscopy. Myotubes were exposed to Ara-C to eliminate contaminating proliferating myoblasts and co-cultured with MMCs (Fig. 4 a, 3). In Fig. 5 (top eight panels), two muscle cells (arrows) are shown fusing together to form a multinucleated myotube. Subsequently, a GFP+ MMC (Fig. 5, arrowhead) fuses with the multinucleated myotube only 5 h after the onset of co-culture (bottom six panels). The entire time-lapse film of this microscopic field is provided with a single channel of phase contrast and GFP and the merged images (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200506123/DC1). Using time-lapse microscopy in two independent experiments, we detected 12 fusion events, 11 of which occurred with multinucleated myotubes and only 1 with a myoblast. Together, these data demonstrate that bone marrow MMCs can fuse in vitro with multinucleated differentiated myotubes.
Figure 5.
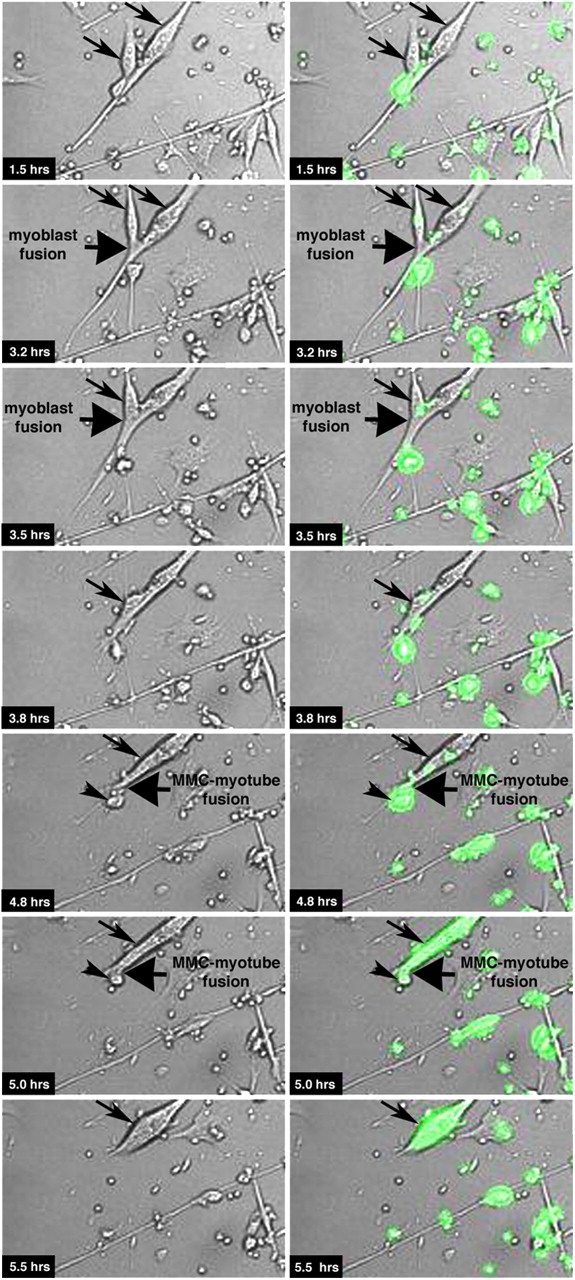
MMCs fuse with differentiated multinucleated myotubes. MMCs were isolated by FACS from fresh bone marrow from GFP transgenic mice, maintained in myoblast GM for 3 d, and co-cultured with Ara-C–treated 3-d-old primary wild-type myotubes, according to Fig. 4 a (3). Immediately after the beginning of the co-culture, we started monitoring the cells using time-lapse confocal microcopy. Arrows indicate muscle cells, and arrowheads indicate MMCs (GFP+) that fuse with each other. The entire time-lapse film of this microscopic field is provided with a single channel of phase contrast and GFP and the merged images (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200506123/DC1).
IGF-I promotes survival of MMCs in vitro
To test the potential role of IGF-I on the fusion of MMCs with myotubes, we had to distinguish its effects on fusion from those on survival. Accordingly, experiments were first designed to determine whether IGF-I affects the survival and proliferation of MMCs. Freshly isolated MMCs were isolated from GFP+ bone marrow by FACS. MMCs were cultured for 3 d in GM with or without IGF-I (100 ng/ml). Cells were then harvested and counted, and cell cycle analysis was performed using propidium iodide and FACS. As shown in Fig. 6, IGF-I led to an increase in the number of cells within this Lin−ckit+Sca1− bone marrow fraction (P < 0.02). By contrast, IGF-I did not lead to a change in the number of Lin+ cells (unpublished data). This potent effect on cell number resulted from an increase in cell survival because no effect of IGF-I on proliferation was observed, as indicated by the cell cycle profile, which was not altered relative to untreated controls. Indeed, 14% of the cells were in S–G2/M phase in each case (Fig. 6 b). As a control, when MMCs were cultured in bone marrow medium in the presence of cytokines (IL3, IL6, and stem cell factor), a medium in which they were highly stimulated to proliferate, 35% of the cells were found in the S–G2/M phase of the cell cycle (Fig. 6 b). Thus, IGF-I increased survival but not proliferation of MMCs under the experimental conditions used here.
Figure 6.

IGF-I promotes survival of MMCs. (a) MMCs were obtained by FACS from freshly isolated bone marrow and 6 × 104 cells plated in myoblast GM for 3 d, with or without 100 ng/ml IGF-I. Cell counts were performed on days 0 and 3. Final cell numbers are expressed as a percentage of the initial number of cells plated on day 0 (± SEM). P value was determined with a t test. *, P < 0.02. (b) Cell cycle profile using propidium iodide for analysis of DNA content in MMCs with or without IGF-I after 3 d of culture in the media indicated. Percentages of the cells in each specific cell cycle phase are indicated on the graphs.
IGF-I increases fusion of MMCs with differentiated myotubes independent of the IGF-I–mediated increase in cell survival
Experiments were designed to test the effect of IGF-I on the fusion of MMCs with myotubes. A dose–response curve for IGF-I effect on fusion was performed (50, 100, 200, and 400 ng/ml; unpublished data), and 100 ng/ml was the optimal IGF-I concentration used for further experiments. GFP+ MMCs that had been exposed in culture to myoblast GM for 3 d were co-cultured with 3-d-old myotubes that had been exposed to the DNA synthesis inhibitor Ara-C for 3 d to eliminate proliferating myoblasts. Co-cultures were maintained for the subsequent 4 d in the presence or absence of IGF-I (Fig. 7 a). As shown in Fig. 7 a, we observed a twofold increase in the number of GFP+ myotubes containing myogenin-expressing nuclei when IGF-I was added to co-cultures (95% confidence interval based on the Poisson distribution). Under these conditions, as shown in Fig. 5, GFP+ myotubes were visible as early as 5 h after co-culture and increased for a period of 4 d thereafter. This effect was not attributable to an increased survival of these cells during co-culture because the number of MMCs does not change in the presence or absence of IGF-I in DM conditioned by myotubes (P > 0.5; Fig. 7 b). An example of a multinucleated GFP+ myotube with all nuclei positive for myogenin is shown in Fig. 7 c. We conclude that IGF-I promotes fusion of MMCs with mature myotubes and that this effect is not attributable to an increase in cell survival under the experimental conditions used.
Figure 7.

IGF-I promotes fusion of MMCs with myotubes. (a) MMCs were isolated by FACS from fresh bone marrow from GFP transgenic mice, maintained in myoblast GM for 3 d, and co-cultured with 3-d-old primary wild-type myotubes, in the presence or absence of 100 ng/ml IGF-I, according to Fig. 4 a (3). Asterisk indicates total numbers of GFP+myogenin+ multinucleated myotubes observed after 4 d, shown with a 95% confidence interval based on the Poisson distribution. (b) For analyses of cell survival, MMCs were isolated and immediately cultured in myotube-conditioned DM in the presence or absence of 100 ng/ml IGF-I. Cell counts were performed on days 0 and 3. Final cell numbers are expressed as percentages of the initial number of cells plated on day 0 (± SEM). P value was determined with a t test. P > 0.5. (c) Laser-scanning confocal image of an example of GFP+myogenin+ multinucleated myotube; immunofluorescence shows nuclear muscle myogenin (red) and MMCs GFP (green).
Discussion
Although HSC derivatives, specifically MMCs, have been shown to incorporate into skeletal muscle after damage, the responsible damage-related molecules have yet to be identified. In this paper, we show that administration to adult muscle of a single growth factor, IGF-I, known to be up-regulated during muscle regeneration substantially increases the contribution of BMDCs to skeletal muscle in vivo in the presence of mild damage. To date, tissue injury caused by myotoxins, sustained exercise, or muscle overload was the only means reported for triggering this response. Although injury is inherent in any delivery method, the extent of damage assessed as the percentage of centrally nucleated myofibers in the three delivery systems used in this study (DNA electrotransfer, myoblast-mediated gene transfer, and recombinant protein injection) ranged from 1 to 5%. Myoblast-mediated gene transfer was the least invasive means of delivery, considerably lower than that obtained when muscles are damaged by notexin (∼75%). Thus, the experimental paradigms used in this study introduced only mild injury to the tissue, and delivery of IGF-I enhanced the contribution of BMDCs to skeletal muscle to the extent that it was on par with notexin damage. The three different delivery methods used show that the effect is specific to IGF-I and not related to a given experimental strategy. Both DNA electrotransfer and genetically engineered myoblasts result in constitutive delivery of IGF-I over time, whereas recombinant protein injection results in a transient exposure of the tissue to IGF-I. These data provide evidence that IGF-I alone can lead to increased incorporation of BMDCs into muscle fibers in vivo. Moreover, the results are novel in that they show that delivery of a single factor increases BMDC contribution to muscle.
IGF-I has been implicated in the fusion of myoblasts with myotubes in tissue culture (Jacquemin et al., 2004). IGF-I has also been shown in transgenic animals to increase the recruitment of BMDCs to damaged muscles (Musaro et al., 2004). However, the fusion of myotubes with bone marrow cell types has not previously been described, except for adherent mesenchymal stem cells derived from the stroma (Shi et al., 2004; Lee et al., 2005). Our data demonstrate that fusion of MMCs with myotubes occurs spontaneously in culture and is also enhanced when IGF-I is present. This is in contrast to a previous study suggesting that HSCs and their derivatives were not able to fuse with myotubes in vitro (Shi et al., 2004). In this paper, we show the formation of heterokaryons between myotubes and MMCs using two different experimental paradigms: (1) GFP+ MMCs in co-culture with wild-type myotubes and time-lapse microscopy and (2) R26R Cre-reporter MMCs in co-culture with Cre-expressing myotubes, which results in DNA recombination and expression of β-gal.
The failure to observe fusion of hematopoietic with myogenic cells by others was likely the result of the time period analyzed, the differentiated state of the cells used (myoblasts), and the absence of growth factor stimulation of hematopoietic cells. In our studies, fusion of MMCs with myotubes occurred within 5 h. Notably, fusion was not seen unless mature myotubes were present and MMCs had been exposed to high-serum medium before co-culture. Thus, the mechanisms that mediate stromal mesenchymal cell fusion with mononucleated myoblasts may well differ from those that mediate MMC fusion with multinucleate-differentiated myotubes.
These results also show that MMCs contribute to skeletal muscle through a direct fusion mechanism and that they do not get converted to myogenic precursors, as muscle-specific gene expression in MMCs in culture was never detected (unpublished data). Cell survival of MMCs could be increased by IGF-I in our studies, in agreement with those of others. However, we show that the increased fusion observed in the presence of IGF-I occurred in the presence of low-serum medium, a condition under which IGF-I had no effect on cell survival. Cell survival increased only when the cells were cultured in GM and not in myotube-conditioned DM, the medium in which fusion was observed.
The in vitro fusion assay described in this paper will allow further characterization of the molecular components involved in the spontaneous formation of heterokaryons in vivo. Several studies performed in Drosophila melanogaster identified proteins involved in the fusion of founder cells and fusion-competent cells during muscle development (Chen and Olson, 2004, 2005). The potential role of these proteins in the fusion of MMCs with myotubes can now be tested. Such experiments will determine whether muscle–muscle homofusion and muscle–MMC heterofusion share similar components.
Materials and methods
Mice and BMT procedure
C57BL/6 wild-type and Cre-reporter R26R (Soriano, 1999) transgenic mice were purchased from The Jackson Laboratory. C57BL/6-GFP transgenic mice (Wright et al., 2001) were a gift from I.L. Weissman (Stanford University, Stanford, CA). For BMT, bone marrow from femurs and tibia of GFP transgenic mice was harvested and cell suspensions were produced. 107 cells were transplanted by tail-vein injection into lethally irradiated 8-wk-old mice. All protocols were approved by the Administrative Panel on Laboratory Animal Care at the Stanford University School of Medicine.
Electrotransfer of DNA into skeletal muscle
IGF-IEa and MGF cDNAs were obtained by RT-PCR from total RNA obtained from damaged mouse skeletal muscles (forward primer for IGF-IEa and MGF, caccatggggaaaatcagc; reverse primer for IGF-IEa, ctacattctgtaggtcttgtt; reverse primer for MGF, ctacttgtgttcttcaaatgta). The PCR amplification of these two products was performed using an annealing temperature of 56°C for 30 cycles. β-Actin control primers were as follows: forward, ccgagcgtggctacagcttcac; reverse, gcacttgcggtgcacgatggag. The PCR amplification of β-actin was performed using an annealing temperature of 64°C for 21 cycles. IGF-I cDNAs were cloned into the pVIVO vector (InvivoGen). Mice that had been GFP–bone marrow transplanted 8 wk earlier were anesthetized with inhaled methoxyfluorane, the skin over their TA muscle was shaved, their leg thickness was measured with a caliber, and 10 μg of plasmid (pVIVO, pVIVO–IGF-IEa, or pVIVO–MGF) in 30 μl of saline was injected (using an insulin syringe, 29-gauge needle) into their TA muscles. Immediately after injection, an electric field was applied to the TA with the Electro Square Porator ECM 830 (BTX; Genetronics) using the following conditions: 200 V/cm, 20-ms pulse field, 176 ms of interval, and eight serial pulses, according to the protocol from the Rhone-Poulenc Company. Muscles were harvested 4 wk after injection and processed as described in Tissue analysis.
Construction of IGF-IEa and MGF retroviral vectors and genetic engineering of primary mouse myoblasts
The IGF-IEa and MGF cDNAs were cloned into the MFG-retroviral vector using the BamHI and XhoI sites, upstream of the internal ribosome entry site–CD8 sequence (Springer et al., 1998). Primary myoblasts isolated from C57Bl/6 mice and previously transduced to express β-gal from a retroviral promoter (Rando and Blau, 1994) were infected again with four successive rounds of MFG–IGF-IEa.CD8, MFG–MGF.CD8, or MFG-CD8 control, yielding to >70% of the myoblasts of each population that was CD8+. Myoblast populations expressing CD8 on their membranes were isolated using a FACStar cell sorter (Becton Dickinson), using 0.2 μg/ml of an anti–CD8-allophycocyanin–conjugated antibody (BD Biosciences). 5 × 105 myoblasts from each population were injected in a 10-μl volume using an insulin syringe (29-gauge needle) into the TA of mice 8 wk after BMT.
For generating Cre-expressing myoblasts, primary myoblasts were infected with a retrovirus carrying the Cre-recombinase gene together with neomycin selection. Cells were subsequently selected with neomycin (1 mg/ml), and these cells were used for the co-culture experiment.
Western blot
Western blot analysis for IGF-I was performed using 4 μg/ml of a goat anti–IGF-I primary antibody (Chemicon) and an anti–goat-HRP–conjugated secondary antibody (1:1,000 dilution; Bio-Rad Laboratories). For tubulin, 2 μg/ml antitubulin antibody (Sigma-Aldrich) and anti–mouse-HRP–conjugated secondary antibody (1:10,000 dilution; Zymed Laboratories) were used.
Injection of recombinant mouse IGF-I
Mice that had been transplanted with GFP-expressing bone marrow 8 wk earlier were anesthetized with inhaled methoxyflurane, were shaved over their TA muscles, and received a single 10-μl dose of 1, 5, or 10 μg of the recombinant mouse IGF-I (carrier free; R&D Systems) in saline by i.m. injection into the TA muscle. As a control, saline alone was injected into the contralateral leg. Muscles were harvested 4 wk after injection and processed as described in Tissue analysis.
Tissue analysis
4 wk after injection, TA muscles were dissected and immersed in PBS/0.5% EM-grade PFA (Polysciences) for 2 h at RT followed by overnight immersion in PBS with 20% sucrose at 4°C. Fixed tissue was embedded in optimal cutting temperature compound (Tissue-Tek; Sakura Finetek), snap-frozen in isobutane and liquid nitrogen, and sectioned at 10 μm thickness. Tissue sections were blocked in PBS/20% normal goat serum/0.3% Triton X-100 (blocking buffer); incubated with primary antibodies (diluted in blocking buffer; 2 μg/ml rabbit anti-GFP [Invitrogen], 3 μg/ml rat anti-laminin-2 [Upstate Biotechnology], or 5 μg/ml rat anti-CD8a [BD Biosciences]); and incubated with 10 μg/ml of the secondary conjugated antibodies anti–rabbit-Alexa488 and anti–rat-Alexa546 (Invitrogen). Sections were then washed overnight and mounted with Fluoromount G. Images of muscle transverse sections were acquired using an epifluorescent microscope (Axioplan2; Carl Zeiss MicroImaging, Inc.), Fluar 20×/0.75 objective lenses, and a digital camera (ORCA-ER C4742-95; Hamamatsu Photonics). The software used for acquisition was OpenLab 4.0.2 (Improvision). All images were composed and edited in Photoshop 7.0 (Adobe). Background was reduced using brightness and contrast adjustments, and color balance was performed to enhance colors. All the modifications were applied to the whole image using Photoshop 7.0 (Adobe). GFP+ muscle fibers were counted using an epifluorescent microscope with a double-bandpass filter that distinguishes between GFP and autofluorescence to ensure that the GFP signal was authentic (Brazelton and Blau, 2005).
Quantification of GFP+ myofibers
To quantitate myofibers, we cut TA muscles, taking sections every 100 μm. All the sections were stained and analyzed, and GFP+ myofibers were counted and expressed as the mean ± SEM. In Figs. 1 and 2, all CD8+ myofibers were counted, ranging from 60 to 500 CD8+ myofibers per cross section. Among them, the percentage of GFP+ myofibers was calculated. In Fig. 3, total GFP+ myofibers per TA were scored (TA muscle contains ∼2,000 myofibers per cross section).
Cell sorting by flow cytometry
Bone marrow from GFP mice was isolated, suspended in FACS buffer (2.5% normal goat serum and 1 mM EDTA in PBS), and stained with 4 μg/ml specific antibodies to ckit and Sca1 directly conjugated to phycoerythrin, allophycocyanin, or phycoerythrin-Cy7 (all from BD Biosciences) and a lineage panel (mixture of biotinylated antibodies to Ter119, B220, CD3, Gr1, and CD11b; 1:30 dilution; BD Biosciences). Antibody-labeled cells were separated using streptavidin coupled to magnetic beads (Miltenyi Biotec) and then counterstained using 10 μg/ml streptavidin Texas red (BD Biosciences). Lin−ckit+Sca1− cells (MMCs) were then fractionated and collected by flow cytometry (DIVA-Van; Becton Dickinson). The purity of the sorted MMC fraction was ∼90%.
Cell cycle analysis
MMCs were collected; resuspended in a solution of 0.1 mg/ml propidium iodide, 0.2% Tween 20, and 0.2 mg/ml RNase in PBS; incubated in the dark at 4°C for 2 h; and analyzed by FACS.
Cell culture experiments
6 × 104 freshly isolated MMCs (as described in Cell sorting by flow cytometry) were co-cultured with 6 × 104 myoblasts or Ara-C–treated 3-d-old myotubes in 24 multiwells coated with laminin (Roche). Muscle differentiation was induced using low-serum DM (DME/2% horse serum) for up to 6 d in the presence or absence of 100 ng/ml human R3–IGF-I (Sigma-Aldrich). As indicated in Results, in some of the experiments MMCs were cultured in myoblast GM (50% DME/10% FBS + 50% F10/20% FBS/50 ng/ml FGF) or in bone marrow medium (αMEM/15% FBS supplemented with IL3, IL6, and stem cell factor cytokines). Cell cultures were then fixed in 1.5% PFA, blocked in blocking buffer, stained for GFP (rabbit; 2 μg/ml) and myogenin (mouse; 0.7 μg/ml; BD Biosciences), and incubated with 10 μg/ml of the secondary conjugated antibodies anti–rabbit-Alexa488 and anti–mouse-Alexa546. Cell cultures were then washed in PBS and left in PBS for imaging acquisition. Images of cell cultures were acquired using a laser-scanning confocal microscope (LSM510; Carl Zeiss MicroImaging, Inc.) using Plan NeoFluar 20×/0.50 objective lenses and maximum optical section with the LSM software. Background was reduced using brightness and contrast adjustments, and color balance was performed to enhance colors. All the modifications were applied to the whole image using Photoshop 7.0. In these co-culture experiments, myotubes that had two or more nuclei and were myogenin+ were scored.
Time lapse
6 × 104 freshly isolated MMCs (as described in Cell sorting by flow cytometry) were cultured for 3 d in myoblast GM and then co-cultured with 6 × 104 Ara-C–treated 3-d-old myotubes in 24 multiwells coated with laminin. Muscle differentiation was induced using low-serum DM. Immediately after the beginning of the co-culture, cells were monitored using a time-lapse apparatus (Carl Zeiss MicroImaging, Inc., CO2: CTI-Controller 3700 Digital; temperature: Tempcontrol 37–2 digital; scanning stage: Incubator XL 100/135 [PECON]) mounted on a laser-scanning confocal microscope. Images of cell cultures were acquired at 37°C every 10 min using a Plan NeoFluar 10×/0.50 objective lenses and maximum optical section with the LSM software. After the acquisition, pictures of the frames were analyzed using Image Ready 7.0 software.
Online supplemental material
Video 1 shows co-cultures of Ara-C–treated 3-d-old wild-type primary myotubes (GFP−) with MMCs (GFP+) kept in myoblast GM for 3 d. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200506123/DC1.
Acknowledgments
We would like to thank Clayton L. Casipit and Kassie Koleckar for their excellent technical support; Adam Palermo for statistical analysis; Andrea Banfi for useful discussion; and Jason Pomerantz, Georges von Degenfeld, and Matthias Lutolf for useful comments on the manuscript.
This work was supported by predoctoral fellowships to M.A. LaBarge (TG AG00259) and by National Institutes of Health grants (AG09521, HD18179, AG20961, and AG24987), a Senior Ellison Medical Foundation award (AG-SS-0817-03), and support from Novartis Institutes for Biomedical Research and the Baxter Foundation to H.M. Blau.
M.A. LaBarge's present address is Lawrence Berkeley National Laboratory, Berkeley, CA 94720.
Abbreviations used in this paper: β-gal, β-galactosidase; BMDC, bone marrow–derived cell; BMT, bone marrow transplant; DM, differentiation medium; GM, growth medium; HSC, hematopoietic stem cell; IGF-I, insulin-like growth factor I; MGF, mechano growth factor; MMC, myelomonocytic precursor; TA, tibialis anterior.
References
- Barr, E., and J.M. Leiden. 1991. Systemic delivery of recombinant proteins by genetically modified myoblasts. Science. 254:1507–1509. [DOI] [PubMed] [Google Scholar]
- Barton-Davis, E.R., D.I. Shoturma, A. Musaro, N. Rosenthal, and H.L. Sweeney. 1998. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc. Natl. Acad. Sci. USA. 95:15603–15607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, G.I., M.M. Stempien, N.M. Fong, and L.B. Rall. 1986. Sequences of liver cDNAs encoding two different mouse insulin-like growth factor I precursors. Nucleic Acids Res. 14:7873–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand, A., V. Ngo-Muller, D. Hentzen, J.P. Concordet, D. Daegelen, and D. Tuil. 2003. Muscle electrotransfer as a tool for studying muscle fiber-specific and nerve-dependent activity of promoters. Am. J. Physiol. Cell Physiol. 285:C1071–C1081. [DOI] [PubMed] [Google Scholar]
- Bittner, R.E., C. Schofer, K. Weipoltshammer, S. Ivanova, B. Streubel, E. Hauser, M. Freilinger, H. Hoger, A. Elbe-Burger, and F. Wachtler. 1999. Recruitment of bone-marrow-derived cells by skeletal and cardiac muscle in adult dystrophic mdx mice. Anat. Embryol. (Berl.). 199:391–396. [DOI] [PubMed] [Google Scholar]
- Brazelton, T.R., and H.M. Blau. 2005. Optimizing techniques for tracking transplanted stem cells in vivo. Stem Cells. 23:1251–1265. [DOI] [PubMed] [Google Scholar]
- Brazelton, T.R., M. Nystrom, and H.M. Blau. 2003. Significant differences among skeletal muscles in the incorporation of bone marrow-derived cells. Dev. Biol. 262:64–74. [DOI] [PubMed] [Google Scholar]
- Burgess, W., K. Jesse, Q. Tang, S.R. Broussard, R. Dantzer, and K.W. Kelley. 2003. Insulin-like growth factor-I and the cytokines IL-3 and IL-4 promote survival of progenitor myeloid cells by different mechanisms. J. Neuroimmunol. 135:82–90. [DOI] [PubMed] [Google Scholar]
- Camargo, F.D., R. Green, Y. Capetanaki, K.A. Jackson, and M.A. Goodell. 2003. Single hematopoietic stem cells generate skeletal muscle through myeloid intermediates. Nat. Med. 9:1520–1527. [DOI] [PubMed] [Google Scholar]
- Chen, E.H., and E.N. Olson. 2004. Towards a molecular pathway for myoblast fusion in Drosophila. Trends Cell Biol. 14:452–460. [DOI] [PubMed] [Google Scholar]
- Chen, E.H., and E.N. Olson. 2005. Unveiling the mechanisms of cell-cell fusion. Science. 308:369–373. [DOI] [PubMed] [Google Scholar]
- Corbel, S.Y., A. Lee, L. Yi, J. Duenas, T.R. Brazelton, H.M. Blau, and F.M. Rossi. 2003. Contribution of hematopoietic stem cells to skeletal muscle. Nat. Med. 9:1528–1532. [DOI] [PubMed] [Google Scholar]
- Dhawan, J., L.C. Pan, G.K. Pavlath, M.A. Travis, A.M. Lanctot, and H.M. Blau. 1991. Systemic delivery of human growth hormone by injection of genetically engineered myoblasts. Science. 254:1509–1512. [DOI] [PubMed] [Google Scholar]
- Dona, M., M. Sandri, K. Rossini, I. Dell'Aica, M. Podhorska-Okolow, and U. Carraro. 2003. Functional in vivo gene transfer into the myofibers of adult skeletal muscle. Biochem. Biophys. Res. Commun. 312:1132–1138. [DOI] [PubMed] [Google Scholar]
- Doyonnas, R., M.A. LaBarge, A. Sacco, C. Charlton, and H.M. Blau. 2004. Hematopoietic contribution to skeletal muscle regeneration by myelomonocytic precursors. Proc. Natl. Acad. Sci. USA. 101:13507–13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus, P.A., F. Chretien, B. Chazaud, Y. Kirova, P. Caramelle, L. Garcia, G. Butler-Browne, and R.K. Gherardi. 2004. Adult bone marrow-derived stem cells in muscle connective tissue and satellite cell niches. Am. J. Pathol. 164:773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari, G., G. Cusella-De Angelis, M. Coletta, E. Paolucci, A. Stornaiuolo, G. Cossu, and F. Mavilio. 1998. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 279:1528–1530. [DOI] [PubMed] [Google Scholar]
- Fukada, S., Y. Miyagoe-Suzuki, H. Tsukihara, K. Yuasa, S. Higuchi, S. Ono, K. Tsujikawa, S. Takeda, and H. Yamamoto. 2002. Muscle regeneration by reconstitution with bone marrow or fetal liver cells from green fluorescent protein-gene transgenic mice. J. Cell Sci. 115:1285–1293. [DOI] [PubMed] [Google Scholar]
- Goldspink, G., and S.Y. Yang. 2004. The splicing of the IGF-I gene to yield different muscle growth factors. Adv. Genet. 52:23–49. [DOI] [PubMed] [Google Scholar]
- Gollins, H., J. McMahon, K.E. Wells, and D.J. Wells. 2003. High-efficiency plasmid gene transfer into dystrophic muscle. Gene Ther. 10:504–512. [DOI] [PubMed] [Google Scholar]
- Gussoni, E., Y. Soneoka, C.D. Strickland, E.A. Buzney, M.K. Khan, A.F. Flint, L.M. Kunkel, and R.C. Mulligan. 1999. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. 401:390–394. [DOI] [PubMed] [Google Scholar]
- Harris, J.B., M.A. Johnson, and E. Karlsson. 1974. Proceedings: histological and histochemical aspects of the effect of notexin on rat skeletal muscle. Br. J. Pharmacol. 52:152P. [PMC free article] [PubMed] [Google Scholar]
- Hill, M., and G. Goldspink. 2003. Expression and splicing of the insulin-like growth factor gene in rodent muscle is associated with muscle satellite (stem) cell activation following local tissue damage. J. Physiol. 549:409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, M., A. Wernig, and G. Goldspink. 2003. Muscle satellite (stem) cell activation during local tissue injury and repair. J. Anat. 203:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley, V., K.M. Jansen, S.T. Mills, and G.K. Pavlath. 2003. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell. 113:483–494. [DOI] [PubMed] [Google Scholar]
- Jacquemin, V., D. Furling, A. Bigot, G.S. Butler-Browne, and V. Mouly. 2004. IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Exp. Cell Res. 299:148–158. [DOI] [PubMed] [Google Scholar]
- Klein-Ogus, C., and J.B. Harris. 1983. Preliminary observations of satellite cells in undamaged fibres of the rat soleus muscle assaulted by a snake-venom toxin. Cell Tissue Res. 230:671–676. [DOI] [PubMed] [Google Scholar]
- Krause, D.S., N.D. Theise, M.I. Collector, O. Henegariu, S. Hwang, R. Gardner, S. Neutzel, and S.J. Sharkis. 2001. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 105:369–377. [DOI] [PubMed] [Google Scholar]
- LaBarge, M.A., and H.M. Blau. 2002. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell. 111:589–601. [DOI] [PubMed] [Google Scholar]
- Lagasse, E., H. Connors, M. Al-Dhalimy, M. Reitsma, M. Dohse, L. Osborne, X. Wang, M. Finegold, I.L. Weissman, and M. Grompe. 2000. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat. Med. 6:1229–1234. [DOI] [PubMed] [Google Scholar]
- Lee, J.H., P.A. Kosinski, and D.M. Kemp. 2005. Contribution of human bone marrow stem cells to individual skeletal myotubes followed by myogenic gene activation. Exp. Cell Res. 307:174–182. [DOI] [PubMed] [Google Scholar]
- Lee, R.J., M.L. Springer, W.E. Blanco-Bose, R. Shaw, P.C. Ursell, and H.M. Blau. 2000. VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation. 102:898–901. [DOI] [PubMed] [Google Scholar]
- LeRoith, D., and C.T. Roberts Jr. 1991. Insulin-like growth factor I (IGF-I): a molecular basis for endocrine versus local action? Mol. Cell. Endocrinol. 77:C57–C61. [DOI] [PubMed] [Google Scholar]
- Liu, J.P., J. Baker, A.S. Perkins, E.J. Robertson, and A. Efstratiadis. 1993. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 75:59–72. [PubMed] [Google Scholar]
- Massengale, M., A.J. Wagers, H. Vogel, and I.L. Weissman. 2005. Hematopoietic cells maintain hematopoietic fates upon entering the brain. J. Exp. Med. 201:1579–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKoy, G., W. Ashley, J. Mander, S.Y. Yang, N. Williams, B. Russell, and G. Goldspink. 1999. Expression of insulin growth factor-1 splice variants and structural genes in rabbit skeletal muscle induced by stretch and stimulation. J. Physiol. 516:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezey, E., K.J. Chandross, G. Harta, R.A. Maki, and S.R. McKercher. 2000. Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science. 290:1779–1782. [DOI] [PubMed] [Google Scholar]
- Minshall, C., S. Arkins, J. Straza, J. Conners, R. Dantzer, G.G. Freund, and K.W. Kelley. 1997. IL-4 and insulin-like growth factor-I inhibit the decline in Bcl-2 and promote the survival of IL-3-deprived myeloid progenitors. J. Immunol. 159:1225–1232. [PubMed] [Google Scholar]
- Mir, L.M., M.F. Bureau, J. Gehl, R. Rangara, D. Rouy, J.M. Caillaud, P. Delaere, D. Branellec, B. Schwartz, and D. Scherman. 1999. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc. Natl. Acad. Sci. USA. 96:4262–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musaro, A., K. McCullagh, A. Paul, L. Houghton, G. Dobrowolny, M. Molinaro, E.R. Barton, H.L. Sweeney, and N. Rosenthal. 2001. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 27:195–200. [DOI] [PubMed] [Google Scholar]
- Musaro, A., C. Giacinti, G. Borsellino, G. Dobrowolny, L. Pelosi, L. Cairns, S. Ottolenghi, G. Cossu, G. Bernardi, L. Battistini, et al. 2004. Stem cell-mediated muscle regeneration is enhanced by local isoform of insulin-like growth factor 1. Proc. Natl. Acad. Sci. USA. 101:1206–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojima, K., A. Uezumi, H. Miyoshi, S. Masuda, Y. Morita, A. Fukase, A. Hattori, H. Nakauchi, Y. Miyagoe-Suzuki, and S. Takeda. 2004. Mac-1(low) early myeloid cells in the bone marrow-derived SP fraction migrate into injured skeletal muscle and participate in muscle regeneration. Biochem. Biophys. Res. Commun. 321:1050–1061. [DOI] [PubMed] [Google Scholar]
- Ozawa, C.R., A. Banfi, N.L. Glazer, G. Thurston, M.L. Springer, P.E. Kraft, D.M. McDonald, and H.M. Blau. 2004. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J. Clin. Invest. 113:516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo, A.T., M.A. Labarge, R. Doyonnas, J. Pomerantz, and H.M. Blau. 2005. Bone marrow contribution to skeletal muscle: a physiological response to stress. Dev. Biol. 279:336–344. [DOI] [PubMed] [Google Scholar]
- Pluskal, M.G., J.B. Harris, R.J. Pennington, and D. Eaker. 1978. Some biochemical responses of rat skeletal muscle to a single subcutaneous injection of a toxin (notexin) isolated from the venom of the Australian tiger snake Notechis scutatus scutatus. Clin. Exp. Pharmacol. Physiol. 5:131–141. [DOI] [PubMed] [Google Scholar]
- Powell-Braxton, L., P. Hollingshead, C. Warburton, M. Dowd, S. Pitts-Meek, D. Dalton, N. Gillett, and T.A. Stewart. 1993. IGF-I is required for normal embryonic growth in mice. Genes Dev. 7:2609–2617. [DOI] [PubMed] [Google Scholar]
- Rando, T.A., and H.M. Blau. 1994. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 125:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schakman, O., H. Gilson, V. de Coninck, P. Lause, J. Verniers, X. Havaux, J.M. Ketelslegers, and J.P. Thissen. 2005. Insulin-like growth factor-I gene transfer by electroporation prevents skeletal muscle atrophy in glucocorticoid-treated rats. Endocrinology. 146:1789–1797. [DOI] [PubMed] [Google Scholar]
- Shavlakadze, T., N. Winn, N. Rosenthal, and M.D. Grounds. 2005. Reconciling data from transgenic mice that overexpress IGF-I specifically in skeletal muscle. Growth Horm. IGF Res. 15:4–18. [DOI] [PubMed] [Google Scholar]
- Sherwood, R.I., J.L. Christensen, I.L. Weissman, and A.J. Wagers. 2004. a. Determinants of skeletal muscle contributions from circulating cells, bone marrow cells, and hematopoietic stem cells. Stem Cells. 22:1292–1304. [DOI] [PubMed] [Google Scholar]
- Sherwood, R.I., J.L. Christensen, I.M. Conboy, M.J. Conboy, T.A. Rando, I.L. Weissman, and A.J. Wagers. 2004. b. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 119:543–554. [DOI] [PubMed] [Google Scholar]
- Shi, D., H. Reinecke, C.E. Murry, and B. Torok-Storb. 2004. Myogenic fusion of human bone marrow stromal cells, but not hematopoietic cells. Blood. 104:290–294. [DOI] [PubMed] [Google Scholar]
- Shimatsu, A., and P. Rotwein. 1987. Mosaic evolution of the insulin-like growth factors. Organization, sequence, and expression of the rat insulin-like growth factor I gene. J. Biol. Chem. 262:7894–7900. [PubMed] [Google Scholar]
- Sjogren, K., J.O. Jansson, O.G. Isaksson, and C. Ohlsson. 2002. A model for tissue-specific inducible insulin-like growth factor-I (IGF-I) inactivation to determine the physiological role of liver-derived IGF-I. Endocrine. 19:249–256. [DOI] [PubMed] [Google Scholar]
- Sommerland, H., M. Ullman, E. Jennische, A. Skottner, and A. Oldfors. 1989. Muscle regeneration. The effect of hypophysectomy on cell proliferation and expression of insulin-like growth factor-I. Acta Neuropathol. (Berl.). 78:264–269. [DOI] [PubMed] [Google Scholar]
- Soriano, P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21:70–71. [DOI] [PubMed] [Google Scholar]
- Springer, M.L., and H.M. Blau. 1997. High-efficiency retroviral infection of primary myoblasts. Somat. Cell Mol. Genet. 23:203–209. [DOI] [PubMed] [Google Scholar]
- Springer, M.L., A.S. Chen, P.E. Kraft, M. Bednarski, and H.M. Blau. 1998. VEGF gene delivery to muscle: potential role for vasculogenesis in adults. Mol. Cell. 2:549–558. [DOI] [PubMed] [Google Scholar]
- Stewart, C.E., and P. Rotwein. 1996. Growth, differentiation, and survival: multiple physiological functions for insulin-like growth factors. Physiol. Rev. 76:1005–1026. [DOI] [PubMed] [Google Scholar]
- Stitt, T.N., D. Drujan, B.A. Clarke, F. Panaro, Y. Timofeyva, W.O. Kline, M. Gonzalez, G.D. Yancopoulos, and D.J. Glass. 2004. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell. 14:395–403. [DOI] [PubMed] [Google Scholar]
- Vilquin, J.T., P.F. Kennel, M. Paturneau-Jouas, P. Chapdelaine, N. Boissel, P. Delaere, J.P. Tremblay, D. Scherman, M.Y. Fiszman, and K. Schwartz. 2001. Electrotransfer of naked DNA in the skeletal muscles of animal models of muscular dystrophies. Gene Ther. 8:1097–1107. [DOI] [PubMed] [Google Scholar]
- Wagers, A.J., R.I. Sherwood, J.L. Christensen, and I.L. Weissman. 2002. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science. 297:2256–2259. [DOI] [PubMed] [Google Scholar]
- Weimann, J.M., C.B. Johansson, A. Trejo, and H.M. Blau. 2003. Stable reprogrammed heterokaryons form spontaneously in Purkinje neurons after bone marrow transplant. Nat. Cell Biol. 5:959–966. [DOI] [PubMed] [Google Scholar]
- Willenbring, H., A.S. Bailey, M. Foster, Y. Akkari, C. Dorrell, S. Olson, M. Finegold, W.H. Fleming, and M. Grompe. 2004. Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nat. Med. 10:744–748. [DOI] [PubMed] [Google Scholar]
- Wolff, J.A., R.W. Malone, P. Williams, W. Chong, G. Acsadi, A. Jani, and P.L. Felgner. 1990. Direct gene transfer into mouse muscle in vivo. Science. 247:1465–1468. [DOI] [PubMed] [Google Scholar]
- Wright, D.E., S.H. Cheshier, A.J. Wagers, T.D. Randall, J.L. Christensen, and I.L. Weissman. 2001. Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood. 97:2278–2285. [DOI] [PubMed] [Google Scholar]
- Yakar, S., J.L. Liu, B. Stannard, A. Butler, D. Accili, B. Sauer, and D. LeRoith. 1999. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA. 96:7324–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S.Y., and G. Goldspink. 2002. Different roles of the IGF-I Ec peptide (MGF) and mature IGF-I in myoblast proliferation and differentiation. FEBS Lett. 522:156–160. [DOI] [PubMed] [Google Scholar]
- Yang, S., M. Alnaqeeb, H. Simpson, and G. Goldspink. 1996. Cloning and characterization of an IGF-1 isoform expressed in skeletal muscle subjected to stretch. J. Muscle Res. Cell Motil. 17:487–495. [DOI] [PubMed] [Google Scholar]