

Abstract
Generation and turnover of phosphoinositides (PIs) must be coordinated in a spatial- and temporal-restricted manner. The small GTPase Rab5 interacts with two PI 3-kinases, Vps34 and PI3Kβ, suggesting that it regulates the production of 3-PIs at various stages of the early endocytic pathway. Here, we discovered that Rab5 also interacts directly with PI 5- and PI 4-phosphatases and stimulates their activity. Rab5 regulates the production of phosphatidylinositol 3-phosphate (PtdIns[3]P) through a dual mechanism, by directly phosphorylating phosphatidylinositol via Vps34 and by a hierarchical enzymatic cascade of phosphoinositide-3-kinaseβ (PI3Kβ), PI 5-, and PI 4-phosphatases. The functional importance of such an enzymatic pathway is demonstrated by the inhibition of transferrin uptake upon silencing of PI 4-phosphatase and studies in weeble mutant mice, where deficiency of PI 4-phosphatase causes an increase of PtdIns(3,4)P2 and a reduction in PtdIns(3)P. Activation of PI 3-kinase at the plasma membrane is accompanied by the recruitment of Rab5, PI 4-, and PI 5-phosphatases to the cell cortex. Our data provide the first evidence for a dual role of a Rab GTPase in regulating both generation and turnover of PIs via PI kinases and phosphatases to coordinate signaling functions with organelle homeostasis.
Introduction
Phosphoinositides (PIs) play a key role in fundamental cell functions, such as signal transduction, cytoskeleton remodeling, cell migration, and membrane trafficking (De Camilli et al., 1996; Fruman et al., 1998; Katso et al., 2001). This multifunctional role is due to cycles of phosphorylation and dephosphorylation at the 3, 4, and 5 positions of the inositol ring in different subcellular compartments. Such process is catalyzed by specific lipid modifying enzymes, PI kinases, and phosphatases, whose recruitment to the membrane and activity is regulated in a temporal- and spatial-dependent manner (Odorizzi et al., 2000; Vanhaesebroeck et al., 2001; Wenk and De Camilli, 2004). Small GTPases, such as Ras, Arf1, Arf6, Rho, as well as Rab, are primary regulators of the recruitment and/or activation of PI kinases (Rodriguez-Viciana et al., 1994; Godi et al., 1999; Honda et al., 1999; Krauss et al., 2003; de Graaf et al., 2004; Weernink et al., 2004). In the endocytic pathway, Rab5 interacts directly with two distinct PI 3-Ks, type I PI 3-K (p85α–p110β; PI3Kβ) and hVps34 (hVps34–p150; Christoforidis et al., 1999b). The latter kinase is responsible for the generation of phosphatidylinositol 3-phosphate (PtdIns[3]P) on the early endosome and the recruitment of a set of PtdIns(3)P binding Rab5 effectors such as EEA1 (Simonsen et al., 1998; Christoforidis et al., 1999b).
Whereas PtdIns(4)P on the TGN and PtdIns(3)P on endosomes exert an essential housekeeping function in organelle homeostasis and membrane transport, other PIs are produced in response to a variety of extracellular stimuli. Phosphatidylinositol 3,4-bisphosphate (PtdIns[3,4]P2) and phosphatidylinositol 3,4,5-trisphosphate (PtdIns[3,4,5]P3) are produced by type I PI 3-K upon stimulation by growth factors or cytokines at the plasma membrane, where they induce morphogenetic changes via the reorganization of actin filaments (Vanhaesebroeck et al., 2001). However, these PI species accumulate only transiently. PtdIns(3,4,5)P3 peaks at 5–6 s after stimulation (Chung et al., 2001) and is rapidly degraded upon phagocytosis and macropinocytosis by PI phosphatases (Marshall et al., 2001; Rupper et al., 2001; Funamoto et al., 2002). Similar to PI kinases, PI phosphatases display exquisite substrate specificity. For example, PI 3-phosphatases such as PTEN (Cantley and Neel, 1999) dephosphorylate PtdIns(3,4,5)P3 to PtdIns(4,5)P2. PI 5-phosphatases such as SHIP, synaptojanin, and type II PI 5-phosphatase, dephosphorylate either PtdIns(3,4,5)P3 or PtdIns(4,5)P2 or both, respectively (Vanhaesebroeck et al., 2001; Mitchell et al., 2002). Two PI 4-phosphatases isoforms exist (types I and II) each having two alternative splicing variants (α and β), which preferentially dephosphorylate PtdIns(3,4)P2 to PtdIns(3)P (Norris et al., 1995, 1997). The functional importance of PI kinases and phosphatases in PI metabolism is underscored by the finding that mutations in genes encoding these proteins are associated with hereditary disorders in humans and induce severe developmental abnormalities in animal model systems, particularly affecting neural function. For example, the gene product deficient in the oculocerebrorenal syndrome of Lowe (OCRL) is an inositol polyphosphate 5-phosphatase (Attree et al., 1992; Zhang et al., 1995) associated with endosomes and Golgi membrane (Ungewickell et al., 2004; Choudhury et al., 2005). A targeted mutation of mouse synaptojanin-1 (Cremona et al., 1999) causes defects in vesicle trafficking and actin dynamics at the synapse. Conditional knockout of PTEN specifically in the brain induces severe alterations in the cerebellum, with decreased cell proliferation and degeneration of Purkinje cells in mice (Backman et al., 2001). The weeble mutant mice bearing a mutation in the gene encoding type I PI 4-phosphatase are characterized by early postnatal neuronal loss in the cerebellum and in the hippocampus, that ultimately results in the death of homozygous animals 2–3 wk after birth (Nystuen et al., 2001). Whereas the identification and characterization of several PI kinases and phosphatases has greatly advanced our understanding of the enzymology of PI metabolism, the mechanisms that coordinate the activity of these enzymes to link PIs function and turnover remain largely unknown. Here, we provide insights into this question through the discovery of novel effectors of Rab5.
Results
Compartmentalization of PIs
Rab5 interacts with two distinct types of PI 3-K, PI3Kβ, and hVps34 (Christoforidis et al., 1999b). To investigate whether such interaction serves to regulate the synthesis of PtdIns(3)P as well as other PIs in the early endocytic pathway we took advantage of the Rab5Q79L activated mutant, which causes the expansion of early endosomes (Fig. 1 A). We examined the intracellular localization of different PIs by expressing GFP fused to various PIs-binding motifs. The 2XFYVE domain of Hrs, which recognizes PtdIns(3)P (Gillooly et al., 2003), accumulated on early endosomes (Fig. 1 A, 2XFYVE). In contrast, even upon Rab5 activation the PH domain of Akt, a probe for PtdIns(3,4)P2 and PtdIns(3,4,5)P3 (Watton and Downward, 1999), localized to the plasma membrane in a polarized fashion and no significant staining was observed on early endosomes (Fig.1 A, Akt1[PH]). The PH domain of phosphatidylinositol 4-phosphate adaptor protein 1 (FAPP1), which recognizes PtdIns(4)P (Dowler et al., 2000), was mainly present on the Golgi complex (Fig.1 A, FAPP1[PH]), where it colocalized with β′-COP (Levine and Munro, 2002; De Matteis and Godi, 2004; unpublished data), whereas the PLCδ PH domain, which binds PtdIns(4,5)P2 (Stauffer et al., 1998) was detected on the plasma membrane and in the nucleus. Thus, among the PIs tested, PtdIns(3)P is the only one that accumulates on early endosomes bearing activated Rab5.
Figure 1.
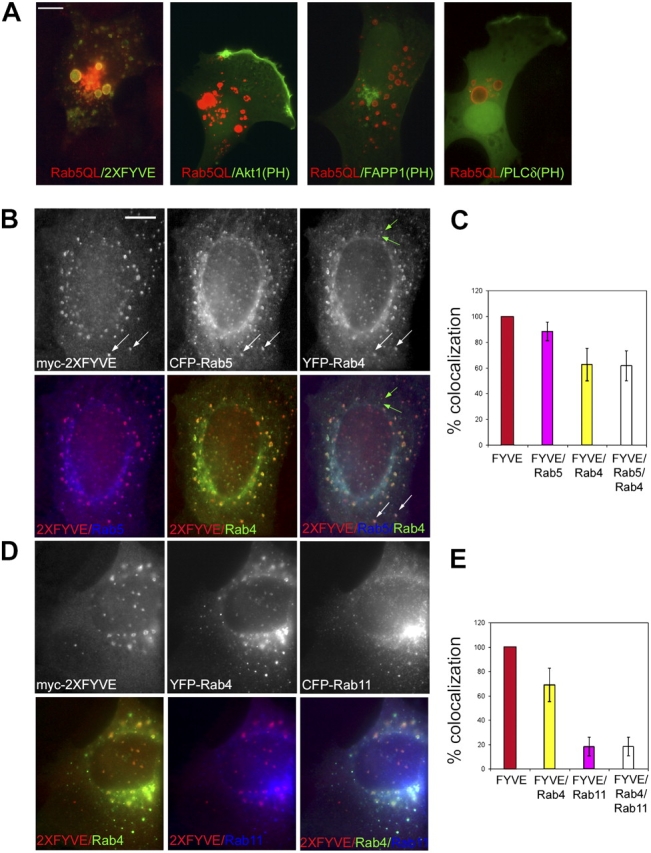
PtdIns(3)P is enriched in the Rab5 domain. (A) NIH3T3 cells expressing Rab5(Q79L) and GFP-tagged 2XFYVE, Akt PH domain, FAPP1 PH domain, or PLCδ PH domain were stained with antibodies against Rab5 (4F11) followed by rhodamine-conjugated secondary antibodies. Bar, 10 μm. A431 cells were microinjected with myc-2XFYVE, CFP–Rab5, and YFP–Rab4 (B) or myc–2XFYVE, YFP–Rab4, and CFP–Rab11 (D) and stained with antibodies against myc (9E10) and Texas red–conjugated secondary antibodies. The extent of colocalization between 2XFYVE, Rab5, and Rab4 (C) or 2XFYVE, Rab4, and Rab11 (E) was quantified. Colors in the histogram correspond to those used in the merge of double or triple fluorescent signals in the panels. White arrows indicate positive for 2XFYVE, Rab5, and Rab4. Green arrows indicate positive for Rab4 but negative for 2XFYVE and Rab5. Bar, 2 μm.
Early and recycling endosomes are structured as a mosaic of subcompartments that harbour distinct Rab GTPases and display a nonstochastic distribution (Sonnichsen et al., 2000). We next investigated whether PtdIns(3)P is not only specifically concentrated in the Rab5 domain within the early endosomes (Gillooly et al., 2003) but also depleted from the other Rab domains along the recycling pathway. Plasmids encoding myc–2XFYVE, CFP–Rab5, and YFP–Rab4 or myc–2XFYVE, YFP–Rab4, and CFP–Rab11 were microinjected in A431 cells (Fig. 1, B and C or D and E) and the overlap between Rab5, Rab4, and Rab11 and myc–2XFYVE was quantified (Fig. 1, C and E), as previously described (Sonnichsen et al., 2000). The 2XFYVE fluorescent probe was expressed at levels that neither caused the displacement of endogenous FYVE proteins such as EEA1 (unpublished data), nor significantly affected the previously reported distribution of Rab5 and Rab4 domains (Sonnichsen et al., 2000). More than 80% of 2XFYVE-positive endosomes colocalized with Rab5. Rab4 was present in ∼60% of these structures, i.e., in the fraction of endosomes containing both Rab5 and Rab4 but never Rab4 alone (Fig. 1 B, f, white arrows; Sonnichsen et al., 2000). In contrast, only 20% of endosomes harbouring Rab11 or both Rab4 and Rab11 were positive for myc–2XFYVE (Fig. 1 D). This fraction either corresponds to the Rab5+Rab4+Rab11 triple-positive compartment (Sonnichsen et al., 2000) or to structures that cannot be resolved by light microscopy techniques. These results suggest that PtdIns(3)P is specifically enriched in the Rab5 domain of early endosomes.
Detection of PIs phosphatase activity in the Rab5 affinity column eluate
Despite the interaction of Rab5 with both PI3Kβ and hVps34, the morphological analysis described above suggests that a mechanism must exist to efficiently segregate their products, PtdIns(3,4,5)P3 and PtdIns(3,4)P2 on the plasma membrane and PtdIns(3)P concentrated in a subdomain of the early endosomes. A clue to this mechanism came from the analysis of the Rab5 effectors purified by affinity chromatography from bovine brain cytosol on a GST–Rab5–GTPγS affinity column (Christoforidis et al., 1999a). Measurements of PI enzymatic activity of the Rab5 affinity column eluate (Fig. 2 A) were consistent with the reported presence of two types of PI 3-Ks (Christoforidis et al., 1999a). First, PtdIns(3)P was generated from PtdIns in a reaction catalyzed by hVps34. Second, PI3Kβ generated PtdIns(3,4)P2 or PtdIns(3,4,5)P3 from PtdIns(4)P and PtdIns(4,5)P2, respectively. Their activity was almost abolished in the presence of 50 nM wortmannin (unpublished data). Surprisingly, in addition to the aforementioned products, additional PI species were generated when PtdIns(4)P and PtdIns(4,5)P2 were used as substrates (Fig. 2 A, asterisks). In the presence of PtdIns(4)P, not only PtdIns(3,4)P2, but also PtdIns(3)P (Fig. 2 A, lane 2, asterisk) was detected. When PtdIns(4,5)P2 was used as substrate, in addition to the expected PtdIns(3,4,5)P3, also PtdIns(3,4)P2 and PtdIns(3)P were generated (Fig. 2 A, lane 3, asterisks). All TLC spots were 3′-phosphorylated PIs (3-PIs) as confirmed by HPLC analysis after excision from the TLC plate (unpublished data). The most straightforward explanation for such complex pattern of PIs is that, in addition to the two PI 3-Ks, the Rab5 affinity column eluate contains PtdIns(3,4,5)P3 5-phosphatase and PtdIns(3,4)P2 4-phosphatase activities. We thus performed PI 5-phosphatase and 4-phosphatase activity assays using PtdIns(3,4,5)P3 and PtdIns(3,4)P2 32P-labeled substrates (see Materials and methods). As shown in Fig. 2 B, PtdIns(3,4,5)P3 5-phosphatase and PtdIns(3,4)P2 4-phosphatase activities were indeed detected in the column eluate from the Rab5–GTPγS but not the Rab5–GDP affinity column. We therefore set out determine the identity of such phosphatases.
Figure 2.

PI phosphatases activity in Rab5–GTP column eluate. (A) PI3-K activity assay on eluates from GST–Rab5–GTPγS affinity columns was performed using PtdIns (PI), PtdIns(4)P (PI[4]P), or PtdIns(4,5)P2 (PI[4,5]P2) as substrates. Asterisks indicate extra product of 3′-phosphorylated PIs (see details in the Results section). (B) The PtdIns(3,4,5)P3 5-phosphatase or PtdIns(3,4)P2 4-phosphatase assay from eluates (15 or 5 μl, respectively) of GST–Rab5–GDP (gray bar) or GST–Rab5–GTPγS (black bar) affinity columns or buffer alone (white bar) was performed using 32P-labeled PtdIns(3,4,5)P3 or PtdIns(3,4)P2 as substrates (see Materials and methods; data are representative of two independent experiments). Higher amounts of Rab5 column eluate were required to measure the activity of PI 5-phosphatase compared with PI 4-phosphatase. (C) The eluate from GST–Rab5–GTPγS affinity column was separated using Superose 6 column and fractions analyzed by SDS-PAGE followed by Coomassie staining (asterisks indicate sequenced phosphatases). (D and E) Every third fraction was analyzed by either PtdIns(3,4,5)P3 5-phosphatase assay or PtdIns(3,4)P2 4-phosphatase assay. The phosphatase activity in peak fractions (fractions 25–29 and 34–36) was determined in two independent experiments.
Identification of PI phosphatases as new Rab5 effectors
The Rab5–GTPγS column eluate was fractionated by Superose-6 gel filtration column chromatography (Christoforidis et al., 1999a; Fig. 2 C) and the PtdIns(3,4,5)P3 5-phosphatase and PtdIns(3,4)P2 4-phosphatase activities measured in every third fraction (Fig. 2, D and E). The phosphatase assay was performed in the presence of 50 nM wortmannin to inhibit the PI 3-K activity during the reaction. Peaks of PI 5- and 4-phosphatase activity were detected in fractions 26–29 and 34–36, respectively, and protein bands from SDS-PAGE of these fractions were subjected to mass spectrometry sequencing. Among these proteins, we identified a type II inositol 5-phosphatase (5-Pase; Jefferson and Majerus, 1995) and a type I α PtdIns(3,4)P2 4-phosphatase (4-Pase; Norris et al., 1995). We cloned the cDNAs encoding both phosphatases, expressed them in Sf9 insect cells using the baculovirus system, purified the recombinant proteins and raised corresponding polyclonal antisera in rabbits. The antibodies against the 5- and 4-Pase detected protein bands corresponding to the expected molecular weight both in brain cytosol and selectively in the Rab5–GTPγS eluate (Fig. 3, A and B) confirming the specificity of the activity assay (Fig. 2, D and E). The anti–5-Pase detected two bands of 105 and 75 kD (Fig. 3 A, asterisks) that peptide sequencing data confirmed to be the 5-Pase and most likely a degradation product, respectively.
Figure 3.
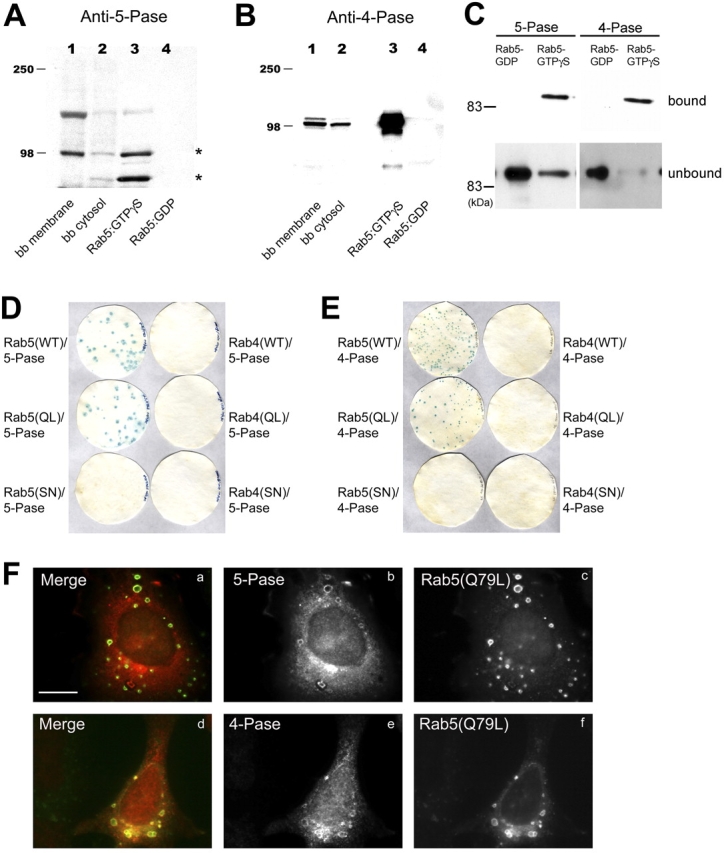
Identification of the 5- and 4-Pases and their direct and specific interaction with Rab5 in vivo and in vitro. (A and B) Eluates from GST–Rab5–GDP and GST–Rab5–GTPγS affinity columns, bovine brain (bb) cytosol, and membrane fractions were analyzed by Western blotting using antibodies against 5- and 4-Pase. (C) Beads with GST–Rab5 preloaded with GDP or GTPγS were incubated with 5 μg recombinant 5- or 4-Pases bound proteins were eluted and, along with unbound material, analyzed by Western blotting using anti–5- or 4-Pase antibodies. (D and E) L40p cotransformants with LexA–Rab5 (WT), –Rab5Q79L (QL), –Rab5S34N (SN), –Rab4 (WT), –Rab4Q67L (QL), or –Rab4S22N (SN) and either pGAD10–5-Pase or –4-Pase were plated on WL dropout media and analyzed by β-galactosidase replica filter assay. (F) HA–5-Pase (a–c) or 4-Pase (d–f) was coexpressed with Rab5(Q79L) or myc–Rab5(Q79L), respectively, in HeLa cells with vaccinia virus expression system. Cells were fixed and stained with antibodies against Rab5 (4F11) and HA (3F10) and then with Alexa 488–conjugated anti–mouse and Texas red–conjugated anti–rat secondary antibodies (a–c) or with anti-myc (9E10) and 4-Pase antibodies and then with Alexa 488–conjugated anti–mouse and rhodamine-conjugated anti–rabbit secondary antibodies (d–f). Bar, 10 μm.
Direct and specific interaction between 5- and 4-Pases and Rab5
The following three experiments demonstrated that both 5- and 4-Pase interact with Rab5 directly and specifically. First, we used a GST–Rab5 pull-down assay. Both recombinant 5- and 4-Pases bound to beads containing GST–Rab5 preloaded with GTPγS but not GDP (Fig. 3 C). Second, the specificity of such interaction was verified using the yeast two-hybrid system. Using the β-galactosidase replica filter assay (Fig. 3, D and E), blue colonies were obtained only upon cotransformation of prey plasmids (5- and 4-Pase) with bait vectors expressing wild-type Rab5 (WT)or activated Rab5 Q79L (QL) but neither from dominant negative Rab5 S34N (SN), nor Rab4 (WT, Q67L, S22N) transformants. Third, we tested the ability of Rab5:GTP to recruit 5- and 4-Pase onto endosomes in vivo. Whereas both endogenous and overexpressed 5- and 4-Pases were predominantly cytosolic, a fraction was recruited onto the membrane of enlarged endosomes specifically upon coexpression of the activated Rab5 Q79L mutant in HeLa cells (Fig. 3 F). Combined with the data of Fig. 2, these results suggest that the 5- and 4-Pases interact in a GTP-dependent manner, directly and specifically with Rab5 both in vitro and in vivo.
Active Rab5 stimulates the catalytic activity of PI3Kβ, 5-, and 4-Pase in vitro
Rab5 interacts with three different enzymes that can be ordered in a pathway to sequentially generate PtdIns(3,4,5)P3 from PtdIns(4,5)P2 (PI3Kβ) and subsequently dephosphorylate it to PtdIns(3,4)P2 (5-Pase) and PtdIns(3)P (4-Pase). To test the hypothesis that conversion of PtdIns(3,4,5)P3 into PI(3)P may indeed be regulated by Rab5, we first investigated whether the interaction of 5- and 4-Pases or PI3Kβ with this GTPase can result in a stimulation of their enzymatic activity. Recombinant 5-Pase, PI3Kβ, or the Superose 6 fraction containing the 4-Pase were incubated with recombinant Rab5 preloaded with GTPγS or GDP before determining the corresponding enzymatic activity. Nonprenylated Rab5 was used in this experiment as it binds these enzymes in vitro (Christoforidis et al., 1999b). PtdIns(4,5)P2 was used as substrate for PI3Kβ activity and 3′-32P-labeled PtdIns(3,4,5)P3 or PtdIns(3,4)P2 as substrates for 5- or 4-Pase activity, as described above (Fig. 2 B). Rab5–GTPγS but not Rab5–GDP dose dependently stimulated the activity of PI3Kβ, 5-Pase, or 4-Pase (Fig. 4, A–C). We conclude that the interaction with Rab5 stimulates the enzymatic activity of PI3Kβ, 5-, and 4-Pases in vitro.
Figure 4.

Rab5 regulates PI(3)P production with its effectors. (A–C) The activity of recombinant protein of PI3Kβ (p85α–p110β, 150 nM) or 5-Pase (25 nM) or 4-Pase fraction (1 μl fraction 36) were analyzed with recombinant Rab5–GDP or–GTPγS in the presence of GDP (white bar) or GTPγS (black bar), as indicated. The concentration of recombinant Rab5 is indicated in each bar. Bars indicate the stimulation of the enzymatic activity expressed in arbitrary units relative to control (see Materials and methods) and each set of data is representative from three independent experiments. Spot areas were quantified by BAS3000 and correspond to the following PIs: (A) refers to the spot area of PI(3,4,5)P3, (B) (PI[3,4]P2/[PI{3,4}P2+PI{3,4,5}P3]), (C) (PI[3]P/[PI{3}P+PI{3,4}P2]). (D) Endosomal fractions were incubated with buffer, 150 nM Rab5–GDI complex alone, 150 nM Rab5–GDI with anti-p110β or anti–4-Pase, or 5 μM HIS–RabGDI in the presence of 100 μM GTP or GDP and [32P]ATP for 5 min at 37°C. (E) Endosomal fractions were incubated with IgG, anti-p110β, anti–4-Pase or anti-hVps34 function blocking Abs and [32P]ATP for 5 min at 37°C. (D and E) Lipids were extracted, deacylated, and analyzed by HPLC (see Materials and methods). Data are means SD of two or three independent experiments.
Rab5 modulates PtdIns(3)P production by two different mechanisms
These findings prompted us to determine whether Rab5 actively regulates the production of PtdIns(3)P on membranes. To address this question, we used endosome enriched fractions from HeLa cells, incubated these membranes with [γ32P] cATP, in the presence or absence of either Rab5–GDI complex or RabGDI alone (see Materials and methods). Lipids were extracted, deacylated, and the PI isomers determined by HPLC analysis. Addition of RabGDI, which efficiently extracted Rab5 from the membrane (Rubino et al., 2000), inhibited PtdIns(3)P synthesis approximately by 60% (Fig. 4 D, compare first bar with second bar). On the other hand, addition of the Rab5–GDI, complex which increases the fraction of Rab5 on the membrane and the recruitment of its effectors (Rubino et al., 2000) stimulated the synthesis of PtdIns(3)P more than 2.5-fold (Fig. 4 D, compare first bar with third bar).
In principle, Rab5 can generate PtdIns(3)P directly through phosphorylation of PI by hVps34. However, because PI3Kβ, 5-, and 4-Pase are also Rab5 effectors, these three enzymes could be functionally linked in the generation of PtdIns(3)P from PtdIns(3,4,5)P3. To test this hypothesis, we used function-blocking antibodies, as shown previously for hVps34 (Siddhanta et al., 1998; Christoforidis et al., 1999b), to specifically block the activity of the PI kinases or phosphatases in the in vitro assay. We succeeded in raising function-blocking antibodies against p110β and 4-Pase (as determined using either recombinant proteins or the Rab5 affinity column eluate as source of enzymes; see Materials and methods). Unfortunately, despite several attempts we could not obtain blocking antibodies against the 5-Pase. The respective affinity-purified antibodies blocked more than 80% of the activity of PI3Kβ (p85α–p110β) or 4-Pase in vitro (unpublished data).
Anti-hVps34 function blocking antibodies reduced the production of PtdIns(3)P in endosomal fractions by 70%, consistent with previous results showing that Vps34 is required for the Rab5-dependent recruitment of EEA1 and early endosome fusion (Siddhanta et al., 1998; Christoforidis et al., 1999b; Hill et al., 2000; Fig. 4 E). Interestingly, anti-p110β function-blocking antibodies also reduced generation of PtdIns(3)P by 30%, and a comparable degree of inhibition was obtained with anti–4-Pase antibodies (Fig. 4 E). The stimulatory effect of Rab5 on PtdIns(3)P production was also dependent on PI3Kβ (p85α–p110β) and 4-Pase. The concomitant addition of anti-p110β or anti–4-Pase to Rab5GDI complex inhibited PtdIns(3)P by 30–40% (Fig. 4 D, compare third bar with fourth and fifth bar), supporting the idea that PI3Kβ and 4-Pase are downstream effectors of Rab5.
The simplest explanation of these results is that 30% of the PtdIns(3)P production in these membrane fractions, which contain early endosomes and some residual plasma membrane fragments, is due to PI3Kβ and to the sequential dephosphorylation of PtdIns(3,4,5)P3 via 5- and 4-Pase activities. Given the established activation of type I phosphoinositide-3-kinase (PI3-K) at the plasma membrane (Stephens et al., 1993; Fig.1), the presence of PI3-Kβ on clathrin-coated vesicles (Christoforidis et al., 1999b) and the enrichment of PI(3)P on early endosomes (Gillooly et al., 2003), these results suggest that Rab5 regulates the maintenance of a gradient of PtdIns(3)P from the plasma membrane to endosomal membranes by a combination of direct synthesis and PtdIns(3,4,5)P3 dephosphorylation.
Down-regulation of the 4- and 5-Pase inhibits transferrin uptake
We next explored the functional role of the 4-Pase in Rab5-mediated endocytic transport, by measuring transferrin internalization. We established experimental conditions to knock-down the 4-Pase using specific small interfering RNA (siRNA) oligonucleotides. 72 h after transfecting the cells with the siRNAs, we observed a dramatic reduction (∼70%) in 4-Pase protein levels as evidenced by Western blot (Fig. 5 A). Strikingly, we observed that knockdown of 4-Pase markedly inhibited transferrin internalization (Fig. 5 B). Silencing of the 5-Pase yielded a similar phenotype (unpublished data). These results therefore suggest a requirement of 4- and 5-Pases for Rab5-dependent receptor-mediated endocytosis.
Figure 5.

Knockdown of 4-Pase decrease transferrin uptake. (A) Reduced levels of 4-Pase 72 h after transfection of HeLa cells with siRNA, as detected by Western blot. (B) Cells transfected with unspecific (diamonds) or 4-Pase–specific (triangles) siRNA were allowed to internalize biotinylated transferrin for the indicated times. The internalized transferrin was quantified as described in Materials and methods, and standardized with respect to total protein concentration of the lysate and expressed as percent of the amount of internalized transferrin in control cells at t = 40 min (n = 4; mean ± SD of two independent experiments).
Translocation of 4- and 5-Pase to cortical ruffles following serum stimulation
We next investigated whether the localization of endogenous 5- and 4-Pase were consistent with the aforementioned model. To this aim we used primary cultures of astrocytes, as pilot experiments revealed significant levels of both proteins in these cells (Fig. 6). PtdIns(3,4,5)P3 production is strongly stimulated by growth factors, which induce recruitment and activation of PI 3-K to the plasma membrane (Stephens et al., 1993). Thus, we examined the localization of 5- and 4-Pase both in serum-starved cells and in starved cells exposed to serum for 15 min. The 4-Pase and the 5-Pase were mainly diffuse in the cytoplasm of serum-starved astrocytes (Fig. 6). However, upon serum stimulation, both enzymes translocated to the cortical ruffles where they colocalized with the actin binding protein cortactin and with a pool of Rab5 also recruited to the cell surface (Fig. 6 A). The localization of Rab5 to the plasma membrane is in good agreement with its participation in the generation of membrane ruffles and lamellipodia (Spaargaren and Bos, 1999; Lanzetti et al., 2004; unpublished data). Furthermore, EEA1-positive early endosomes also accumulated underneath the regions of the ruffling plasma membrane that were enriched in the 5- and the 4-Pase (Fig. 6 B). However, the two phosphatases did not accumulate on these structures although we occasionally observed the presence of the 4-Pase (unpublished data). Thus, both 5- and 4-Pases colocalized primarily with the cell surface but not the endosomal pool of Rab5. These results indicate that, under stimulatory conditions, the sites where PtdIns(3,4,5)P3 is generated (the plasma membrane) and the cell compartment enriched in PtdIns(3)P (early endosomes) are brought in close proximity of each other, and that Rab5, the 5- and 4-Pase are also enriched at these sites. These data support the hypothesis that a function of the enzymatic cascade controlled by Rab5 may be to rapidly and efficiently convert newly generated PtdIns(3,4,5)P3 into PtdIns(3)P.
Figure 6.
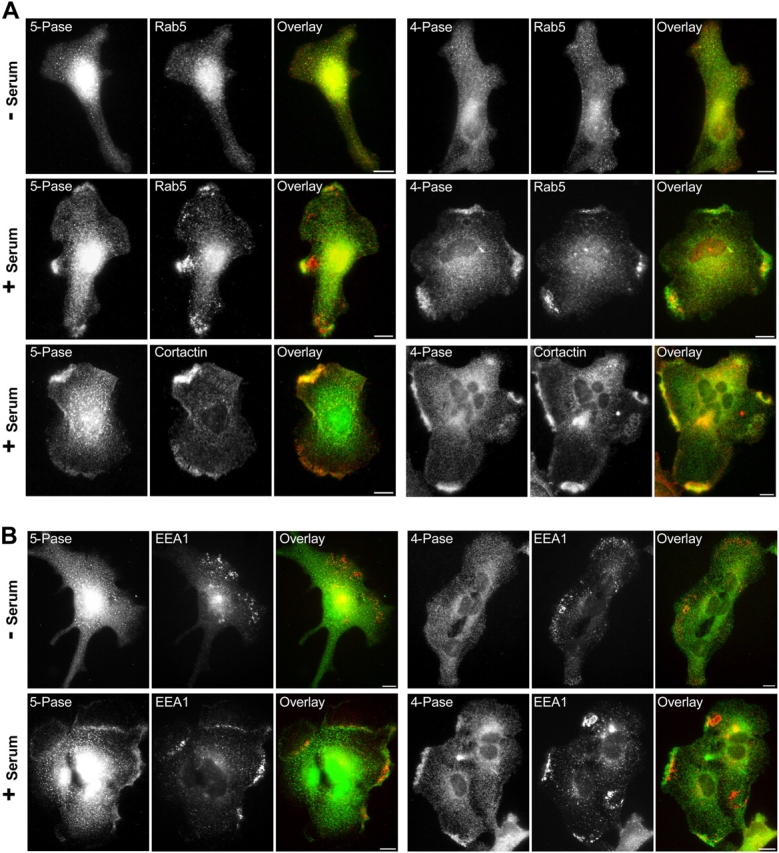
Localization of 5- and 4-Pase in cultured astrocytes. Cultured astrocytes starved overnight in serum-free medium were stimulated with 10% FCS-containing medium for 15 min at 37°C, stained by immunofluorescence with rabbit polyclonal antibodies against 4- or 5-Pase (green), and counterstained (red) with mouse antibodies directed against Rab5 (4F11) and cortactin or human autoantibodies directed against EEA1. Bars, 10 μm.
Functional consequences of loss of 4-Pase in brain
To provide evidence for the importance of the 4-Pase in a pathway leading to the generation of PtdIns(3)P in vivo, we searched for potential alterations of PI metabolism in cells of weeble mutant mice. Weeble mice carry a loss of function mutation in the gene encoding the 4-Pase (Nystuen et al., 2001). The 4-Pase was found to be present at the highest level in cerebellar Purkinje cells (Fig. 7 B), where it had a diffuse distribution throughout the cell body, dendrites, axons, and axon terminals (Fig. 7, B and D; Fig. 7, F–H shows colocalization of 4-Pase with the nerve termimal marker VAMP2–synaptobrevin 2 in Purkinje cell nerve terminals in the deep cerebellar nuclei). This result is consistent with high expression of 4-Pase mRNA in Purkinje cells and with the finding that Purkinje cells of the cerebellum are the most severely affected cell population in the weeble mice brain (Nystuen et al., 2001). A qualitatively similar immunostaining pattern, but of lower intensity, was observed in cells of the hippocampus (Fig. 7 I). An even weaker, but similar, staining pattern was observed in most other regions of the brain (unpublished data). The staining was completely absent in brain of weeble mice (Fig. 7, C, E, and J), indicating that the antibody specifically detects the 4-Pase protein, as confirmed by Western blot analysis (Fig. 7 A).
Figure 7.

Immunohistochemistry and PIs analysis in weeble mice. (A) Western blot for 4-Pase in +/+ and −/− mice and, as control, for β-tubulin. (B–J) Fluorescence microscopy analysis of 4-Pase on cerebellar and brain sections from 12-d-old wt (+/+; B, D, F, and I) and weeble (−/−; C, E, and J) mice. In +/+ mice, 4-Pase immunoreactivity was highly enriched in Purkinje cells and present throughout their cell bodies and dendrites (B) as well as axons and axon terminals in the deep cerebellar nuclei (D). (F) 4-Pase positive (green) Purkinje cell nerve terminals that outline the profile of a neuron in the deep cerebellar nuclei were counterstained with anti-VAMP2/synaptobrevins 2 antibodies (red). Green not overlapping with red signal represent nonsynaptic portions of Purkinje cell axons. (I) shows 4-Pase immunoreactivity in the hippocampus of +/+ mice. In −/− mice there was no immunoreactivity for 4-Pase in either the cerebellum (C and E), or the hippocampus (j). Bars: (A–I) 50 μm; (insets) 25 μm. ML, molecular layer; GL, granular layer; N, nucleus. (K) Production of free phosphate was measured with mice brain cytosol in the presence of the indicated substrates (see Materials and methods). The PI(3,4)P2 phosphatase activity is specifically impaired in the weeble mutant (−/−) mice. (L) Cultured astrocytes were labeled with [3H]myo-inositol and then incubated with FCS-free ([3H]myo-inositol containing) medium. After the cells were stimulated with 10% FCS for 15 min at 37°C, total lipids were extracted and analyzed by HPLC. Data show the percentages of fold increase compared with the unstimulated wt astrocytes and are means ± SD of three independent experiments. The symbols indicate the results of t test analysis; *, P < 0.05, **, P < 0.01 compared with the unstimulated wt astrocytes.
We investigated whether the absence of 4-Pase in weeble mutant mice produces changes in PI metabolism similar to those caused by function-blocking antibodies in endosome-enriched fractions (Fig. 4, D and E). First, we measured PI phosphatase activities in cytosol extracts from wild-type (wt), heterozygous, and homozygous weeble mutant mouse brains. Fig. 7 K shows that PtdIns(3,4)P2 4-phosphatase activity was specifically decreased in cytosol extracts from brain of weeble compared with wt mice. Second, we measured the level of various PI species after metabolic labeling of primary cultures of brain cells from wt and weeble mice. Given that immunofluorescence analysis of primary cultures from mouse cerebral cortices revealed significant expression of 4-Pase both in neurons and glia (Fig. 6), we used primary cultures of astrocytes for further experiments, as these cells grow very efficiently in vitro.
We compared levels of 3-PIs in serum-starved and serum-stimulated wt and mutant cells. Cultured astrocytes were metabolically labeled with [3H]inositol for 48 h followed by overnight incubation in serum-free medium in the continued presence of [3H]inositol. Cells were subsequently incubated in the absence or presence of serum for 15 min at 37°C, after which PIs were analyzed by HPLC. Strikingly, an approximate twofold increase in the levels of PtdIns(3,4)P2 was observed in astrocytes from weeble mice compared with cells from wt animals, under stimulated conditions (Fig. 7 L). Reduction in the levels of PtdIns(3,4,5)P3 in stimulated cells was also observed (Fig. 7 L), possibly reflecting some compensatory feed-back mechanisms. Consistent with the data of Fig. 4 (D and E), the levels of PtdIns(3)P were also reduced under both resting and stimulated conditions (27% and 28%, respectively; Fig. 7 L, inset) in the cells lacking 4-Pase as compared with wt cells. Altogether, these data indicate that the absence of 4-Pase specifically leads to an accumulation of PtdIns(3,4)P2 and a reduction of PtdIns(3)P presumably due a defect of PtdIns(3,4,5)P3 turnover, in agreement with the measurements on HeLa cells in vitro (Fig. 4 E).
Discussion
In this study we report that Rab5 coordinates a cascade of PI enzymes including the two previously described PI 3-Ks (hVps34 and PI3Kβ) and two new Rab5 effectors, the type II inositol 5-Pase (Jefferson and Majerus, 1995) and the type I α PtdIns(3,4)P2 4-Pase (Norris et al., 1995). Our data uncover a mechanism whereby a single GTPase, Rab5, regulates both synthesis and turnover of PIs through an enzymatic cascade of effectors, thus coupling the function of PIs in signal transduction to the requirements of endocytic trafficking.
Rab5 regulates PI synthesis and turnover in the endocytic pathway
Rab5 regulates a complex network of effector proteins that are recruited on the early endosome membrane by binding to PtdIns(3)P (Simonsen et al., 1998; Christoforidis et al., 1999b; Nielsen et al., 2000; Schnatwinkel et al., 2004). PtdIns(3)P is an important molecular hallmark of the endocytic pathway (Wurmser and Emr, 1998). It is required for early endosome fusion and motility along microtubules (Christoforidis et al., 1999a; Hoepfner et al., 2005), phagosome maturation (Vieira et al., 2001), multivesicular body formation (Futter et al., 2001), and signaling (Tsukazaki et al., 1998). The findings that the lipid kinase that generates PtdIns(3)P, hVps34, is also a Rab5 effector (Christoforidis et al., 1999b) and that the effectors form large oligomeric complexes on the early endosome membrane (McBride et al., 1999), led us to propose that Rab5 regulates the formation of a membrane domain on the early endosome enriched in PtdIns(3)P and containing the various effector proteins required for early endosome tethering, fusion, and motility (Zerial and McBride, 2001). Our present results strengthen this model with the demonstration that the synthesis of PtdIns(3)P is regulated by Rab5 itself.
Among the PIs tested, PtdIns(3)P was the major 3-PI present on early endosomes, despite the generation of other PI species on the plasma membrane and a continuous membrane flow toward the early endosomes. Furthermore, PtdIns(3)P is enriched in the Rab5 domain and less abundant or absent from other subcompartments of early and recycling (Rab4- and Rab11-positive) endosomes. In addition to hVps34, Rab5 interacts also with PI3Kβ, a type I PI 3-K involved in the generation of PtdIns(3,4,5)P3 and PtdIns(3,4)P2 upon stimulation at the plasma membrane. Even in the presence of the constitutively active Rab5Q79L mutant, PtdIns(3,4,5)P3 was localized to the plasma membrane but not early endosomes, suggesting that turnover of this PI must occur early to maintain the specificity of PtdIns(3)P localization in the endosomal system. The discovery that Rab5 interacts with, and stimulates the enzymatic activity of, 5- and 4-Pase, provides an explanation for how such synthesis and turnover can be coordinated.
We propose that the synthesis of PtdIns(3,4,5)P3 at the plasma membrane is coupled either to the dephosphorylation of the 3′ position by PTEN, thus leading to PtdIns(4,5)P2, or to the sequential dephosphorylation by 5- and 4-Pase, leading to PtdIns(3)P. Such enzymatic cascade could initiate at the plasma membrane, where under certain stimulatory conditions pools of PtdIns(3)P can be detected (Maffucci et al., 2003), and continue along transport to the early endosomes, given that PI3Kβ is detected in clathrin-coated vesicles (Christoforidis et al., 1999b), presumably until PtdIns(3,4,5)P3 and PtdIns(3,4)P2 are depleted. Other 5-phosphatases, in particular SHIP, that prefers PtdIns(3,4,5)P3 as a substrate, may cooperate with 5-Pase in the first of the two dephosphorylation reactions. This model is consistent with a study by Ivetac et al. (2005), published while this manuscript was in revision, that reported the association of 4-Pase with early and recycling endosomes in COS-1 cells.
Interestingly, the 5-Pase was identified in a search for PtdIns(3,4,5)P3-binding proteins (Krugmann et al., 2002) and the 4-Pase was recovered in a complex with PI3-K (Munday et al., 1999) and binds PtdIns(3,4)P2 via its C2 domains (Ivetac et al., 2005). These observations raise the interesting possibility that, at the plasma membrane, Rab5 may regulate the recruitment of various effectors by a combinatorial principle similar to the one operating on early endosomes. Whereas on early endosomes Rab5 regulates the recruitment of FYVE proteins (e.g., EEA1) in combination with PtdIns(3)P, at the plasma membrane it may cooperate with PtdIns(3,4,5)P3 in the recruitment of other effector proteins. Specifically, activated Rab5 would bind and stimulate PI3Kβ activity, thus eliciting in a positive feedback mechanism the production of its “co-receptor” PtdIns(3,4,5)P3. Both Rab5 and PtdIns(3,4,5)P3 would then serve as binding sites to recruit the 5-Pase, resulting in dephosphorylation of PtdIns(3,4,5)P3 to PtdIns(3,4)P2. PtdIns(3,4)P2 would then recruit the 4-Pase, which, activated by Rab5-GTP, would dephosphorylate PtdIns(3,4)P2 to PtdIns(3)P.
The extent to which such enzymatic cascade operates along the pathway probably depends on cell type and growth conditions. Under steady-state, it may contribute only a lesser (<30%) fraction of PtdIns(3)P production in comparison with direct phosphorylation of PtdIns by hVps34. However, it may constitute an important regulatory system ensuring endocytic transport and organelle homeostasis under various PI 3-K–dependent signaling conditions. Rab5 itself is activated both at the plasma membrane and on EEA1-positive early endosomes upon EGF stimulation (Di Fiore and De Camilli, 2001). The stimulatory activity of Rab5 on PI3Kβ could thus contribute to the generation of 3-PIs at the cell surface in response to various signals, thus inducing morphogenetic changes (Spaargaren and Bos, 1999; Lanzetti et al., 2004). The finding that both 5- and 4-Pase are recruited along with Rab5 to the cell cortex upon serum stimulation (this study and Ivetac et al., 2005) strongly supports the view that the PtdIns(3,4,5)P3 produced is subjected to Rab5-regulated turnover to restrict it to the plasma membrane and to restore the production of PtdIns(3)P before, or at arrival into, the early endosomes that accumulate underneath the ruffling region. This mechanism must operate with high efficiency early in the pathway as both 5- and 4-Pase do not accumulate on early endosomes (Fig. 6), and PtdIns(3,4,5)P3 and PtdIns(3,4)P2 could not be detected on this compartment (Fig. 1). Accordingly, PtdIns(3,4,5)P3 was observed in the phagocytic cup (Marshall et al., 2001) but not on phagosomes, which were instead enriched in PtdIns(3)P (Vieira et al., 2001). In addition, Rab5 may regulate the activity of other PI Pases, as we have recently detected the inositol polyphosphate 5-phosphatase OCRL (Attree et al., 1992; Zhang et al., 1995) in the eluate from the Rab5 affinity column (unpublished data). By converting PtdIns(4,5)P2 into PtdIns(4)P, OCRL may contribute to the Rab5-dependent regulation of PIs on endosomal receptor trafficking and sorting (Ungewickell et al., 2004; Choudhury et al., 2005).
Although dephosphorylation at the 3′ position of the inositol ring by PI 3-Pases such as PTEN (Leslie and Downes, 2002) may contribute to termination of PI(3,4,5)P3 signaling, a distinguished feature of the enzymatic cascade described in this study is the generation of other 3-PIs that have signaling functions of their own. Unlike Ivetac et al. (2005), we could not consistently observe endosomal abnormalities in HeLa cells lacking 4-Pase or primary cultures of weeble neurons (unpublished data). The expression of inositol 4-phosphatase type II may partially compensate for the lack of 4-Pase (the type I isoform; Majerus et al., 1999) and the presence of Vps34 ensures the bulk of production of PtdIns(3)P. However, unexpectedly we could detect alterations in receptor-mediated endocytosis. Because inhibition of PI3-K with wortmannin does not dramatically impair transferrin internalization (Martys et al., 1996; Shpetner et al., 1996; Spiro et al., 1996), it is plausible that the accumulation of PtdIns(3,4,5)P3 and PI(3,4)P2 rather than reduced production of PI(3)P on endosomes by 4-Pase RNAi may exert an inhibitory effect on the endocytic process.
Unbalance in PI metabolism and neuronal degeneration in 4-Pase–deficient mice
That the aforementioned turnover of PIs is of high physiological importance is underscored by the phenotypic analysis of weeble mutant mice (Nystuen et al., 2001). When we inspected the PIs levels in cultured astrocytes of weeble mice, we detected an enhancement of PtdIns(3,4)P2 as well as significant (∼30%) reduction in PtdIns(3)P, under both resting and stimulatory conditions. These data are consistent with the measurements on PI synthesis on HeLa membrane fractions in vitro and the view that dephosphorylation of PtdIns(3,4,5)P3 to PtdIns(3)P is impaired in weeble mutants due to a selective block of the 4-Pase reaction.
The precise mechanism leading to early neuronal loss in weeble mutant mice remain to be established. We suggest that impaired PtdIns(3,4)P2 dephosphorylation may cause an imbalance in both signaling and endocytosis due to excess PtdIns(3,4)P2 signaling, lowered PtdIns(3)P production or both. Several scenarios, including the ones listed below, can be hypothesized to explain neuronal death.
First, abnormal PI signaling may affect the balance between factors that promote cell survival and cell death. During postnatal neural development, cell proliferation, apoptosis, and differentiation are regulated by complex and accurately orchestrated signaling pathways, triggered by various neurotrophic factors (Segal and Greenberg, 1996). PIs play an essential role in these processes (Fruman et al., 1998) and the unbalance in PI turnover, primarily the accumulation of PtdIns(3,4)P2, may have important disrupting effects of growth factor receptor signaling. For example, neurons that are forced to reenter the cell cycle by an expressed oncogene undergo apoptosis rather than divide (Feddersen et al., 1992).
Second, the endocytic alterations due to loss of 4-Pase may produce directly or indirectly an impact on neuronal function. For example, a chronic impairment in glutamate receptor endocytosis may lead to excess excitatory signaling with resulting cell death (Garthwaite and Garthwaite, 1991; Doughty et al., 2000).
Third, as 4-Pase can also act on inositol polyphosphates (Norris et al., 1995, 1997; Majerus et al., 1999), actions mediated by abnormal metabolic flux through the various inositol polyphosphate metabolites cannot be excluded. Besides Ins(1,4,5)P3, which controls Ca2+ signaling, inositol polyphosphates also have important effects on nuclear function (Steger et al., 2003; York, 2003).
In conclusion, our data underscore the importance of enzymatic networks regulating PI synthesis and turnover to coordinate both signaling and trafficking functions. The Rab5 network of PI kinases and phosphatases appears to be fundamental under stimulatory conditions, particularly during maturation of the nervous system. It will be important to further elucidate how Rab5 and its effectors may actively participate in signal transduction in neurons and other highly differentiated cell types.
Materials and methods
Reagents and cell lines
Phospholipids were purchased from Sigma-Aldrich, [γ-32P]ATP from Amersham Biosciences, and Silica Gel 60 TLC plates (20 × 20 cm) from Merck. [3H]PtdIns, [3H]PtdIns(4)P, and [3H]PtdIns(4,5)P2 were purchased from NEN Life Science Products. Most HPLC grade organic chemicals and water were purchased from Fluka or Merck. GST–p85α baculovirus was a gift of Dr. Waterfield (Ludwig Institute for Cancer Research, London, UK), the polyclonal human anti-EEA1 antibody, GFP-2XFYVE (Hrs) and myc-2XFYVE plasmids of Dr. Stenmark (Norwegian Radium Hospital, Oslo, Norway). A431, NIH3T3, HeLa cells, and astrocytes from weeble mice were cultured under standard conditions.
Antibodies and plasmids
Anti-p110β or hVps34 function blocking antibodies were raised against synthetic peptides (C)KVNWMAHTVRKDYRS or AVVEQIHKFAQYWRK (Siddhanta et al., 1998; Hill et al., 2000) and anti-5 or -4 Pase antibodies against recombinant full length proteins (see Preparation of recombinant PI3Kβ, 5-Pase, 4-Pase, and Rab5). The affinity-purified anti-p110β and anti–4-Pase antibodies blocked >80% activity of the recombinant proteins in 1:2 or 1:4 antigen/antibody molar ratio. Anti-hVps34 antibody was used as previously reported (Siddhanta et al., 1998; Christoforidis et al., 1999b). Mouse monoclonal antibody against Rab5a was 4F11, monoclonal mouse anti-cortactin, and rat anti-HA (3F10) antibodies, and HRP- and fluorescent-conjugated secondary antibodies were purchased from Upstate Biotechnology, Roche, Dianova, and Molecular Probes, respectively.
The human cDNA encoding human p110β, a gift of Dr. Maier (Freie University, Berlin, Germany), was subcloned into modified baculovirus expression vector pFastBacGST. The human cDNAs encoding 5- and 4-Pase obtained by RT-PCR were subcloned into pcDNA3 (Invitrogen), pGAD10, and pFastBacGST. Plasmids expressing ECFP- and EYFP-tagged Rab and Rab5Q79L were as previously described (Stenmark et al., 1994; Sonnichsen et al., 2000). cDNAs encoding the PH domain of human Akt was cloned by RT-PCR and mouse PLCδ, human FAPP1 were purchased from IMAGE consortium and subcloned into EGFP-c3 vectors (Clontech).
Yeast two-hybrid and pull down assay
Yeast transformation and two-hybrid analysis were performed according to the MATCHMAKER instructions (Clontech). In brief, a yeast strain L40 was cotransformed with a pLexA-based bait vector and a pGAD10-based prey vector and plated on a medium lacking Trp and Leu. After 2–3 d of incubation colonies were tested for β-galactosidase activity by the replica filter assay. GST–Rab5 pull-down assay using recombinant 5- and 4-Pase proteins (5 μg each) was performed as previously described (Christoforidis et al., 1999a).
Cell transfection, microinjection, and immunofluorescence analysis
Plasmids were transfected to NIH3T3 cells with FuGENE6 (Roche). Expression of proteins in HeLa cells grown to 60% confluence using the T7 RNA polymerase recombinant vaccinia virus was as in (Stenmark et al., 1994).
For the colocalization of 2XFYVE with Rab proteins, a mixture of 50 ng/μl plasmid DNA for myc-2XFYVE, EYFP-Rab4, and ECFP-Rab5 or ECFP-Rab11 was injected into the nucleus of A431 cells with an Eppendorf micromanipulator and transjector. Immunofluorescence analysis (Stenmark et al., 1994) was performed using an LSM 510 station confocal microscope (Carl Zeiss MicroImaging, Inc.). Quantification of colocalization was performed as in Sonnichsen et al. (2000).
Cultured astrocytes serum-starved overnight were incubated in MEM with or without 10% FCS for 15 min at 37°C, fixed in 4% PFA in phosphate buffer, and then stained by immunofluorescence by standard procedures. Primary antibodies were visualized with goat anti–rabbit IgGs conjugated to Oregon green, anti–mouse IgGs conjugated to Alexa 594, and anti–human IgGs conjugated to Texas red.
Immunohistochemistry of brain sections
Weeble mice and wt 12- or 19-d-old littermates were fixed by transcardiac perfusion with 1% PFA in 120 mM sodium phosphate buffer. Brains were collected, fixed for an additional 3 h in the same solution, cryoprotected by infiltration with sucrose 30%, and frozen. 12-μm cryostat sections were immunolabeled as previously described (De Camilli et al., 1983) with rabbit anti–4-Pase antibodies and monoclonal antibodies to VAMP2-synaptobrevin 2 (Synaptic Systems). Goat anti–rabbit IgGs conjugated to Alexa 568 or Oregon green and goat anti–mouse IgGs conjugated to Texas red were used as secondary antibodies.
Preparation of recombinant PI3Kβ, 5-Pase, 4-Pase, and Rab5
Recombinant Pases were expressed as GST-tagged proteins in High Five insect cells according to the manufacturer's instructions (BD Biosciences) with the use of pFAST Bac GST-5-Pase or GST-4-Pase. To produce recombinant p85α–p110β complex, High Five cells were coinfected with GST-tagged p85α and untagged p110β baculoviruses, and the proteins purified by a single-step on a glutathione–agarose column followed by cleavage of GST with Precission protease (GE Healthcare). Recombinant GST–Rab5 was purified as previously described (Christoforidis et al., 1999a) and GST was cleaved with Factor Xa.
PI 3-K activity assay
PI 3-K activity in the GST–Rab5/GTPγS column eluate of bovine brain cytosol (Christoforidis et al., 1999a) or by recombinant PI3Kβ was assayed in buffer containing PtdIns (0.2 mg/ml), PtdIns(4)P (0.2 mg/ml), or PtdIns(4,5)P2 (0.23 mg/ml) in 50 μl of 20 mM Hepes, pH 7.4, 1 mM EGTA, 5 mM MgCl2, 50 μM ATP, 10 μCi [γ32P]ATP, 0.23 mg/ml phosphatidylserine, 0.2 mM adenosine. The reaction was run for 10 min at 37°C (unless stated) and arrested with 50 μl of 1 N HCl, extracted with 100 μl of CHCl3/MeOH (1:1) and washed twice with 1 N HCl/MeOH (1:1). Dried lipids were resuspended in 15 μl of CHCl3/MeOH/H2O (75:25:2, vol/vol/vol), separated by TLC using CHCl3/acetone/MeOH/glacial acetic actid/H2O (80:30:26:24:14,vol/vol/vol/vol/vol), analyzed using BAS3000 bioimaging analyzer (Fuji) and the corresponding spots were quantified by Image Guage software (Fuji). TLC plates were pretreated with (1% potassium oxalate/2 mM EDTA)/MeOH (1:1, vol/vol) and dried overnight.
Production of 32P-labeled PIs and Pase activity assay
Recombinant PI3Kβ (p85α/p110β) was used to produce [32P]PtdIns(3,4)P2 and [32P]PtdIns(3,4,5)P3 using the described methods above. The mixture of PtdIns(4)P/[32P]PtdIns(3,4)P2 or PtdIns(4,5)P2/[32P]PtdIns(3,4,5)P3 was then extracted, dried under nitrogen, and stored at −20°C. Dried [32P]PtdIns(3,4)P2 were dissolved in 50 mM MOPS, pH 6.8, 200 mM NaCl, 0.3% β-octylglucoside for 4-Pase activity and [32P]PtdIns(3,4,5)P3 in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.3% β-octylglucoside for 5-Pase activity assays by sonication. The assay was started upon addition of either GST–Rab5 column eluate or recombinant phosphatases, incubated for 5 min at 37°C and the reaction was blocked by addition of 1 M HCl. Upon extraction the inositol lipids were then separated by TLC and quantified, as described above. The 5- and 4-Pase activities were calculated as PtdIns(3,4)P2/(PtdIns[3,4]P2+PtdIns[3,4,5]P3) or PtdIns(3)P/(PtdIns[3]P+PtdIns[3,4]P2) ratio, respectively.
For phosphate release assay, the cytosolic protein preparations were obtained by homogenization of brains from 10-d-old mice in 25 mM Tris, pH 8.0, 250 mM sucrose, 500 mM KCl, 10 mM MgCl2, 2 mM EGTA, 1 mM DTT containing Complete Mini, EDTA-free protease inhibitor cocktail (Roche Applied Sciences). Homogenates were ultrafuged 45 min at 50,000 rpm at 4°C in a TLA 100.2 rotor (Beckman Coulter). Supernatants were passed over NAP 5 columns (Amersham Biosciences) and eluted with 30 mM Hepes, pH 7.4, 1 mM EGTA, 1 mM MgCl2, and 100 mM KCl. Two independent wt, heterozygous, and knock out mice were assayed in triplicate with and without addition of synthetic PIs (Echelon Research Laboratories). Production of free phosphate was assayed as described previously (Harder et al., 1994). 5–10 μg cytosol protein were incubated with 1 μg lipid substrate for 10–30 min at 37°C. Free Pi generated was measured by malachite green assay using a microplate reader at 620 nm. Data were corrected for background activity by subtracting −PIP absorbance from +PIP absorbance for each reaction.
Analysis of [32P]PIs production in isolated endosomes
Endosome fractions (20 μg) prepared as described by Gorvel et al. (1991) were incubated in reaction buffer (50 μl of 20 mM Hepes, pH 7.4, 1 mM EGTA, 5 mM MgCl2, 5 mM MnCl2, 20 μM ATP, 70 μCi [γ32P]ATP) for 5 min at 37°C. The membrane fraction was incubated with 50 μM GTP and purified 164 nM of Rab5–GDI complex, 50 μM GDP, and 10 μM of His–RabGDI from Escherichia coli (Ullrich et al., 1995) or preincubated with IgG or affinity purified anti-p110β or anti–4-Pase Ab. The reaction was arrested by adding 50 μl of 1 N HCl and lipids were extracted, dried under nitrogen gas, and stored at −20°C until use. To isolate PI isomers, [32P]phospholipids mixture was deacylated and analyzed by anion exchange PartiSphere 5 SAX column (Whatman) connected to HPLC (Waters 2690) and radioactive detector (Biostep) according to Serunian et al. (1991). Relevant peaks were identified by coelution with commercially available 3H-labeled standards or by comparing with the retention time of 3′-32P-labeled products from PI3-K assay as described above. The recorded peak by radioactive detector was calculated as a peak area by Millenium software (Waters) or the radioactivity in each fraction counted by β-scintillation.
Metabolic labeling of cultured astrocytes
Cultured astrocytes were incubated with [3H]myo-inositol (20 μCi/ml) for 48 h in medium containing FCS and subsequently starved with FCS-free ([3H]myo-inositol–containing) medium overnight at 37°C. Cells were then stimulated with 10% FCS for 15 min at 37°C. Total lipids were extracted by adding 50% MeOH/1 M HCl and chloroform, deacylated, and analyzed by HPLC according to Serunian et al. (1991). Radioactivity was assayed with an on-line counter (Packard). Peaks were identified using internal standards (Cremona et al., 1999; Nemoto et al., 2000).
siRNA tranfection and biotinylated transferrin internalization
Duplex siRNA (4-Pase: 5′-GGAAAUAUACAAGACCCAGTT and 5′-CUGGGUCUUGUAUAUUUCCTG; 5-Pase: 5′-CGCUCUCUUCCUCUAUACGTT and 5′-CGUAUAGAGGAAGAGAGCGTG) were purchased from Ambion. For the transferrin internalization assay, HeLa cells were transfected with specific siRNAs for various phosphatases or GFP (mock) using oligofectamine (Invitrogen). At 72 h (siRNA) after transfection, cells were starved for 2 h in CO2-independent DME medium containing 0.2% BSA and then allowed to internalize biotinylated transferrin (10 μg/ml) for the indicated times. Cells were placed on ice, washed, lysed, and the total cell extract was incubated with affinity-purified, Ruthenium-labeled, sheep anti–human transferrin antibodies (SAPU) and subsequently analyzed using an ECL-Analyser System from Igen Inc.
Acknowledgments
We are indebted to A. Giner for excellent technical assistance, Drs. M. Miaczynska, C. Schnatwinkel, and A. Schenck for valuable comments on the paper, Dr. K. Nakayama for encouragement and discussions, and G. Di Paolo, T. Itoh, and L. Lucast for discussions and advice.
This work was supported by grants from the The Human Frontier Science Program (HFSP; RG-0260/1999-M), the European Union (HPRN-CT-2000-00081), the Max Planck Society, the National Institutes of Health (NS36251, DK45735, NS31348, and NS43927), and the Yale Center for Genomics and Proteomics. H.-W. Shin was a recipient of fellowships from the Alexander von Humboldt Foundation and Max Planck Society and M. Hayashi of a long-term fellowship from the HFSP.
Abbreviations used in this paper: 4-Pase, a type I α PtdIns(3,4)P2 4-phosphatase; 5-Pase, type II inositol 5-phosphatase; OCRL, oculocerebrorenal syndrome of Lowe; PI, phosphoinositide; PI3-K, phosphoinositide-3-kinase; PtdIns, phosphatidylinositol; wt, wild-type.
S. Uttenweiler-Joseph's present address is IPBS, CNRS, 31077 Toulouse Cedex, France.
H.-W. Shin's present address is Dept. of Physiological Chemistry, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan.
M.R. Wenk's present address is Depts. of Biochemistry and Biological Sciences, National University of Singapore, Singapore 117597, Republic of Singapore.
References
- Attree, O., I.M. Olivos, I. Okabe, L.C. Bailey, D.L. Nelson, R.A. Lewis, R.R. McInnes, and R.L. Nussbaum. 1992. The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 358:239–242. [DOI] [PubMed] [Google Scholar]
- Backman, S.A., V. Stambolic, A. Suzuki, J. Haight, A. Elia, J. Pretorius, M.S. Tsao, P. Shannon, B. Bolon, G.O. Ivy, and T.W. Mak. 2001. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat. Genet. 29:396–403. [DOI] [PubMed] [Google Scholar]
- Cantley, L.C., and B.G. Neel. 1999. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA. 96:4240–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury, R., A. Diao, F. Zhang, E. Eisenberg, A. Saint-Pol, C. Williams, A. Konstantakopoulos, J. Lucocq, L. Johannes, C. Rabouille, et al. 2005. Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol. Biol. Cell. 10.1091/mbc.E05-02-0120. [DOI] [PMC free article] [PubMed]
- Christoforidis, S., H. McBride, D. Burgoyne, and M. Zerial. 1999. a. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 397:621–625. [DOI] [PubMed] [Google Scholar]
- Christoforidis, S., M. Miaczynska, K. Ashman, M. Wilm, L. Zhao, S.C. Yip, M.D. Waterfield, J.M. Backer, and M. Zerial. 1999. b. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1:249–252. [DOI] [PubMed] [Google Scholar]
- Chung, C.Y., S. Funamoto, and R.A. Firtel. 2001. Signaling pathways controlling cell polarity and chemotaxis. Trends Biochem. Sci. 26:557–566. [DOI] [PubMed] [Google Scholar]
- Cremona, O., G. Di Paolo, M.R. Wenk, A. Luthi, W.T. Kim, K. Takei, L. Daniell, Y. Nemoto, S.B. Shears, R.A. Flavell, et al. 1999. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 99:179–188. [DOI] [PubMed] [Google Scholar]
- De Camilli, P., S.M. Harris Jr., W.B. Huttner, and P. Greengard. 1983. Synapsin I (Protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J. Cell Biol. 96:1355–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli, P., S.D. Emr, P.S. McPherson, and P. Novick. 1996. Phosphoinositides as regulators in membrane traffic. Science. 271:1533–1539. [DOI] [PubMed] [Google Scholar]
- de Graaf, P., W.T. Zwart, R.A.J. van Dijken, M. Deneka, T.K.F. Schulz, N. Geijsen, P.J. Coffer, B.M. Gadella, A.J. Verkleij, P. van der Sluijs, and P.M.P. van Bergen en Henegouwen. 2004. Phosphatidylinositol 4-kinaseβ is critical for functional association of rab11 with the Golgi complex. Mol. Biol. Cell. 15:2038–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis, M.A., and A. Godi. 2004. PI-loting membrane traffic. Nat. Cell Biol. 6:487–492. [DOI] [PubMed] [Google Scholar]
- Di Fiore, P.P., and P. De Camilli. 2001. Endocytosis and signaling. An inseparable partnership. Cell. 106:1–4. [DOI] [PubMed] [Google Scholar]
- Doughty, M.L., P.L. De Jager, S.J. Korsmeyer, and N. Heintz. 2000. Neurodegeneration in Lurcher mice occurs via multiple cell death pathways. J. Neurosci. 20:3687–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowler, S., R.A. Currie, D.G. Campbell, M. Deak, G. Kular, C.P. Downes, and D.R. Alessi. 2000. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J. 351:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feddersen, R.M., R. Ehlenfeldt, W.S. Yunis, H.B. Clark, and H.T. Orr. 1992. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 9:955–966. [DOI] [PubMed] [Google Scholar]
- Fruman, D.A., R.E. Meyers, and L.C. Cantley. 1998. Phosphoinositide kinases. Annu. Rev. Biochem. 67:481–507. [DOI] [PubMed] [Google Scholar]
- Funamoto, S., R. Meili, S. Lee, L. Parry, and R.A. Firtel. 2002. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 109:611–623. [DOI] [PubMed] [Google Scholar]
- Futter, C.E., L.M. Collinson, J.M. Backer, and C.R. Hopkins. 2001. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J. Cell Biol. 155:1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite, G., and J. Garthwaite. 1991. Mechanisms of AMPA neurotoxicity in rat brain slices. Eur. J. Neurosci. 3:729–736. [DOI] [PubMed] [Google Scholar]
- Gillooly, D.J., C. Raiborg, and H. Stenmark. 2003. Phosphatidylinositol 3-phosphate is found in microdomains of early endosomes. Histochem. Cell Biol. 120:445–453. [DOI] [PubMed] [Google Scholar]
- Godi, A., P. Pertile, R. Meyers, P. Marra, G. Di Tullio, C. Iurisci, A. Luini, D. Corda, and M.A. De Matteis. 1999. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1:280–287. [DOI] [PubMed] [Google Scholar]
- Gorvel, J.-P., P. Chavrier, M. Zerial, and J. Gruenberg. 1991. Rab5 controls early endosome fusion in vitro. Cell. 64:915–925. [DOI] [PubMed] [Google Scholar]
- Harder, K.W., P. Owen, L.K. Wong, R. Aebersold, I. Clark-Lewis, and F.R. Jirik. 1994. Characterization and kinetic analysis of the intracellular domain of human protein tyrosine phosphatase beta (HPTP beta) using synthetic phosphopeptides. Biochem. J. 298:395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, K., S. Welti, J. Yu, J.T. Murray, S.-C. Yip, J.S. Condeelis, J.E. Segall, and J.M. Backer. 2000. Specific requirement for the p85-p110α phosphatidylinositol 3-kinase during epidermal growth factor-stimulated actin nucleation in breast cancer cells. J. Biol. Chem. 275:3741–3744. [DOI] [PubMed] [Google Scholar]
- Hoepfner, S., F. Severin, A. Cabezas, B. Habermann, A. Runge, D. Gillooly, H. Stenmark, and M. Zerial. 2005. Modulation of receptor recycling and degradation by the endosomal kinesin KIF16B. Cell. 121:437–450. [DOI] [PubMed] [Google Scholar]
- Honda, A., M. Nogami, T. Yokozeki, M. Yamazaki, H. Nakamura, H. Watanabe, K. Kawamoto, K. Nakayama, A.J. Morris, M.A. Frohman, and Y. Kanaho. 1999. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 99:521–532. [DOI] [PubMed] [Google Scholar]
- Ivetac, I., A.D. Munday, M.V. Kisseleva, X.-M. Zhang, S. Luff, T. Tiganis, J.C. Whisstock, T. Rowe, P.W. Majerus, and C.A. Mitchell. 2005. The type Iα inositol polyphosphate 4-phosphatase generates and terminates phosphoinositide 3-kinase signals on endosomes and the plasma membrane. Mol. Biol. Cell. 16:2218–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson, A.B., and P.W. Majerus. 1995. Properties of type II inositol polyphosphate 5-phosphatase. J. Biol. Chem. 270:9370–9377. [DOI] [PubMed] [Google Scholar]
- Katso, R., K. Okkenhaug, K. Ahmadi, S. White, J. Timms, and M.D. Waterfield. 2001. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 17:615–675. [DOI] [PubMed] [Google Scholar]
- Krauss, M., M. Kinuta, M.R. Wenk, P. De Camilli, K. Takei, and V. Haucke. 2003. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Iγ. J. Cell Biol. 162:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugmann, S., K.E. Anderson, S.H. Ridley, N. Risso, A. McGregor, J. Coadwell, K. Davidson, A. Eguinoa, C.D. Ellson, P. Lipp, et al. 2002. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol. Cell. 9:95–108. [DOI] [PubMed] [Google Scholar]
- Lanzetti, L., A. Palamidessi, L. Areces, G. Scita, and P.P. Di Fiore. 2004. Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature. 429:309–314. [DOI] [PubMed] [Google Scholar]
- Leslie, N.R., and C.P. Downes. 2002. PTEN: the down side of PI 3-kinase signalling. Cell. Signal. 14:285–295. [DOI] [PubMed] [Google Scholar]
- Levine, T.P., and S. Munro. 2002. Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and -independent components. Curr. Biol. 12:695–704. [DOI] [PubMed] [Google Scholar]
- Maffucci, T., A. Brancaccio, E. Piccolo, R.C. Stein, and M. Falasca. 2003. Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J. 22:4178–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerus, P.W., M.V. Kisseleva, and F.A. Norris. 1999. The role of phosphatases in inositol signaling reactions. J. Biol. Chem. 274:10669–10672. [DOI] [PubMed] [Google Scholar]
- Marshall, J.G., J.W. Booth, V. Stambolic, T. Mak, T. Balla, A.D. Schreiber, T. Meyer, and S. Grinstein. 2001. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fcγ receptor-mediated phagocytosis. J. Cell Biol. 153:1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martys, J.L., C. Wjasow, D.M. Gangi, M.C. Kielian, T.E. McGraw, and J.M. Backer. 1996. Wortmannin-sensitive trafficking pathways in Chinese hamster ovary cells. J. Biol. Chem. 271:10953–10962. [DOI] [PubMed] [Google Scholar]
- McBride, H.M., V. Rybin, C. Murphy, A. Giner, R. Teasdale, and M. Zerial. 1999. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell. 98:377–386. [DOI] [PubMed] [Google Scholar]
- Mitchell, C.A., R. Gurung, A.M. Kong, J.M. Dyson, A. Tan, and L.M. Ooms. 2002. Inositol polyphosphate 5-phosphatases: lipid phosphatases with flair. IUBMB Life. 53:25–36. [DOI] [PubMed] [Google Scholar]
- Munday, A.D., F.A. Norris, K.K. Caldwell, S. Brown, P.W. Majerus, and C.A. Mitchell. 1999. The inositol polyphosphate 4-phosphatase forms a complex with phosphatidylinositol 3-kinase in human platelet cytosol. Proc. Natl. Acad. Sci. USA. 96:3640–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto, Y., B.G. Kearns, M.R. Wenk, H. Chen, K. Mori, J.G. Alb Jr., P. De Camilli, and V.A. Bankaitis. 2000. Functional characterization of a mammalian Sac1 and mutants exhibiting substrate-specific defects in phosphoinositide phosphatase activity. J. Biol. Chem. 275:34293–34305. [DOI] [PubMed] [Google Scholar]
- Nielsen, E., S. Christoforidis, S. Uttenweiler-Joseph, M. Miaczynska, F. Dewitte, M. Wilm, B. Hoflack, and M. Zerial. 2000. Rabenosyn-5, a novel Rab5 effector, is complexed with hVPS45 and recruited to endosomes through a FYVE finger domain. J. Cell Biol. 151:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, F.A., V. Auethavekiat, and P.W. Majerus. 1995. The isolation and characterization of cDNA encoding human and rat brain inositol polyphosphate 4-phosphatase. J. Biol. Chem. 270:16128–16133. [DOI] [PubMed] [Google Scholar]
- Norris, F.A., R.C. Atkins, and P.W. Majerus. 1997. The cDNA cloning and characterization of inositol polyphosphate 4-phosphatase type II. Evidence for conserved alternative splicing in the 4-phosphatase family. J. Biol. Chem. 272:23859–23864. [DOI] [PubMed] [Google Scholar]
- Nystuen, A., M.E. Legare, L.D. Shultz, and W.N. Frankel. 2001. A null mutation in inositol polyphosphate 4-phosphatase type I causes selective neuronal loss in weeble mutant mice. Neuron. 32:203–212. [DOI] [PubMed] [Google Scholar]
- Odorizzi, G., M. Babst, and S.D. Emr. 2000. Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem. Sci. 25:229–235. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., P.H. Warne, R. Dhand, B. Vanhaesebroeck, I. Gout, M.J. Fry, M.D. Waterfield, and J. Downward. 1994. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 370:527–532. [DOI] [PubMed] [Google Scholar]
- Rubino, M., M. Miaczynska, R. Lippe, and M. Zerial. 2000. Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J. Biol. Chem. 275:3745–3748. [DOI] [PubMed] [Google Scholar]
- Rupper, A., K. Lee, D. Knecht, and J. Cardelli. 2001. Sequential activities of phosphoinositide 3-kinase, PKB/Aakt, and Rab7 during macropinosome formation in Dictyostelium. Mol. Biol. Cell. 12:2813–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnatwinkel, C., S. Christoforidis, M.R. Lindsay, S. Uttenweiler-Joseph, M. Wilm, R.G. Parton, and M. Zerial. 2004. The Rab5 effector Rabankyrin-5 regulates and coordinates different endocytic mechanisms. PLoS Biol. 2:E261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal, R.A., and M.E. Greenberg. 1996. Intracellular signaling pathways activated by neurotrophic factors. Annu. Rev. Neurosci. 19:463–489. [DOI] [PubMed] [Google Scholar]
- Serunian, L.A., K.R. Auger, and L.C. Cantley. 1991. Identification and quantification of polyphosphoinositides produced in response to platelet-derived growth factor stimulation. Methods Enzymol. 198:78–87. [DOI] [PubMed] [Google Scholar]
- Shpetner, H., M. Joly, D. Hartley, and S. Corvera. 1996. Potential sites of PI-3 kinase function in the endocytic pathway revealed by the PI-3 kinase inhibitor, wortmannin. J. Cell Biol. 132:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddhanta, U., J. McIlroy, A. Shah, Y. Zhang, and J.M. Backer. 1998. Distinct roles for the p110alpha and hVPS34 phosphatidylinositol 3′-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J. Cell Biol. 143:1647–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.-M. Gaullier, A. Brech, J. Callaghan, B.-H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links phosphatidylinositol 3-kinase function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Sonnichsen, B., S. De Renzis, E. Nielsen, J. Rietdorf, and M. Zerial. 2000. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaargaren, M., and J.L. Bos. 1999. Rab5 induces Rac-independent lamellipodia formation and cell migration. Mol. Biol. Cell. 10:3239–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro, D., W. Boll, T. Kirchhausen, and M. Wessling-Resnick. 1996. Wortmannin alters the transferrin receptor endocytic pathway in vivo and in vitro. Mol. Biol. Cell. 7:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer, T.P., S. Ahn, and T. Meyer. 1998. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 8:343–346. [DOI] [PubMed] [Google Scholar]
- Steger, D.J., E.S. Haswell, A.L. Miller, S.R. Wente, and E.K. O'Shea. 2003. Regulation of chromatin remodeling by inositol polyphosphates. Science. 299:114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark, H., R.G. Parton, O. Steele-Mortimer, A. Lutcke, J. Gruenberg, and M. Zerial. 1994. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 13:1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, L.R., T.R. Jackson, and P.T. Hawkins. 1993. Agonist-stimulated synthesis of phosphatidylinositol(3,4,5)-trisphosphate: a new intracellular signalling system? Biochim. Biophys. Acta. 1179:27–75. [DOI] [PubMed] [Google Scholar]
- Tsukazaki, T., T.A. Chiang, A.F. Davison, L. Attisano, and J.L. Wrana. 1998. SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell. 95:779–791. [DOI] [PubMed] [Google Scholar]
- Ullrich, O., H. Horiuchi, K. Alexandroy, and M. Zerial. 1995. Use of RabGDI for solubilization and delivery of Rab proteins to biological membranes in streptolysin-O permeabilized cells. Methods Enzymol. 257:243–253. [DOI] [PubMed] [Google Scholar]
- Ungewickell, A., M.E. Ward, E. Ungewickell, and P.W. Majerus. 2004. The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc. Natl. Acad. Sci. USA. 101:13501–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck, B., S.J. Leevers, K. Ahmadi, J. Timms, R. Katso, P.C. Driscoll, R. Woscholski, P.J. Parker, and M.D. Waterfield. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 70:535–602. [DOI] [PubMed] [Google Scholar]
- Vieira, O.V., R.J. Botelho, L. Rameh, S.M. Brachmann, T. Matsuo, H.W. Davidson, A. Schreiber, J.M. Backer, L.C. Cantley, and S. Grinstein. 2001. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 155:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watton, S.J., and J. Downward. 1999. Akt/PKB localisation and 3′ phosphoinositide generation at sites of epithelial cell-matrix and cell-cell interaction. Curr. Biol. 9:433–436. [DOI] [PubMed] [Google Scholar]
- Weernink, P.A.O., K. Meletiadis, S. Hommeltenberg, M. Hinz, H. Ishihara, M. Schmidt, and K.H. Jakobs. 2004. Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J. Biol. Chem. 279:7840–7849. [DOI] [PubMed] [Google Scholar]
- Wenk, M.R., and P. De Camilli. 2004. Protein-lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc. Natl. Acad. Sci. USA. 101:8262–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser, A.E., and S.D. Emr. 1998. Phosphoinositide signaling and turnover: PtdIns(3)P, a regulator of membrane traffic, is transported to the vacuole and degraded by a process that requires lumenal vacuolar hydrolase activities. EMBO J. 17:4930–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York, J.D. 2003. Handbook of Cell Signaling. Elsevier Science, Amsterdam. 229–232.
- Zerial, M., and H. McBride. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2:107–117. [DOI] [PubMed] [Google Scholar]
- Zhang, X., A.B. Jefferson, V. Auethavekiat, and P.W. Majerus. 1995. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc. Natl. Acad. Sci. USA. 92:4853–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]