

Abstract
The HIV-1 Gag protein recruits the cellular factor Tsg101 to facilitate the final stages of virus budding. A conserved P(S/T)AP tetrapeptide motif within Gag (the “late domain”) binds directly to the NH2-terminal ubiquitin E2 variant (UEV) domain of Tsg101. In the cell, Tsg101 is required for biogenesis of vesicles that bud into the lumen of late endosomal compartments called multivesicular bodies (MVBs). However, the mechanism by which Tsg101 is recruited from the cytoplasm onto the endosomal membrane has not been known. Now, we report that Tsg101 binds the COOH-terminal region of the endosomal protein hepatocyte growth factor–regulated tyrosine kinase substrate (Hrs; residues 222–777). This interaction is mediated, in part, by binding of the Tsg101 UEV domain to the Hrs 348PSAP351 motif. Importantly, Hrs222–777 can recruit Tsg101 and rescue the budding of virus-like Gag particles that are missing native late domains. These observations indicate that Hrs normally functions to recruit Tsg101 to the endosomal membrane. HIV-1 Gag apparently mimics this Hrs activity, and thereby usurps Tsg101 and other components of the MVB vesicle fission machinery to facilitate viral budding.
Keywords: virus budding; virions; ubiquitin; vacuolar protein sorting; multivesicular body
Introduction
The HIV Gag protein orchestrates viral assembly and budding, and forms the structural shell of the immature virus (for review see Göttlinger, 2001). Even in the absence of any other viral proteins, HIV-1 Gag can form extracellular virus-like particles (VLPs) that resemble authentic HIV virions. Efficient release of HIV-1 virions and Gag VLPs from most cell types requires the presence of a “late domain” located in the COOH-terminal Gag p6 region (Göttlinger et al., 1991; Huang et al., 1995; Demirov et al., 2002b). All HIV-1 strains contain the late domain tetrapeptide motif P(S/T)AP (where the second position can be either Ser or Thr; Fig. 1), which is a docking site for the cellular protein, tumor susceptibility gene 101 (Tsg101; for review see Pornillos et al., 2002c). Tsg101 appears to facilitate viral budding by recruiting additional cellular factors that can catalyze release of the enveloped virion. In addition to the P(S/T)AP sequence found in HIV-1 Gag, distinct late domain sequences have also been identified and characterized in several other enveloped RNA viruses. The best characterized of these is the PPXY motif (where X is any amino acid), which binds the Nedd4 protein family of ubiquitin E3 ligases (Xiang et al., 1996; Harty et al., 1999, 2001; Strack et al., 2000; Kikonyogo et al., 2001; Yasuda et al., 2002; Timmins et al., 2003).
Figure 1.

Domain organization of the HIV-1 Gag and human Tsg101 and Hrs proteins. The HIV Gag and Hrs proteins are aligned to emphasize their similarities, with the NH2-terminal membrane-binding domains separated from the COOH-terminal protein–protein interaction domains by a vertical dashed line. UEV, ubiquitin E2 variant; PRD, proline-rich domain; COIL, putative coiled-coil; SBOX, “steadiness box/Vps28 binding site” (Feng et al., 2000). Locations of P(S/T)AP and PPEY motifs are also indicated.
The P(S/T)AP late domains of HIV-1, HIV-2, and Ebola virus bind to the NH2-terminal ubiquitin E2 variant (UEV) domain of Tsg101 (Garrus et al., 2001; Martin-Serrano et al., 2001; VerPlank et al., 2001; Demirov et al., 2002a; Myers and Allen, 2002; Pornillos et al., 2002b; Timmins et al., 2003). UEV domains also bind ubiquitin and are structurally related to ubiquitin E2–conjugating enzymes, but lack the active site cysteine residue required for ubiquitin transfer (Moraes et al., 2001; VanDemark et al., 2001; Pornillos et al., 2002a). Tsg101 UEV also differs from the canonical E2 enzymes in that it displays a hydrophobic groove that specifically recognizes P(S/T)AP sequences (Pornillos et al., 2002a). All four P(S/T)AP residues are required for late domain activity (Huang et al., 1995), and all four make important contacts within the Tsg101 UEV binding site (Pornillos et al., 2002a,b). However, despite this sequence specificity, the Tsg101 UEV domain presumably did not evolve to bind viral P(S/T)AP sequences, and it is therefore reasonable to speculate that the Tsg101 UEV domain may bind P(S/T)AP elements found in cellular proteins.
In the cell, Tsg101 (yeast Vps23p) normally functions as a subunit of the endosomal sorting complex required for transport-I (ESCRT-I) protein complex (Katzmann et al., 2001). Tsg101 and ESCRT-I perform essential roles in the vacuolar protein sorting (Vps) pathway, in which integral membrane proteins such as cell surface receptors are targeted for destruction in the lysosome (for review see Katzmann et al., 2002). In this pathway, monoubiquitylated proteins are delivered to endosomal membranes where they are sorted into microdomains that ultimately bud as small vesicles into late endosomal compartments to form multivesicular bodies (MVBs). MVBs can then fuse with lysosomes and release these vesicles and their protein cargos into the lumen of the lysosome, where they are degraded by hydrolases and lipases.
ESCRT-I is one of a series of soluble protein complexes that are recruited from the cytoplasm onto the surface of the endosome during MVB biogenesis. Once on the membrane, Tsg101/ESCRT-I appears to perform at least two essential functions: (1) it binds ubiquitylated cargo proteins via direct interactions between the Tsg101 UEV domain and the ubiquitin modifications (Katzmann et al., 2001); and (2) it helps to recruit the downstream ESCRT-II, ESCRT-III, Vps4, and other proteins and complexes required to complete protein sorting and vesicle formation (Babst et al., 2002a,b). Thus, recruitment of Tsg101 to the membrane surface represents a key step in the commitment to vesicle formation. However, the mechanism by which Tsg101/ESCRT-I is recruited to the membrane has not been elucidated.
Although interactions between Tsg101 and cellular P(S/T)AP motifs have not been described, we reasoned that such interactions might occur and play a role in the recruitment of Tsg101 to the endosomal membrane. Protein database searches revealed several Vps proteins with PTAP or PSAP motifs (Garrus et al., 2001), including (1) Tsg101 itself; and (2) hepatocyte growth factor–regulated tyrosine kinase substrate (Hrs; yeast Vps27p; Raiborg et al., 2001a; Raiborg and Stenmark, 2002). Hrs is an attractive candidate for the factor that recruits Tsg101 to endosomal membranes because it is required for receptor down-regulation and MVB biogenesis (Piper et al., 1995; Odorizzi et al., 1998; Lloyd et al., 2002; Shih et al., 2002; Bache et al., 2003b) and because it interacts with ubiquitylated cargo proteins on the early endosome (and therefore appears to function upstream of Tsg101; Bilodeau et al., 2002; Bishop et al., 2002; Raiborg et al., 2002; Bache et al., 2003b).
The domain organization and biochemical properties of the Hrs and HIV-1 Gag proteins exhibit several intriguing similarities (Fig. 1). First, both proteins contain amino-proximal membrane-targeting domains. In HIV-1 Gag, the N-myristoylated MA domain binds membranes, and contains signals that target Gag to the plasma membrane in some cell lines (Göttlinger, 2001; Raposo et al., 2002; Pelchen-Matthews et al., 2003). In Hrs, the FYVE domain binds PI(3)P and thereby targets Hrs to endosomal membranes (Komada et al., 1997; Odorizzi et al., 1998; Raiborg et al., 2001b; Katzmann et al., 2003). Second, both proteins contain P(S/T)AP motifs within proline-rich (and apparently unstructured) regions. Third, both Hrs and HIV-1 Gag proteins are monoubiquitylated. Although a functional role for HIV Gag ubiquitylation in retrovirus budding has not been demonstrated, ubiquitin transfer is important for virus budding (for review see Vogt, 2000), and viral late domains have been shown to recruit ubiquitin ligase activities (Strack et al., 2000, 2002). Hrs ubiquitylation is essential for receptor down-regulation through the MVB pathway, and requires a cis-acting sequence motif called the ubiquitin-interacting motif (UIM; Young et al., 1998; Hofmann and Falquet, 2001; Bishop et al., 2002; Lloyd et al., 2002; Polo et al., 2002; Raiborg et al., 2002; Shih et al., 2002). Other UIM-containing proteins are substrates for the Nedd4 (yeast Rsp5p) family of ubiquitin ligases (Katz et al., 2002; Polo et al., 2002), and although it is not yet known whether Nedd4 ubiquitylates Hrs, the protein does harbor a PPEY motif. This sequence matches the consensus sites for cellular Nedd4 substrates and for PPXY viral late domains (Rotin et al., 2000; Freed, 2002; Pornillos et al., 2002c).
The preceding observations are consistent with a model in which (1) Tsg101 is normally recruited to the endosomal membrane through a direct interaction with Hrs; and (2) the HIV-1 Gag protein has evolved to mimic the Tsg101-recruiting functions of Hrs, and thereby redirect the machinery of MVB vesicle formation to sites of viral budding on the plasma membrane. Experiments described in this paper were designed to test key aspects of these models for Hrs function and viral mimicry.
Results
Tsg101 UEV binds the P(S/T)AP motifs from HIV-1 Gag, Tsg101, and Hrs
First, we tested whether the UEV domain of Tsg101 could bind to isolated P(S/T)AP motifs from HIV-1NL4–3 Gag and from the human Tsg101 and Hrs proteins. The Tsg101 UEV binding epitope is small, and the domain binds with the same affinity to a nine-amino acid peptide as to the intact HIV-1 p6 protein (∼25 μM; Pornillos et al., 2002b). Therefore, 10-amino acid peptides spanning the central P(S/T)AP motifs from Gag, Tsg101, and Hrs were tested for Tsg101 UEV domain binding in BIAcore biosensor experiments. As expected, Tsg101 UEV exhibited concentration-dependent binding to the Gag PTAP peptide, and the equilibrium responses fit a simple 1:1 binding model with a dissociation constant (Kd 20°C) of 21 ± 1 μM (Fig. 2). This interaction was specific, as the Tsg101 UEV domain did not bind to a control peptide in which the second Pro residue in the PTAP motif was mutated to Leu (Fig. 2).
Figure 2.

The Tsg101 UEV domain binds the P(S/T)AP motifs from HIV-1 Gag, Tsg101, and Hrs. Biosensor binding isotherms showing the concentration-dependent binding of purified recombinant Tsg101 UEV domain to immobilized fusion peptides spanning the P(S/T)AP motifs of HIV-1 Gag, Tsg101, and Hrs, as well as a mutant form of the HIV-1 Gag PTAP motif (negative control). Solid lines show the optimal fits to simple 1:1 binding models used to obtain binding affinities. Note that substitution of S for T at the second position of the P(S/T)AP motif does not significantly affect the Tsg101 binding affinity (not depicted), but that sequences flanking the central P(S/T)AP tetrapeptides can modulate Tsg101 binding affinity significantly.
The Tsg101 UEV domain also bound to P(S/T)AP peptides from Tsg101 and from Hrs (Fig. 2). However, the binding affinities were fourfold (Tsg101) and sevenfold (Hrs) weaker than Tsg101 UEV binding to the HIV-1 PTAP peptide. Hence, the Tsg101 UEV domain can bind the P(S/T)AP motifs of all three proteins, but sequences flanking the central P(S/T)AP motif can modulate the absolute binding affinities significantly. Indeed, we have examined some isolated P(S/T)AP peptide sequences that exhibited almost undetectable Tsg101 UEV binding (Fisher, R., personal communication).
Interaction of full-length Tsg101 and Hrs proteins
The interaction of full-length Tsg101 and wild-type Hrs proteins was examined in directed yeast two-hybrid assays. Plasmids expressing a Tsg101–Gal4p binding domain fusion (Tsg101-BD) and an Hrs–Gal4p activation domain fusion (Hrs-AD) were cotransfected into the reporter yeast strain J693, resulting in significant levels of β-galactosidase activity (Fig. 3 A, top left). In contrast, control experiments in which one of the two plasmids encoded only the Gal4p BD or AD alone did not produce significant β-galactosidase activity (Fig. 3 A). Similarly, in semi-quantitative liquid culture assays, the binding of full-length Hrs-AD to full-length Tsg101-BD typically stimulated β-galactosidase activity ∼100-fold above background (Fig. 3, B–D). Therefore, we conclude that the full-length Tsg101 and Hrs proteins interact in the yeast two-hybrid assay.
Figure 3.

Hrs/Tsg101 yeast two-hybrid binding assays. (A) Filter paper colony lift assay for β-galactosidase reporter gene activity. Yeast cells were cotransformed with pairs of plasmids expressing the indicated Gal4 DNA-binding domain (BD) and activation domain (AD) fusion constructs. Cells were grown on synthetic agar media lacking the appropriate amino acids and were analyzed for β-galactosidase activity (blue) as described in Materials and methods. (B) Binding of Hrs to wild-type and mutant (N45A) Tsg101. Binding experiments in B–D show yeast two-hybrid interactions (together with appropriate controls) as measured in semi-quantitative CPRG β-galactosidase activity assays. Bars depict the average absorbance (595 nm) and SDs from three independent measurements. (C) Binding of wild-type and mutant (ΔPSAP) Hrs to wild-type and mutant (M95A) Tsg101. (D) Binding of Tsg101 to Hrs deletion mutants.
Requirements for Tsg101/Hrs binding
Based on the known binding properties of the Tsg101 UEV domain, we hypothesized that Tsg101 might interact with Hrs via (1) an interaction between the Tsg101 UEV domain and the Hrs PSAP motif; and/or (2) an interaction between the Tsg101 UEV domain and a ubiquitin modification on Hrs. Tsg101 UEV binds P(S/T)AP peptides and ubiquitin at different sites, and distinct Tsg101 point mutations eliminate PTAP (M95A) or Ub (N45A) binding (Pornillos et al., 2002a). Therefore, these mutations were tested for their effects on the Tsg101/Hrs two-hybrid interaction (Fig. 3, B and C).
The ubiquitin-binding mutation in Tsg101 UEV (N45A) had no effect on the Hrs/Tsg101 interaction (Fig. 3 B). Likewise, alanine substitutions in the Hrs PPEY sequence had no significant effect on Tsg101/Hrs binding (unpublished data). These experiments indicated that the Tsg101/Hrs two-hybrid interaction was not mediated through ubiquitin.
In contrast, the PSAP-binding mutation in Tsg101 (M95A) reduced Hrs binding significantly (approximately sixfold; Fig. 3 C). In complementary experiments, alanine substitution of the Hrs 348PSAP351 sequence (HrsΔPSAP) also reduced wild-type Tsg101 binding, albeit to a lesser extent (approximately twofold). The greater reduction in binding seen for the Tsg101 M95A mutation may reflect weak Tsg101 UEV binding to several additional PSAP-like motifs (583PSGP586 and 620PSMP623) that are conserved from human Hrs to yeast Vps27p (Emr, S., personal communication). As expected, the HrsΔPSAP mutation did not further reduce binding of the Tsg101 M95A mutation, consistent with the idea that these mutations affected the two sides of the same protein–protein interface.
Surprisingly, even in the absence of the Tsg101 UEV/Hrs PSAP interaction, Hrs and Tsg101 exhibited significant residual binding. Therefore, directed two-hybrid experiments were used to define the complete Hrs region required for full affinity binding. As summarized in Fig. 3 D, Tsg101 did not bind the NH2-terminal VHS and FYVE domains of Hrs (residues 1–221). These domains were also completely dispensable for the Tsg101/Hrs interaction, as Tsg101 bound equally well to a COOH-terminal fragment spanning residues 222–777 (HrsΔN) as to full-length Hrs. In contrast, deletion of COOH-terminal Hrs residues 565–777 reduced (but did not eliminate) Hrs binding, and a further six-amino acid deletion that removed the entire COOH-terminal Pro/Gln-rich region of Hrs (HrsΔNΔC; Hrs residues 222–559) reduced Tsg101 binding to nearly background levels. A murine Hrs construct (residues 287–573) that extended ∼15 residues into the Pro/Gln-rich region bound well (Bache et al., 2003a), and we therefore conclude that Hrs residues 560–573 contribute to Tsg101 binding. Tsg101 bound only weakly (or not at all) to a series of smaller Hrs fragments that spanned just the Hrs PSAP element (e.g., Hrs 1–450) or the coiled-coil Pro/Gln-rich region (e.g., Hrs 451–777; see Fig. 3 D). Thus, we conclude that full affinity Tsg101 binding requires the presence of two (or more) different Hrs regions: (1) the PSAP motif; and (2) a second region spanning the putative coiled-coil and at least part of the Pro/Gln-rich region.
Hrs late domain activity
Next, we tested whether the protein-recruiting functions of Hrs were sufficient to support the budding of VLPs from cultured human cells. Expression of HIV Gag–GFP fusion proteins in human cell lines recapitulates many aspects of viral assembly and budding (Hermida-Matsumoto and Resh, 2000), including the requirement for Tsg101 and other components of the MVB pathway (Garrus et al., 2001).
Human embryonic kidney 293T cells were transfected with HIV Gag expression constructs, and analyzed for protein expression and for VLP release 24–28 h later (Fig. 4). VLP release was analyzed by Western blotting of culture supernatants after sucrose cushion pelleting (Fig. 4, B and C; VLP). Intracellular Gag protein expression levels were analyzed in Western blots of cytoplasmic extracts (Fig. 4, B and C; Cell). As expected, the control Gag–GFP fusion protein expressed well and formed VLPs efficiently (Fig. 4 B, lane 1; Hermida-Matsumoto and Resh, 2000; Garrus et al., 2001). VLP release was dependent on the presence of a functional PTAP late domain, as mutation of the PTAP motif to LIRL (GagΔPTAP–GFP; Fig. 4 B, lane 2) severely attenuated particle release (Göttlinger et al., 1991; Huang et al., 1995; Garrus et al., 2001).
Figure 4.
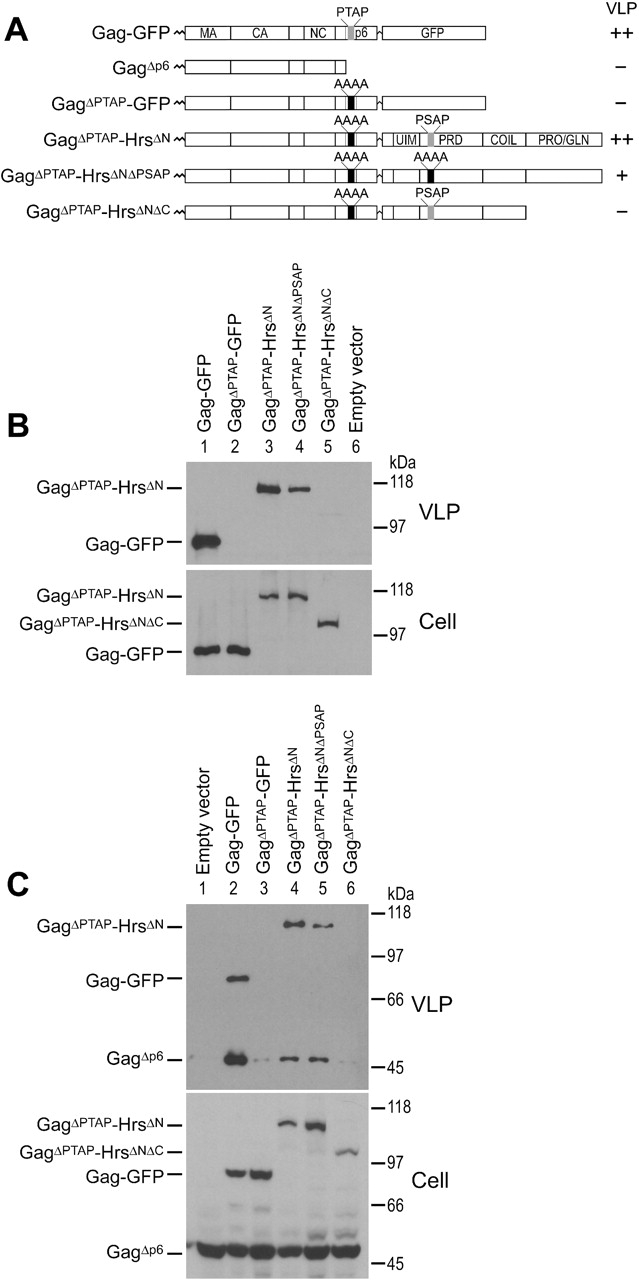
Hrs can substitute for late domain functions of HIV-1 Gag p6. (A) Summary of the Gag and Gag–Hrs fusion constructs and their VLP-budding phenotypes. (B) The HrsΔN polypeptide rescues the budding arrest caused by mutation of the Gag PTAP late domain. Top, Western blot analysis of VLP release by the Gag–GFP and Gag–Hrs proteins. Bottom, Western blot showing cytoplasmic expression of the Gag–GFP and Gag–Hrs fusion proteins. 293T cells were transfected with 0.5 μg plasmid encoding Gag–GFP (lane 1), GagΔPTAP–GFP (lane 2), GagΔPTAP–HrsΔN (lane 3), GagΔPTAP–HrsΔNΔPSAP (lane 4), GagΔPTAP–HrsΔNΔC (lane 5), or empty vector (lane 6). (C) Trans complementation of deficient GagΔp6 budding. Top, Western blot analyzing VLP release. Bottom, Western blot showing cytoplasmic expression of the different Gag constructs. Cells were cotransfected with 1.5 μg plasmid DNA encoding GagΔp6 and 0.5 μg empty vector (lane 1), Gag–GFP (lane 2), GagΔPTAP–GFP (lane 3), GagΔPTAP–HrsΔN (lane 4), GagΔPTAP–HrsΔNΔPSAP (lane 5), or GagΔPTAP–HrsΔNΔC (lane 6).
To test whether the protein recruitment activities of Hrs could replace the late domain activity of the cis-acting Gag PTAP motif, the GFP polypeptide of GagΔPTAP-GFP was replaced with the HrsΔN polypeptide (denoted GagΔPTAP–HrsΔN; Fig. 4 A). As described in the previous section, the HrsΔN fragment is missing the NH2-terminal domains that normally target Hrs to the endosomal membrane, but contains both the 348PSAP351 motif and the Pro/Gln-rich region, and binds Tsg101 (Fig. 3 D). As shown in Fig. 4 B (top, lane 3), the GagΔPTAP–HrsΔN fusion protein was efficiently released into the culture supernatant. Indeed, the late domain activity of the HrsΔN polypeptide (51% of total Gag protein released) was nearly as strong as the native Gag PTAP motif (57% of total Gag protein released). The HrsΔN construct also rescued particle release when fused to a Gag protein that was missing the entire p6 domain (GagΔp6, unpublished data and see below).
Analogous results were obtained in experiments designed to test whether Gag–Hrs fusion proteins could rescue the budding of defective Gag constructs in trans (Fig. 4 C). This experiment took advantage of the fact that defective Gag proteins can be released from cells by co-assembling with Gag proteins that are competent for budding (Martin-Serrano et al., 2001). Control experiments demonstrated that wild-type Gag–GFP could rescue the release of GagΔp6 when the two proteins were coexpressed in the same cell (Fig. 4 C, top, compare lane 1 and lane 2), and that rescue required the Gag–GFP PTAP sequence (Fig. 4 C, top, compare lane 2 and 3). GagΔp6 release was also efficiently rescued in trans by coexpression with GagΔPTAP–HrsΔN (Fig. 4 C, top, lane 4). Hence, we conclude that the HrsΔN polypeptide has potent late domain activity and can support HIV-1 Gag budding both in cis and in trans.
In addition to rescuing VLP formation in trans, some Gag fusion proteins with late domain activities can also rescue the infectivity of HIV-1 proviral constructs with budding defects (Martin-Serrano et al., 2001). For example, the wild-type Gag–GFP protein can rescue both the budding and infectivity of HIV-1 proviral constructs that harbor mutations in the PTAP-coding region of the Gag gene. However, the GagΔPTAP–HrsΔN construct did not efficiently rescue infectivity of the R9ΔPTAP proviral construct in a single cycle replication assay (unpublished data), and we speculate that this may reflect alterations in particle morphology caused by incorporation of the Hrs222–777 polypeptide (see below).
Sequence requirements for Hrs late domain activity
To test the sequence requirements for Hrs late domain activity, HrsΔN constructs missing either the PSAP motif (GagΔPTAP–HrsΔNΔPSAP) or the Pro/Gln-rich region (GagΔPTAP–HrsΔNΔC) were also tested in the two VLP release assays. As shown in Fig. 4 B, GagΔPTAP–HrsΔNΔC was not released from cells (Fig. 4 B, lane 5), whereas GagΔPTAP–HrsΔNΔPSAP was released (Fig. 4 B, lane 4), but with reduced efficiency as compared with the GagΔPTAP–HrsΔN control (27 vs. 51% total Gag protein released). Similarly, GagΔp6 release was not efficiently rescued in trans by the GagΔPTAP–HrsΔNΔC protein (Fig. 4 C, lane 6), although in some repetitions of this experiment, GagΔp6 was released at low levels (unpublished data). The low levels of release, when seen, presumably reflected weak residual late domain activity of the PSAP sequence within the HrsΔNΔC polypeptide. The GagΔPTAP–HrsΔNΔPSAP protein again rescued the release of GagΔp6 to some degree (Fig. 4 C, lane 5), but was again less efficient than GagΔPTAP–HrsΔN (Fig. 4 C, compare lane 4 and lane 5). Also, we tested the release of a GagΔPTAP–HrsΔN construct with a mutation in the UIM (265LA266 → AL). This mutation abolishes Hrs ubiquitin binding and ubiquitylation of Hrs itself (Polo et al., 2002), but did not diminish VLP release. Indeed, the 265LA266 → AL mutation actually appeared to enhance GagΔp6–HrsΔN release slightly (unpublished data). Overall, we conclude that full Hrs late domain activity requires the 348PSAP351 element and COOH-terminal coiled-coil Pro/Gln-rich region, but not the UIM. These requirements correlate well with the requirements for Tsg101 binding.
EM analyses of VLPs
Transmission EM was used to confirm that the GagΔPTAP–HrsΔN protein was released in the form of VLPs and to examine the phenotypes of the Gag constructs that failed to bud efficiently. In control experiments, Gag–GFP VLPs appeared as enveloped, spherical particles that generally resembled immature HIV virions in both appearance and size (100–200 nm in diameter; Fig. 5 A). However, the GFP polypeptide appeared to create discontinuities in the Gag–GFP layer, in contrast to the evenly distributed ring of Gag density that is normally observed beneath the membranes of immature HIV-1 virions and Gag VLPs.
Figure 5.

Electron microscopic analyses of Gag–GFP and Gag–Hrs budding. (A and B) Transmission electron micrographs of thin-sectioned VLPs released from 293T cells transfected with plasmids encoding Gag–GFP (A) and GagΔPTAP–HrsΔN (B). Note that in both cases, the proteins formed enveloped, spherical VLPs that resembled authentic immature virions except that (1) the electron-dense Gag layer beneath the VLP membrane often appeared discontinuous, suggesting that the COOH-terminal GFP and Hrs polypeptides perturbed the underlying Gag lattice; and (2) VLPs formed by GagΔPTAP–HrsΔN exhibited greater size variation and were, on average, somewhat larger than authentic immature virions. (C–E) Transmission electron micrographs of thin-sectioned 293T cells transfected with plasmids expressing GagΔPTAP–GFP (C), GagΔPTAP–HrsΔNΔC (D), and GagΔPTAP–HrsΔNΔPSAP (E). The top images show clusters of particles associated with the cellular surface, and the bottom images show examples of classic “late domain” phenotypes in which the assembled particles remained tethered to the plasma membrane (or to one another). Bars, 100 nm.
As shown in Fig. 5 B, the GagΔPTAP–HrsΔN protein was also released from cells in the form of enveloped VLPs that were similar in appearance to the Gag–GFP particles, but were larger and more heterogeneous in size (up to 500 nm in diameter). The Gag “discontinuity” phenotype was also quite pronounced in the GagΔPTAP–HrsΔN particles. Thus, GagΔPTAP–HrsΔN formed enveloped VLPs as expected, but the HrsΔN fusion protein altered particle morphology to some extent.
EM images of thin-sectioned cells were also examined to determine the phenotypes of the Gag fusion proteins that were not released efficiently (Fig. 5, C–E). In cells expressing GagΔPTAP–GFP, GagΔPTAP–HrsΔNΔC, and GagΔPTAP–HrsΔNΔPSAP, clusters of enveloped spherical particles associated with the cell surface were frequently observed (Fig. 5, C–E; top images). Although it can be difficult to establish membrane connectivities in thin-sectioned images, many of the assembling VLPs clearly remained tethered to the plasma membrane via thin membrane stalks (Fig. 5, C–E, arrows in bottom images). Also, we observed images in which it appeared that vacuolar structures filled with assembled VLPs were in the process of (or had recently) fused with the plasma membrane (unpublished data). These images suggested that at least some of the VLPs budded intracellularly, in good agreement with recent experiments showing that HIV-1 can bud into MVB compartments in macrophages (Raposo et al., 2002; Pelchen Matthews et al., 2003).
Overall, our EM analyses established that although the GagΔPTAP–GFP, GagΔPTAP–HrsΔNΔC, and GagΔPTAP–HrsΔNΔPSAP proteins were not released from cells efficiently, they did associate with membranes and initiate spherical particle assembly. Thus, the block to particle release in these constructs occurred at a late stage in the budding process, and therefore reflected the defect(s) in late domain activity.
The late domain activity of Hrs is dependent on Tsg101
Next, we examined whether the late domain activity of the HrsΔN polypeptide required the presence of Tsg101. Cellular Tsg101 can be efficiently depleted using RNA interference (Garrus et al., 2001; Fig. 6, bottom, compare lane 1 and lane 2). As shown in Fig. 6, release of GagΔp6–HrsΔN VLPs from 293T cells was blocked when Tsg101 was depleted from the producer cells (Fig. 6, top, compare lane 1 and lane 2). VLP release was restored when an exogenous RNA interference–resistant Tsg101–FLAG protein (denoted Tsg101*) was expressed (Fig. 6, compare lane 2 and lane 3). Therefore, this experiment confirmed that the late domain activity of HrsΔN is dependent on Tsg101.
Figure 6.

Budding of Gag Δp6 –Hrs ΔN is dependent on the P(S/T)AP-binding activity of Tsg101. Top, Western blot showing relative levels of Gag–HrsΔN VLP release. Middle, Western blot showing cytoplasmic expression levels of Gag–HrsΔN. Bottom, Anti-Tsg101 Western blot showing depletion of endogenous Tsg101 protein with siRNA, and reexpression of exogenous, siRNA-resistant Tsg101-Flag proteins (Tsg101*). 293T cells were cotransfected as described in the Materials and methods with the following exceptions: (1) lane 1 received no RNA; and (2) lane 5 was a mock transfection.
As mentioned earlier in this paper, mutation of Tsg101 residue Met95 to Ala blocks the P(S/T)AP-binding activity of the UEV domain (Pornillos et al., 2002a). Interestingly, the Tsg101* M95A mutation also blocked the release of the Gag–HrsΔN protein (Fig. 6, lane 4). This absolute block was somewhat unexpected because the Tsg101 M95A protein could still bind Hrs (Fig. 3 C) and because the GagΔPTAP–HrsΔNΔPSAP mutant protein was released from cells (Fig. 4 B). However, Hrs binding was diminished by the Tsg101 M95A mutation, and VLP release was reduced by the HrsΔPSAP mutation, and it is therefore possible that the reduction in Hrs binding caused by the Tsg101 M95A mutation is sufficient to block VLP budding under conditions where Tsg101* levels are limiting. Alternatively, simply recruiting Tsg101 to the site of budding may not be sufficient to support virus budding because the NH2-terminal domain of Tsg101 UEV may perform additional functions that are inhibited by the M95A mutation. In either case, the result again emphasizes the functional significance of P(S/T)AP binding by the UEV domain of Tsg101.
Discussion
Implications for cellular Hrs function
The molecular events underlying MVB biogenesis are of interest because this pathway mediates a number of important biological processes including development (Kramer, 2002), receptor down-regulation (Katzmann et al., 2002; Sorkin and Von Zastrow, 2002), and MHC-II presentation (Murk et al., 2002). We have shown that full-length Tsg101 binds Hrs (and COOH-terminal fragments), and that COOH-terminal Hrs fragments can support Tsg101-dependent plasma membrane vesicle fission in a heterologous VLP system. These experiments, together with complementary experiments from the Emr and Stenmark laboratories (Bache et al., 2003a; Katzmann et al., 2003), indicate that Hrs recruits Tsg101/ESCRT-I from the cytoplasm onto the surface of the endosome, where it interacts with protein cargos and recruits downstream factors required for protein sorting and MVB formation. Although analyses of the cellular pathway are clearly essential for understanding MVB biogenesis, the complementary Gag protein release assay provides an effective functional test, in mammalian cells, for the vesicle fission activities required for both MVB biogenesis and virus budding.
Sequential recruitment of proteins onto the endosomal membrane
An intriguing aspect of MVB biogenesis is that a series of soluble complexes must be sequentially recruited from the cytoplasm onto subdomains of the endosomal membrane where ubiquitylated protein cargos accumulate and are eventually sorted into MVB vesicles. Hrs is the first of the soluble MVB factors to bind, and its recruitment is mediated, at least in part, by FYVE domain binding to PI(3)P molecules displayed on the endosomal membrane (Odorizzi et al., 1998; Urbe et al., 2000; Raiborg et al., 2001b; Katzmann et al., 2003). Hrs localization may also be cargo-dependent (e.g., through interactions between the Hrs UIM motif and the ubiquitin modifications on cargo proteins; Raiborg et al., 2002). Finally, interactions with other endosomal proteins may also play important roles in Hrs localization (Urbe et al., 2000; Raiborg et al., 2001b; Bache et al., 2002).
On the membrane, Hrs is an essential subunit of a multiprotein complex that includes Hrs, ubiquitylated protein cargos, STAM1, STAM2, Eps15, and clathrin (Asao et al., 1997; Raiborg et al., 2002; Yamada et al., 2002; Bache et al., 2003b). This complex next recruits the soluble Tsg101/ESCRT-I complex, and our experiments indicate that this occurs, at least in part, through a direct binding interaction between Hrs and Tsg101. The nature of the “switch” that allows Tsg101 recruitment only after Hrs is bound to the membrane is still unclear, but plausible possibilities include (1) avidity effects (as multiple copies of Hrs assemble at the membrane, and Tsg101 can also oligomerize; Martin-Serrano et al., 2003); (2) cooperative binding of Tsg101 to Hrs and other endosomal membrane protein(s) (e.g., ubiquitylated protein cargos); and/or (3) membrane-dependent Hrs conformational changes or phosphorylation events (Urbe et al., 2000; Bache et al., 2002).
In the next stages of MVB biogenesis, the soluble ESCRT-II and -III proteins are recruited from the cytoplasm onto the membrane (Babst et al., 2002b). Again, it seems likely that the membrane-bound ESCRT-I must be “activated” in some fashion to allow the recruitment of these downstream factors only after ESCRT-I is bound to the membrane.
Possible mechanisms of Tsg101 recruitment and activation
Unexpectedly, our binding experiments suggested that full affinity Tsg101 binding requires at least two Hrs elements, a first which involves the Hrs PSAP sequence binding to the Tsg101 UEV domain, and a second that includes downstream Hrs elements that spans the putative coiled-coil region and at least part of the Pro/Gln-rich region. Both interactions were functionally important, although mutations that disrupted the Tsg101 UEV/PSAP interaction did not completely eliminate complex formation and VLP budding in all contexts, indicating that the downstream Hrs elements may play the dominant role in Tsg101 recruitment.
The use of multiple contact sites could simply serve to increase the affinity and specificity of the Hrs/Tsg101 interaction. However, an attractive alternative model is that the downstream Hrs site serves primarily to recruit Tsg101 to sites of vesicle budding, whereas the Hrs PSAP element serves primarily as the switch that “activates” Tsg101 for ESCRT-II and/or MVB cargo binding (or for other essential events in MVB biogenesis). In this model, we envision that the soluble cytoplasmic ESCRT-I protein may exist primarily in an autoinhibited conformation in which the Tsg101 UEV domain binds its own PTAP element (i.e., in cis; Fig. 7 A). Hrs binding could then drive a conformational change in which the Tsg101 UEV domain switches to bind the PSAP element of Hrs (i.e., in trans). This activation event might also involve the ubiquitin-binding site on the UEV domain interacting with ubiquitin modifications on protein cargos, which would provide a cooperative binding mechanism for insuring that activation occurs only in the presence of both Hrs and ubiquitylated protein cargos. Although this model remains to be tested rigorously, we note that analogous autoinhibition/conformational switching mechanisms are used by other classes of proteins, such as protein kinases, to create directionality in other cellular pathways (Francis et al., 2002; Pellicena and Miller, 2002).
Figure 7.
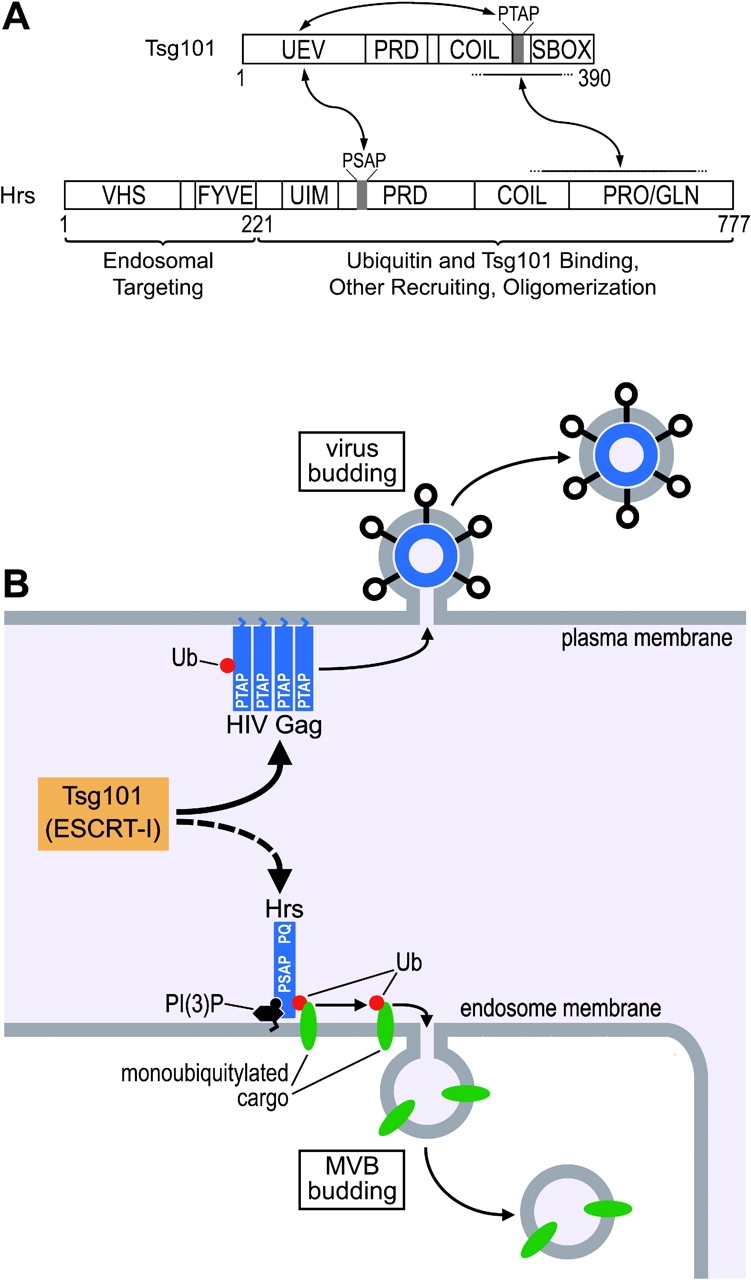
Models for Tsg101 recruitment and activation during MVB and HIV budding. (A) Model illustrating sites of Tsg101/Hrs interaction and a possible activation mechanism for Tsg101 (see text for details). Note that our data allow the possibility that other proteins might bridge or contribute to the interaction between the COOH-terminal regions of Tsg101 and Hrs. (B) Model illustrating how HIV Gag mimics the Tsg101-recruiting function of Hrs and redirects Tsg101 and the ESCRT-I complex to the plasma membrane to facilitate viral budding.
Viral recruitment of Tsg101
Our analyses also indicate that the structural proteins of HIV-1, HIV-2, and Ebola have evolved to mimic the normal protein-recruiting functions of the cellular Hrs protein in order to hijack the machinery that normally catalyzes MVB vesicle formation and budding (Fig. 7 B). The higher affinity P(S/T)AP element of HIV-1 Gag apparently allows the virus to compete effectively for Tsg101 binding against the lower affinity Hrs PSAP sequence. Viruses and cells may also interact differently with Tsg101 because viruses have presumably evolved to exit cells as efficiently as possible, whereas cells must carefully regulate the flux of cargo into MVB vesicles. This raises the intriguing possibility that by binding tightly to the PTAP-binding site in the UEV domain, the virus may simultaneously recruit Tsg101 and effect a conformational change that activates the protein to perform subsequent steps necessary for vesicle formation and membrane fission.
Materials and methods
Plasmid constructs
A Tsg101 mammalian expression vector was constructed by fusing the Flag epitope to the 3′ end of the full-length Tsg101 gene using PCR amplification with a downstream primer encoding the Flag sequence. The amplified product was cloned into pIRES2-EGFP (CLONTECH Laboratories, Inc.) to give pIRES-TsgFlag (Garrus et al., 2001). Seven silent mutations that render pIRES-TsgFlag resistant to siRNA (denoted pIRES-Tsg*Flag) were introduced into the wild-type Tsg101-coding region 413AACCTCCAGTCTTCTCTCGTC433 (mutated nucleotides are underlined) using Kunkel mutagenesis (Garrus et al., 2001). N45A and M95A mutations were also introduced into pIRES-Tsg*Flag using Kunkel mutagenesis.
All Gag and Gag–Hrs constructs were based on pGag–GFP, which contains the Rev-independent HIV-1HBX2 Gag sequence fused to EGFP (Hermida-Matsumoto and Resh, 2000; a gift from Marilyn Resh, Sloan-Kettering Institute, New York, NY). GagΔPTAP–GFP was created by mutating the Gag PTAP motif to LIRL as described previously (Garrus et al., 2001). GagΔPTAP–Hrs fusion constructs were created from the GagΔPTAP–GFP construct by replacing the last three amino acids of Gag and the GFP-coding region with the appropriate Hrs sequences. GagΔp6 and GagΔp6–Hrs constructs were created by replacing the p6- and GFP-coding regions with either a stop codon or the appropriate Hrs sequences. Hrs constructs were PCR amplified from an EST (GenBank/EMBL/DDBJ accession no. BE276844; from American Type Culture Collection, Manassas, VA).
For yeast two-hybrid assays, Tsg101 constructs were PCR amplified from pIRES-TsgFlag and subcloned into the plasmid MP30 (Myriad Genetics), which encodes the Gal4p DNA-binding domain (BD). Hrs constructs were subcloned into the plasmid MP29 (Myriad Genetics). The resulting plasmids encoded in-frame fusions of Tsg101 with the Gal4p BD and of Hrs with the Gal4p activation domain (AD). All constructs were verified by DNA sequencing. Full cloning details are available from the authors.
Biosensor binding assays
BIAcore biosensor measurements of Tsg101 UEV binding to immobilized GST–peptide fusion constructs were performed as described previously (Garrus et al., 2001), and purified recombinant Tsg101 UEV domain and GST–peptide fusion constructs were also obtained as described previously (Jenkins et al., 2001; Pornillos et al., 2002a).
Yeast two-hybrid binding assays
Yeast strain J693 (MATα ade2 his3 leu2 trp1 cyh2 ura3::GAL1p-LacZ gal4 gal80 lys2::GAL1p-HIS3; Bendixen et al., 1994) was cotransformed with various pairs of BD and AD constructs (using empty BD and AD vectors as controls), and plated on synthetic media lacking the appropriate amino acids. To identify colonies producing β-galactosidase, cells were lifted onto filter paper disks, lysed by freeze-thawing in liquid nitrogen, and assayed with X-gal (5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside). For quantitation of β-galactosidase activity, colonies (∼20 per plate) were selected at random, pooled, grown on synthetic liquid media, and assayed in triplicate with CPRG (chlorophenol red β-d-galactopyranoside) or ONPG (o-nitrophenyl-β-d-galactopyranoside) as described previously (Garrus et al., 2001). These experiments were repeated a total of six times.
Tissue culture and VLP-budding assays
Human embryonic kidney 293T cells were grown in 2 ml Dulbecco's minimum essential medium (6-well plates) and were transfected with expression plasmids as indicated in the figure legends using LipofectAMINE™ 2000 (Invitrogen; Garrus et al., 2001). VLP-budding assays were performed essentially as described previously (Garrus et al., 2001). In brief, plasmid DNA encoding Gag–GFP and Gag–Hrs fusion proteins was transfected into 293T cells. Supernatants and cytoplasmic lysates were harvested 24–28 h later and analyzed by Western blotting (see below).
Functional siRNA knockout and rescue of Tsg101 expression were performed as described previously (Elbashir et al., 2001; Garrus et al., 2001). Synthetic 21-nt siRNA duplexes were designed to target Tsg101 at coding nucleotides 413–433. siRNA sequences: sense, CCU CCA GUC UUC UCU CGU CdTdT; antisense, 5′ GAC GAG AGA AGA CUG GAG GdTdT. 293T cells were cotransfected twice at 24-h intervals. The first transfection was with 50 nM siRNA duplexes and either 2 μg pIRES2-EGFP or 2 μg wild-type or mutant RNAi-resistant pIRES-Tsg*Flag expression vector (Tsg101*). The second transfection was performed with the same 50-nM siRNA duplexes and 1 μg GagΔp6–HrsΔN expression vector. VLP-containing supernatants and cytoplasmic lysates were harvested after an additional 24–30 h and analyzed by Western blotting (see below).
Western blots
VLPs from 1.2 ml of supernatants from transfected cells were pelleted through a 20% sucrose cushion in a microcentrifuge for 90 min at 13,000 rpm (4°C) and resuspended in 25–30 μl of 1× SDS gel loading buffer. 5–8-μl samples were separated by SDS-PAGE, transferred, blocked, blotted with antisera, and protein bands were detected by ECL (Pierce Chemical Co.). Transfected 293T cells (one well from a 6-well plate) were harvested directly into 30–35 μl RIPA buffer (10 mM TrisCl, pH 7.0, 150 mM NaCl, 1% NP-40, and 0.1% SDS) and incubated on ice for 4 min. Samples were clarified by microcentrifugation for 4 min at 13,000 rpm (4°C), and resuspended in an equal volume of 2× SDS gel loading buffer. 5–8-μl aliquots were resolved by SDS-PAGE and blotted for ECL. To detect Gag proteins, rabbit anti-HIV CA antibody (1:2,000; from Hans-Georg Krausslich, Heidelberg, Germany) was mixed with rabbit anti-HIV MA (1:25,000; from Didier Trono, Geneva, Switzerland). Murine monoclonal anti-Tsg101–4A10 (1:1,000; GeneTex, Inc.) was used to detect Tsg101.
Electron microscopy
VLPs from 80 ml pooled culture supernatant were pelleted through 20% sucrose cushions and prepared for thin-section EM as described previously (von Schwedler et al., 1998). Cells were prepared for thin-section EM as described previously (Garrus et al., 2001). Samples were imaged on a transmission electron microscope (Tecnai-12; Philips).
Acknowledgments
We thank Rebecca Rich and David Myszka for help with the BIAcore experiments; Nancy Chandler and Barbie Ganser for help with electron microscopy; and members of the Sundquist lab for technical assistance and advice. We also thank the Stenmark and Emr laboratories for communicating their results before publication.
This work was supported by an National Institutes of Health grant to W.I. Sundquist.
O. Pornillos, D.S. Higginson, and K.M. Stray contributed equally to this paper.
O. Pornillos' present address is The Scripps Research Institute, Department of Molecular Biology, 10550 North Torrey Pines Road, La Jolla, CA 92037.
Abbreviations used in this paper: ESCRT, endosomal sorting complex required for transport; Hrs, hepatocyte growth factor–regulated tyrosine kinase substrate; MVB, multivesicular body; Tsg101, tumor susceptibility gene 101; UEV, ubiquitin E2 variant; UIM, ubiquitin-interacting motif; VLP, virus-like particle; Vps, vacuolar protein sorting.
References
- Asao, H., Y. Sasaki, T. Arita, N. Tanaka, K. Endo, H. Kasai, T. Takeshita, Y. Endo, T. Fujita, and K. Sugamura. 1997. Hrs is associated with STAM, a signal-transducing adaptor molecule. Its suppressive effect on cytokine-induced cell growth. J. Biol. Chem. 272:32785–32791. [DOI] [PubMed] [Google Scholar]
- Babst, M., D. Katzmann, E. Estepa-Sabal, T. Meerloo, and S. Emr. 2002. a. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell. 3:271–282. [DOI] [PubMed] [Google Scholar]
- Babst, M., D. Katzmann, W. Snyder, B. Wendland, and S. Emr. 2002. b. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev. Cell. 3:283–289. [DOI] [PubMed] [Google Scholar]
- Bache, K.G., C. Raiborg, A. Mehlum, I.H. Madshus, and H. Stenmark. 2002. Phosphorylation of Hrs downstream of the epidermal growth factor receptor. Eur. J. Biochem. 269:3881–3887. [DOI] [PubMed] [Google Scholar]
- Bache, K.G., A. Brech, A. Mehlum, and H. Stenmark. 2003. a. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J. Cell Biol. 162:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache, K.G., C. Raiborg, A. Mehlum, and H. Stenmark. 2003b. STAM and Hrs are subunits of a multivalent ubiquitin-binding complex on early endosomes. J. Biol. Chem. 278:12513–12521. First published on January 27, 2003; 10.1074/jbc.M210843200. [DOI] [PubMed]
- Bendixen, C., S. Gangloff, and R. Rothstein. 1994. A yeast mating-selection scheme for detection of protein-protein interactions. Nucleic Acids Res. 22:1778–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilodeau, P.S., J.L. Urbanowski, S.C. Winistorfer, and R.C. Piper. 2002. The Vps27p Hse1p complex binds ubiquitin and mediates endosomal protein sorting. Nat. Cell Biol. 4:534–539. [DOI] [PubMed] [Google Scholar]
- Bishop, N., A. Horman, and P. Woodman. 2002. Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 157:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirov, D.G., A. Ono, J.M. Orenstein, and E.O. Freed. 2002. a. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA. 99:955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirov, D.G., J.M. Orenstein, and E.O. Freed. 2002. b. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J. Virol. 76:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir, S.M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 411:494–498. [DOI] [PubMed] [Google Scholar]
- Feng, G.H., C.J. Lih, and S.N. Cohen. 2000. TSG101 protein steady-state level is regulated posttranslationally by an evolutionarily conserved COOH-terminal sequence. Cancer Res. 60:1736–1741. [PubMed] [Google Scholar]
- Francis, S.H., C. Poteet-Smith, J.L. Busch, R. Richie-Jannetta, and J.D. Corbin. 2002. Mechanisms of autoinhibition in cyclic nucleotide-dependent protein kinases. Front. Biosci. 7:d580–d592. [DOI] [PubMed] [Google Scholar]
- Freed, E.O. 2002. Viral late domains. J. Virol. 76:4679–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrus, J.E., U.K. von Schwedler, O.W. Pornillos, S.G. Morham, K.H. Zavitz, H.E. Wang, D.A. Wettstein, K.M. Stray, M. Cote, R.L. Rich, et al. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 107:55–65. [DOI] [PubMed] [Google Scholar]
- Göttlinger, H.G. 2001. The HIV-1 assembly machine. AIDS. 15:S13–S20. [DOI] [PubMed] [Google Scholar]
- Göttlinger, H.G., T. Dorfman, J.G. Sodroski, and W.A. Haseltine. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA. 88:3195–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty, R.N., J. Paragas, M. Sudol, and P. Palese. 1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 73:2921–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty, R.N., M.E. Brown, J.P. McGettigan, G. Wang, H.R. Jayakar, J.M. Huibregtse, M.A. Whitt, and M.J. Schnell. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75:10623–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermida-Matsumoto, L., and M.D. Resh. 2000. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol. 74:8670–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, K., and L. Falquet. 2001. A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem. Sci. 26:347–350. [DOI] [PubMed] [Google Scholar]
- Huang, M., J.M. Orenstein, M.A. Martin, and E.O. Freed. 1995. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 69:6810–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins, Y., O. Pornillos, R.L. Rich, D.G. Myszka, W.I. Sundquist, and M.H. Malim. 2001. Biochemical analyses of the interactions between human immunodeficiency virus type 1 Vpr and p6(Gag). J. Virol. 75:10537–10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz, M., K. Shtiegman, P. Tal-Or, L. Yakir, Y. Mosesson, D. Harari, Y. Machluf, H. Asao, T. Jovin, K. Sugamura, and Y. Yarden. 2002. Ligand-independent degradation of epidermal growth factor receptor involves receptor ubiquitylation and Hgs, an adaptor whose ubiquitin-interacting motif targets ubiquitylation by Nedd4. Traffic. 3:740–751. [DOI] [PubMed] [Google Scholar]
- Katzmann, D.J., M. Babst, and S.D. Emr. 2001. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 106:145–155. [DOI] [PubMed] [Google Scholar]
- Katzmann, D.J., G. Odorizzi, and S.D. Emr. 2002. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3:893–905. [DOI] [PubMed] [Google Scholar]
- Katzmann, D.J., C.J. Stefan, M. Babst, and S.D. Emr. 2003. Vps27 recruits/activates ESCRT machinery during endosomal sorting. J. Cell Biol. 162:413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikonyogo, A., F. Bouamr, M.L. Vana, Y. Xiang, A. Aiyar, C. Carter, and J. Leis. 2001. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. USA. 98:11199–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada, M., R. Masaki, A. Yamamoto, and N. Kitamura. 1997. Hrs, a tyrosine kinase substrate with a conserved double zinc finger domain, is localized to the cytoplasmic surface of early endosomes. J. Biol. Chem. 272:20538–20544. [DOI] [PubMed] [Google Scholar]
- Kramer, H. 2002. Sorting out signals in fly endosomes. Traffic. 3:87–91. [DOI] [PubMed] [Google Scholar]
- Lloyd, T.E., R. Atkinson, M.N. Wu, Y. Zhou, G. Pennetta, and H.J. Bellen. 2002. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell. 108:261–269. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano, J., T. Zang, and P.D. Bieniasz. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313–1319. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano, J., T. Zang, and P.D. Bieniasz. 2003. Role of ESCRT-I in retroviral budding. J. Virol. 77:4794–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes, T.F., R.A. Edwards, S. McKenna, L. Pastushok, W. Xiao, J.N. Glover, and M.J. Ellison. 2001. Crystal structure of the human ubiquitin conjugating enzyme complex, hMms2-hUbc13. Nat. Struct. Biol. 8:669–673. [DOI] [PubMed] [Google Scholar]
- Murk, J.L., W. Stoorvogel, M.J. Kleijmeer, and H.J. Geuze. 2002. The plasticity of multivesicular bodies and the regulation of antigen presentation. Semin. Cell Dev. Biol. 13:303–311. [DOI] [PubMed] [Google Scholar]
- Myers, E.L., and J.F. Allen. 2002. Tsg101, an inactive homologue of ubiquitin ligase e2, interacts specifically with human immunodeficiency virus type 2 gag polyprotein and results in increased levels of ubiquitinated gag. J. Virol. 76:11226–11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi, G., M. Babst, and S.D. Emr. 1998. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 95:847–858. [DOI] [PubMed] [Google Scholar]
- Pelchen-Matthews, A., B. Kramer, and M. Marsh. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162:443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicena, P., and W.T. Miller. 2002. Coupling kinase activation to substrate recognition in SRC-family tyrosine kinases. Front. Biosci. 7:d256–d267. [DOI] [PubMed] [Google Scholar]
- Piper, R.C., A.A. Cooper, H. Yang, and T.H. Stevens. 1995. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae. J. Cell Biol. 131:603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo, S., S. Sigismund, M. Faretta, M. Guidi, M.R. Capua, G. Bossi, H. Chen, P. De Camilli, and P.P. Di Fiore. 2002. A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature. 416:451–455. [DOI] [PubMed] [Google Scholar]
- Pornillos, O., S. Alam, R.L. Rich, D.G. Myszka, D.R. Davis, and W.I. Sundquist. 2002. a. Structure and functional interactions of the Tsg101 UEV domain. EMBO J. 21:2397–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos, O., S.L. Alam, D.R. Davis, and W.I. Sundquist. 2002. b. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat. Struct. Biol. 9:812–817. [DOI] [PubMed] [Google Scholar]
- Pornillos, O.P., J.E. Garrus, and W.I. Sundquist. 2002. c. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 12:569–579. [DOI] [PubMed] [Google Scholar]
- Raiborg, C., K.G. Bache, A. Mehlum, and H. Stenmark. 2001. a. Function of Hrs in endocytic trafficking and signalling. Biochem. Soc. Trans. 29:472–475. [DOI] [PubMed] [Google Scholar]
- Raiborg, C., B. Bremnes, A. Mehlum, D.J. Gillooly, A. D'Arrigo, E. Stang, and H. Stenmark. 2001. b. FYVE and coiled-coil domains determine the specific localisation of Hrs to early endosomes. J. Cell Sci. 114:2255–2263. [DOI] [PubMed] [Google Scholar]
- Raiborg, C., and H. Stenmark. 2002. Hrs and endocytic sorting of ubiquitinated membrane proteins. Cell Struct. Funct. 27:403–408. [DOI] [PubMed] [Google Scholar]
- Raiborg, C., K.G. Bache, D.J. Gillooly, I.H. Madshus, E. Stang, and H. Stenmark. 2002. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat. Cell Biol. 4:394–398. [DOI] [PubMed] [Google Scholar]
- Raposo, G., M. Moore, D. Innes, R. Leijendekker, A. Leigh-Brown, P. Benaroch, and H. Geuze. 2002. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic. 3:718–729. [DOI] [PubMed] [Google Scholar]
- Rotin, D., O. Staub, and R. Haguenauer-Tsapis. 2000. Ubiquitination and endocytosis of plasma membrane proteins: role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J. Membr. Biol. 176:1–17. [DOI] [PubMed] [Google Scholar]
- Shih, S.C., D.J. Katzmann, J.D. Schnell, M. Sutanto, S.D. Emr, and L. Hicke. 2002. Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat. Cell Biol. 4:389–393. [DOI] [PubMed] [Google Scholar]
- Sorkin, A., and M. Von Zastrow. 2002. Signal transduction and endocytosis: close encounters of many kinds. Nat. Rev. Mol. Cell Biol. 3:600–614. [DOI] [PubMed] [Google Scholar]
- Strack, B., A. Calistri, M.A. Accola, G. Palu, and H.G. Göttlinger. 2000. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA. 97:13063–13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack, B., A. Calistri, and H.G. Göttlinger. 2002. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J. Virol. 76:5472–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins, J., G. Schoehn, S. Ricard-Blum, S. Scianimanico, T. Vernet, R.W. Ruigrok, and W. Weissenhorn. 2003. Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J. Mol. Biol. 326:493–502. [DOI] [PubMed] [Google Scholar]
- Urbe, S., I.G. Mills, H. Stenmark, N. Kitamura, and M.J. Clague. 2000. Endosomal localization and receptor dynamics determine tyrosine phosphorylation of hepatocyte growth factor-regulated tyrosine kinase substrate. Mol. Cell. Biol. 20:7685–7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDemark, A.P., R.M. Hofmann, C. Tsui, C.M. Pickart, and C. Wolberger. 2001. Molecular insights into polyubiquitin chain assembly. Crystal structure of the Mms2/Ubc13 deterodimer. Cell. 105:711–720. [DOI] [PubMed] [Google Scholar]
- VerPlank, L., F. Bouamr, T.J. LaGrassa, B. Agresta, A. Kikonyogo, J. Leis, and C.A. Carter. 2001. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA. 98:7724–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt, V.M. 2000. Ubiquitin in retrovirus assembly: actor or bystander? Proc. Natl. Acad. Sci. USA. 97:12945–12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler, U.K., T.L. Stemmler, V.Y. Klishko, S. Li, K.H. Albertine, D.R. Davis, and W.I. Sundquist. 1998. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 17:1555–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, Y., C.E. Cameron, J.W. Wills, and J. Leis. 1996. Fine mapping and characterization of the Rous sarcoma virus Pr76gag late assembly domain. J. Virol. 70:5695–5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, M., N. Ishii, H. Asao, K. Murata, C. Kanazawa, H. Sasaki, and K. Sugamura. 2002. Signal-transducing adaptor molecules STAM1 and STAM2 are required for T-cell development and survival. Mol. Cell. Biol. 22:8648–8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, J., E. Hunter, M. Nakao, and H. Shida. 2002. Functional involvement of a novel Nedd4-like ubiquitin ligase on retrovirus budding. EMBO Rep. 3:636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, P., Q. Deveraux, R.E. Beal, C.M. Pickart, and M. Rechsteiner. 1998. Characterization of two polyubiquitin binding sites in the 26 S protease subunit 5a. J. Biol. Chem. 273:5461–5467. [DOI] [PubMed] [Google Scholar]