

Abstract
Mutations in the X-linked Plp gene lead to dysmyelinating phenotypes and oligodendrocyte cell death. Here, we exploit the X inactivation phenomenon to show that a hierarchy exists in the influence of different mutant Plp alleles on oligodendrocyte survival. We used compound heterozygote mice to study the long-term fate of oligodendrocytes expressing either the jimpy or rumpshaker allele against a background of cells expressing a Plp-null allele. Although mutant and null oligodendrocytes were generated in equal numbers, the proportion expressing the mutant allele subsequently declined, but whereas those expressing the rumpshaker allele formed a reduced but stable population, the number of jimpy cells fell progressively. The age of decline in the jimpy cells in different regions of the CNS correlated with the temporal sequence of myelination. In compound heterozygotes expressing rumpshaker and jimpy alleles, oligodendrocytes expressing the former predominated and were more abundant than when the rumpshaker and null alleles were in competition. Thus, oligodendrocyte survival is not determined solely by cell intrinsic factors, such as the conformation of the misfolded PLP, but is influenced by neighboring cells, possibly competing for cell survival factors.
Keywords: myelin protein; mutation; oligodendrocyte; proteolipid protein; heterozygote
Introduction
The highly conserved X-linked Plp gene, encodes for proteolipid protein (PLP),* the major CNS myelin protein, and its minor isoprotein DM20 (Nave et al., 1987). PLP/DM20 is believed to regulate the structure of the intraperiod line and may be involved in glial/axonal interactions (Nave et al., 1987; Boison and Stoffel, 1994; Klugmann et al., 1997; Griffiths et al., 1998b) and oligodendrocyte survival. Mutations of the PLP/Plp gene in man and animals cause dysmyelinating diseases ranging in severity from mild to lethal (for reviews see Griffiths et al., 1998a; Werner et al., 1998; Garbern et al., 1999; Yool et al., 2000). In the mouse, the disparity in phenotype is illustrated by the jimpy (Plp jp) and rumpshaker (Plp jp-rsh) mutations. Plp jp causes a lethal disorder with severe hypomyelination, premature death of many oligodendrocytes, and astrocytosis (Knapp et al., 1986; Vela et al., 1998). On the other hand, Plp jp-rsh is a relatively benign condition when expressed in the C3H/101 strain (Griffiths et al., 1990; Schneider et al., 1992). rumpshakers have normal longevity, produce more myelin than jimpy mice, and have fewer numbers of dying oligodendrocytes.
The Plp gene, like the majority of X-linked genes, is subject to random inactivation of one allele in the female. Because X-linked genes in glia exhibit the predicted 50% maternal/paternal allele inactivation (Tan et al., 1995), 50% of the oligodendrocytes in Plp mutant heterozygotes should express the mutant allele. In reality, the defect, as assessed by the degree of dysmyelination, never affects 50% of the total oligodendrocyte population (Skoff and Montgomery, 1981; Bartlett and Skoff, 1986; Duncan et al., 1987; Fanarraga et al., 1991). This strongly suggests that cells expressing the mutant allele are at a disadvantage through their expression of the mutant allele or their failure to express the wild-type allele. Alternatively, neighboring cells expressing the wild-type allele may hinder the mutant cells' development or survival. The exact fate of these mutant cells is uncertain because they are difficult to identify against the wild-type background.
We, therefore, used Plp knockout mice to generate Plp jp/− or Plp jp-rsh/− compound heterozygotes in which the jimpy or rumpshaker cells can be identified against the PLP-negative background. This allowed us to assess the survival of oligodendrocytes expressing different mutant alleles, when in competition with those expressing the null allele. The results demonstrate that in adult compound heterozygotes, fewer cells express the missense alleles compared with oligodendrocytes expressing the null allele. The number of cells expressing the jimpy allele decreases markedly over the first months of life, whereas the number of oligodendrocytes expressing the rumpshaker allele remains relatively constant for several months. When cells expressing either the jimpy or the rumpshaker allele are in direct competition, the latter predominate. Thus, the various alleles of the murine Plp gene demonstrate a hierarchy in their influence on oligodendrocyte survival and myelination from wild type > null > rumpshaker > jimpy. Our results show that survival depends not only on the “intrinsic severity” of an individual mutation but also on the allele being expressed by neighboring cells.
Results
PLP does confer an advantage for myelination
Our previous studies established that oligodendrocytes expressing a null allele of the Plp gene survived in normal numbers and elaborated sheaths so that the vast majority of larger-diameter axons were myelinated (Klugmann et al., 1997; Yool et al., 2001). Absence of PLP/DM20 resulted in ∼26–30% of axons <2 μm remaining nonmyelinated (Yool et al., 2001). Because of X chromosome inactivation, heterozygotes are chimeras in which oligodendrocytes lacking PLP/DM20 are in competition with those expressing the proteins. If the presence of PLP/DM20 confers no advantage or disadvantage, then 50% of cells and their myelin sheaths should express each allotype. In fact, ∼66% of sheaths were PLP positive, a value significantly different from 50% (Yool et al., 2001). The proportion of PLP+ sheaths was similar throughout the CNS and was stable between postnatal day (P) 20 and 1 yr of age. In general, the distribution of PLP+ and PLP− sheaths showed an intermingling mosaic pattern throughout the CNS, with the exception of the optic nerve, where, commonly, a large number of contiguous myelin sheaths were of a single immunotype, thus forming a “patch” (Fig. 1) .
Figure 1.
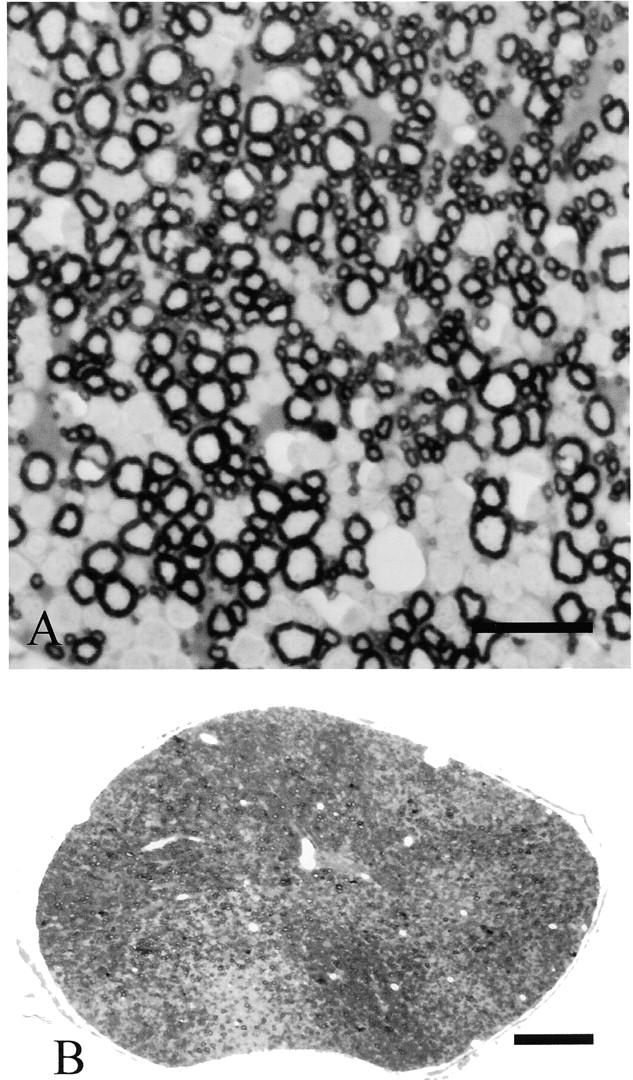
Heterozygote, 4-mo-old mouse CNS, expressing wild-type and null alleles of the Plp gene, immunostained for PLP/DM20. (A) Ventral column of spinal cord to show mosaic of wild-type myelin (positively stained) and myelin formed by oligodendrocytes expressing a null allele (no staining); the former predominate. (B) Optic nerve to show mosaic of positive and negative sheaths in conjunction with patches predominantly of one immunotype. Bars: (A) 20 μm; (B) 50 μm.
These studies established that although oligodendrocytes expressing a Plp-null allele survive, the presence of PLP/DM20 does confer an advantage for myelination, probably in the ability of the cell to associate with axons. The strategy for the present studies is to allow oligodendrocytes expressing a mutant allele to compete with those expressing a null allele, thus providing a “PLP-negative” background on which the former cells are detectable. A further benefit of this strategy is that because the null cell does not have the “advantage” conferred by wild-type PLP, any deviation from the predicted 50% survival can be linked to the mutant protein. If mutant alleles have a similar influence as the null allele on cell survival and myelination, the compound heterozygotes should contain equal numbers of oligodendrocytes and sheaths of each allotype.
No PLP is better than misfolded PLP
The rumpshaker allele causes the least severe phenotype of any of the murine Plp mutants and also results in a relatively mild disorder in humans (Kobayashi et al., 1994). Oligodendrocyte numbers in affected males (Plp jp-rsh/Y) are not reduced compared with wild type, and, with time, the majority of axons in the spinal cord acquire a PLP/DM20+ myelin sheath (Fanarraga et al., 1992, 1993).
Females heterozygous for both the rumpshaker and null alleles were clinically normal; lacking the tremor associated with affected rumpshaker males. We used in situ hybridization to identify oligodendrocytes expressing the mutant allele in cervical spinal cord of compound heterozygotes and their affected male rumpshaker littermates at P20, P50, and P100. By visual inspection, the number of positive cells in the heterozygotes was <50% of those in the male littermates at all ages (Fig. 2) . When we immunostained sections from the compound heterozygotes (Plp jp-rsh/−), the majority of myelin sheaths throughout the CNS at ages from P20 to P200 were PLP−, indicating their origin from oligodendrocytes expressing the null allele (Fig. 3) . In the white matter, the distribution of the PLP+ fibers varied from single isolated sheaths through to large patches of immunopositive fibers, the latter being particularly prominent in the optic nerves (Fig. 3, C and D). Small islands of PLP+ sheaths were present in the cerebral cortex, suggesting their origin from a single cell or a small cluster of oligodendrocytes expressing the rumpshaker allele (Fig. 3, G and H). By light microscopy, we detected small numbers of dysmyelinated axons with thin myelin sheaths, corresponding to the immunostained sheaths (unpublished data). We quantified the proportions of PLP+ and PLP− myelin sheaths in 1-μm resin sections from the ventral columns of thoracic spinal cord in compound heterozygotes at P20, P50, and P100. The proportion of PLP+ (rumpshaker) sheaths changed from 26 ± 5% (mean ± SEM, n = 4) at P20 to 19 ± 2% at P50 to 16 ± 3% at P100, values that were not significantly different (Fig. 4) . However, at all ages, the percentage of PLP+ myelin sheaths was significantly <50% (P = 0.0286) (Fig. 4).
Figure 2.

In situ hybridization for Plp/Dm20 in spinal cords of rumpshaker male mice (rsh/Y) and female littermates expressing a rumpshaker and a null allele of the Plp gene (rsh/−). Animals aged P20 and P100 are shown. The affected males indicate the total number of oligodendrocytes capable of expressing the rumpshaker allele. At both P20 and P100 the number of positive cells in the heterozygotes is considerably <50% of those in the male littermates. However, the proportion of positive cells appears very similar at both ages in the heterozygotes. The bright signal to the lower left of the P100 heterozygote cord is an artifact. Bars, 0.5 mm.
Figure 3.

Female compound heterozygotes expressing rumpshaker and null alleles of the Plp gene, immunostained for PLP/DM20. Myelin sheaths formed by oligodendrocytes expressing the rumpshaker allele are positively stained, whereas those supported by cells expressing the null allele remain unstained. (A and B) Resin sections of ventral columns of spinal cord from mice aged P20 and P100, respectively. There is a small reduction in the number of positive sheaths between the two ages. Bars, 20 μm. (C and D) Resin sections of optic nerves from mice aged P20 and P50, respectively. Both mice show patches of rumpshaker myelin (arrows) comprising a minority of the myelin sheaths. Bars, 50 μm. (E and F) Paraffin sections of the dorsal columns of spinal cord from a heterozygote aged P200 (rsh/−) and her male rumpshaker littermate (rsh/Y), respectively. The affected male (F) indicates the amount of PLP/DM20 that can be present if all oligodendrocytes express the rumpshaker allele. The heterozygote contains considerably <50% positive sheaths although their concentration is higher in the corticospinal tracts (CST) than elsewhere in the dorsal columns. Bars, 50 μm. (G) Paraffin section of cerebral cortex from a heterozygote aged P200 to show several small regions containing rumpshaker myelin, suggesting single oligodendrocytes or a small clone of cells expressing the mutant allele. Bar, 100 μm. (H) Higher magnification of one of the patches of rumpshaker myelin shown in G. Bar, 20 μm.
Figure 4.

Thoracic spinal cord from compound heterozygotes expressing a rumpshaker and a null allele of the Plp gene was immunostained for PLP/DM20. The proportion of myelin sheaths formed by oligodendrocytes expressing the rumpshaker allele (PLP/DM20+) was determined in the ventral funiculi at different ages. At all ages, the proportion of rumpshaker myelin sheaths is significantly <50% (P = 0.0286, Mann-Whitney test). There is no significant difference between the groups at the three ages (P = 0.1498, ANOVA). Data are mean ± SEM, n = 4.
Phenotypically severe mutations result in a progressive loss of oligodendrocytes
The jimpy allele causes one of the most severe phenotypes of any Plp gene mutation, one characteristic of which is the increased apoptosis of oligodendrocytes (Knapp et al., 1986). Indirect assessments suggest that in natural heterozygotes, there is a progressive loss of cells expressing the mutant allele, when in competition with oligodendrocytes expressing a wild-type allele (Skoff and Ghandour, 1995). To examine the long-term survival and distribution of such oligodendrocytes, we generated compound heterozygotes in which oligodendrocytes could express either a null or jimpy allele.
Cells expressing the jimpy allele were detectable by a positive in situ hybridization signal using a probe recognizing Plp/Dm20 and the absence of any signal when hybridized with a Plp exon 5–specific probe (unpublished data). Spinal cords and hindbrains were examined by in situ hybridization at P5, 20, 35, 50, and 100 to determine the approximate proportion of positive cells (Fig. 5 A). The additional time points at P5 and P35 were taken because preliminary studies suggested a more dynamic change in the cell population in the jimpy compared with the rumpshaker mouse. At P5, the number of Plp + cells appeared similar to those found in the Plp −/+ heterozygotes. By P20, the number of Plp + (jimpy) cells was reduced in the ventral and lateral columns compared with P5. In the dorsal columns, the positive cells were predominantly in the areas of the fasciculus gracilis and corticospinal tracts. At P35 and subsequently, the proportion of Plp + cells was markedly reduced throughout the entire transverse section compared with earlier ages, although there was little further change between P35 and P100. The surviving cells were present in white and gray matter and appeared random in distribution. A similar loss of positive cells was observed in hindbrain regions, although foci of jimpy oligodendrocytes were present at P100 (Fig. 5 B).
Figure 5.

Female compound heterozygotes expressing jimpy and null alleles of the Plp gene hybridized for Plp/Dm20 mRNA. (A) Dark field autoradiograms of cervical spinal cord from mice at P5, P20, P35, and P100. At P5 and P20, many positive cells are present, but by P35 and P100, their numbers have reduced considerably. Bars, 1 mm. (B) Brightfield autoradiogram, counterstained with haematoxylin, from the corpus callosum of a P100 mouse with a several positive cells present. Bar, 50 μm.
Using PLP-specific antibodies, we immunolabeled the cell bodies and major processes of oligodendrocytes expressing the mutant allele. As the mutant PLP is largely retained in the cell body (Gow et al., 1994; Gow and Lazzarini, 1996; Jung et al., 1996), any myelin sheaths formed by these oligodendrocytes remain unstained. For the purpose of quantification, the number of jimpy (PLP+) cells was expressed as a percentage of the total number of mature oligodendrocytes (adenomatous polyposis coli [APC]+). The results mirrored those produced by in situ hybridization with an age-related decrease in jimpy oligodendrocytes (Fig. 6) . By analyzing three different CNS regions, we showed that the age at which the loss of cells occurred varied and appeared to correlate with the temporal differences in myelination pattern. Thus, in the cervical spinal cord, which normally myelinates early, the proportion of jimpy cells declined significantly between P10 and P20 (Fig. 6, A, B, and E), whereas in the optic nerve and corpus callosum, where myelination occurs later, differences were delayed until P50 (Fig. 6, C, D, and E). As with the hybridization studies, jimpy cells were still detectable in the optic nerve and forebrain at P100 and even P280 (unpublished data). The oligodendrocytes expressing the jimpy allele were clearly identifiable within the myelinated white matter generated by the Plp-null cells (Fig. 7 A). Foci of immunopositive oligodendrocytes were often observed, particularly in the optic nerve (Fig. 7, B and C), and in some instances, the density of jimpy cells was sufficient to result in a focus of dysmyelination (Fig. 7 C).
Figure 6.
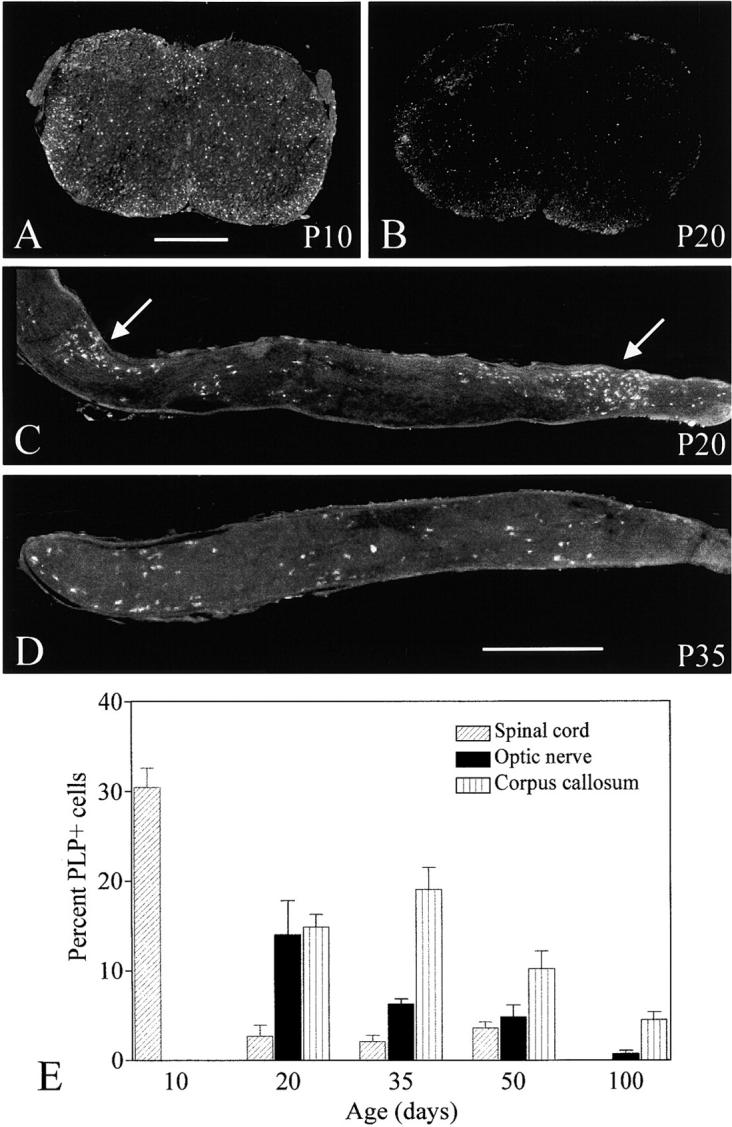
Female compound heterozygotes expressing jimpy and null alleles of the Plp gene. (A and B) Cervical spinal cords at P10 and P20, respectively, immunostained for PLP to identify those oligodendrocytes expressing the jimpy allele. There is a marked decrease in cells between the two ages. Whereas at P10 most of the positive cells are distributed in white matter, at P20, there are relatively more in the gray matter. (C and D) Optic nerves from mice aged P20 and P35, respectively, also immunostained to show the oligodendrocytes expressing the jimpy allele. The P20 nerve (C) shows two clusters of jimpy cells (arrows) together with more dispersed cells, whereas at P35, the cells are mainly dispersed. Bars, 0.5 mm. (E) Quantification of oligodendrocytes expressing the jimpy allele in different regions of the CNS of the compound heterozygotes. The results (mean ± SEM) are expressed as the percentage of PLP+ cells relative to the number of APC+ cells. The APC antigen marks mature oligodendrocytes. The age of the decline in jimpy cells varies between regions, corresponding to the order in which the regions normally myelinate. In the spinal cord, there is a significant decrease between P10 and P20 (P < 0.001), in the optic nerve between P20 and P50 (P < 0.05), and in the corpus callosum between P35 and P50 (P < 0.05). Group sizes are between four and six.
Figure 7.
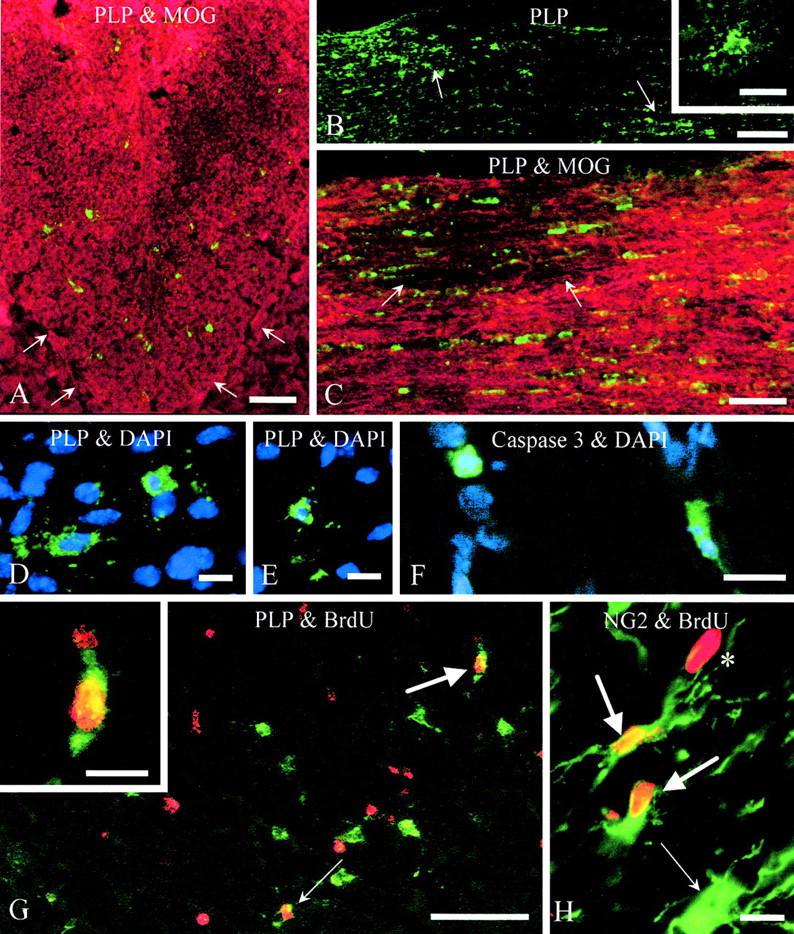
Female compound heterozygotes expressing jimpy and null alleles of the Plp gene. (A) Dorsal column from P20 mouse immunostained for PLP (green) to show oligodendrocytes expressing the jimpy allele and for MOG (red). Scattered jimpy oligodendrocytes are present in the white matter, although they represent considerably <50% of the expected total oligodendrocyte population. The base of the dorsal columns is outlined by arrows. Bar, 50 μm. (B) Optic nerve from P20 mouse immunostained for PLP to show clusters of oligodendrocytes (arrows) expressing the jimpy allele. Bar, 100 μm. (Inset) Higher magnification of individual jimpy oligodendrocyte showing immunostained cell body and processes but no obvious labeling of any myelin sheaths. Bar, 20 μm. (C) Optic nerve from P20 mouse stained for PLP (green) and MOG (red) to show a small dysmyelinated region (arrows) of deficient MOG staining, associated with oligodendrocytes expressing the jimpy allele. Other jimpy cells are scattered through the myelinated region of the nerve. Bar, 50 μm. (D and E) Optic nerve from P20 mouse immunostained for PLP (green) to identify oligodendrocytes expressing the jimpy allele and DAPI (blue) to show three jimpy cells with pyknotic nuclei indicating apoptosis. Bars, 10 μm. (F) Corpus callosum from P20 mouse immunostained for caspase 3 (green) to show two dying putative jimpy oligodendrocytes. Nuclei are stained with DAPI. Bar, 10 μm. (G) Corpus callosum from P34 mouse that was injected with BrdU on three successive days from P30 to P32 and the tissue was immunostained for PLP (green) and BrdU (red). Many PLP+ jimpy oligodendrocytes are present, only two of which have incorporated BrdU into their nuclei (arrows). One of these (larger arrow) is shown at higher magnification in the inset. Many other cells of unknown phenotype have also incorporated BrdU. Bar, 50 μm. Inset bar, 10 μm. (H) Optic nerve from P34 mouse injected with BrdU, as above, and coimmunostained for NG2; two double-labeled cells (large arrows) are present and an NG2+ cell with an unlabeled nucleus (small arrow). A BrdU-labeled nucleus from another cell type (*) is also evident. Bar, 10 μm.
Are the jimpy oligodendrocytes replaced?
The age-related decrease in the number of jimpy oligodendrocytes in the compound heterozygotes suggested that many were dying, as evidenced by examples of PLP+ (jimpy) oligodendrocytes with pyknotic nuclei in brain and optic nerve at P20 (Fig. 7, D and E). Cells in white matter tracts, with a similar distribution to jimpy oligodendrocytes, immunostained for caspase 3 (Fig. 7 F), a key effector caspase in the apoptotic pathway (Casaccia-Bonnefil, 2000). As our anticaspase and anti-PLP antibodies were raised in the same species, we were unable to double label for both markers.
To determine whether jimpy cells were continuing to be generated in mice older than 1 mo, we injected animals with BrdU between P30 and P32 and immunostained for PLP at P34. Only a small minority of cells in brain white matter were PLP+/BrdU+ (Fig. 7 G). In the optic nerve, for example, 2.8 ± 1.5% (mean ± SEM, n = 3) of jimpy oligodendrocytes had incorporated BrdU. Numerous PLP−/BrdU+ cells were present throughout the CNS (Fig. 7 G); in the optic nerve, 51 ± 10% (mean ± SEM, n = 2) of BrdU-labeled cells immunostained for NG2 (Fig. 7 H), 26 ± 6% for APC, and 12.7 ± 5% stained for CD45. We were unable to identify any BrdU-labeled cells costained for caspase 3.
A hierarchy in the survival of oligodendrocytes expressing different Plp alleles
The results from the various compound heterozygotes indicated that oligodendrocytes expressing the rumpshaker allele fared better than those expressing the jimpy allele when in competition with PLP-deficient cells. This suggested a possible hierarchy for survival of oligodendrocytes expressing these Plp alleles. To test this further, we generated compound heterozygotes whose oligodendrocytes could express either rumpshaker or jimpy alleles, with the expectation that the former cells would survive better. Female mice (Plp jp/jp-rsh) developed a marked tremor that persisted for at least 100 d (the longest time point used in the study) and appeared more severe than that seen in rumpshaker female homozygotes (Plp jp-rsh/jp-rsh). The prolonged survival of these compound heterozygotes expressing jimpy and rumpshaker alleles is in marked distinction to jimpy males (Plp jp/Y), which die at around P30; this provides further evidence for the dominance of the rumpshaker allele in the heterozygotes. Tissue from mice aged from P20 to P100 was immunostained with a PLP COOH-terminal antibody that recognizes the rumpshaker, but not the jimpy, products. At all ages and locations, the vast majority of the myelin sheaths and oligodendrocyte processes were positively stained, indicating a rumpshaker origin (Fig. 8 A), and contrasted markedly with the paucity of rumpshaker cells and myelin when they competed with those expressing a null allele (Fig. 8 B).
Figure 8.

Female compound heterozygotes expressing either rumpshaker and jimpy alleles or rumpshaker and null alleles of the Pil gene. (A) Compound female heterozygote mouse aged P50 expressing rumpshaker and jimpy alleles of the Plp gene (rsh/jp). A section of spinal cord showing the dorsal and ventral columns and central gray matter is immunostained with an antibody against the COOH terminus of PLP/DM20. As this region of the protein is deleted in the jimpy product, the staining detects only the myelin sheaths and oligodendrocytes expressing the rumpshaker allele. The vast majority of the cells and myelin are of rumpshaker origin. (B) Similarly stained section from a P50 compound heterozygote expressing rumpshaker and null alleles (rsh/−). In contrast to the previous animal, the majority of cells and sheaths express the null allele. Bar, 100 μm.
Competing alleles are generated in equal proportions but have nonuniform distribution
The above data obtained from juvenile and adult animals suggest that the various Plp alleles affect oligodendrocyte survival differentially in competitive situations. To confirm that cell survival and not cell production was compromised, we determined whether the initial populations of oligodendrocytes expressing the competing alleles were equal in the heterozygotes. To achieve this, we examined the PLP+ cell population in the ventral cervical spinal cord, the earliest region to myelinate, on the first day of life (P1) in compound heterozygotes expressing null and rumpshaker alleles and compared this with the male rumpshaker littermates. This age is before when apoptosis of mutant oligodendrocytes occurs; the rumpshaker PLP is partially retained in the cell bodies, making them easy to distinguish from myelin sheaths or cell processes. The number of PLP+ cells in the compound heterozygote was 125 ± 25 cells/mm2 (mean ± SEM, n = 4), a value not significantly different from 50% of the population in male littermates, 112 ± 16. We used the same method to show that the number of PLP+ cells in wild-type/null heterozygotes was not different from the 50% value in wild-type male littermates at P1. Visual appraisal of tissue from early myelinating optic nerve (Fig. 9 , A–F) and spinal cord (Fig. 9, G–I) of compound heterozygotes expressing null and rumpshaker alleles also suggested that starting cell populations were approximately equal. We also examined the spinal cord of compound heterozygotes expressing jimpy or null alleles at P5 (jimpy cells are detected with a PLP-specific antibody [Table I] as opposed to wild-type or rumpshaker cells, which are detected with an anti-PLP/DM20 antibody; the low level of the PLP isoform in the cells at P1 precluded quantification of the jimpy cells at this age). The number of PLP+ jimpy cells was 133 ± 13 (mean ± SEM, n = 3), compared with 121 ± 5, which represented 50% of the total APC+ cells; these values are not significantly different.
Figure 9.

Distribution of competing cell populations in compound heterozygotes expressing null and rumpshaker alleles during early myelination of the optic nerve and spinal cord. (A and B) Optic nerves at P9 from compound heterozygote (rsh/−) and affected male littermate (rsh/Y) stained for PLP/DM20, showing nonuniform distribution of PLP+ (rumpshaker) cells in the heterozygote. (C–F) Optic nerves from compound heterozygotes at P10 and P14, respectively, showing the patches of PLP+ rumpshaker cells compared with the uniform MBP staining of the same nerve. (G–I) Spinal cords from compound heterozygotes and an affected male littermate at P1, stained for PLP. The heterozygotes show small clumps (arrows) of rumpshaker oligodendrocytes and myelin, in contrast to the male, in which the distribution is relatively uniform in the ventral white matter. The size and number of patches are less marked than in the optic nerves. Bars, 200 μm.
Table I. Strategy for detecting oligodendrocytes expressing different alleles of the Plp gene in the various heterozygotes.
| Genotype of heterozygotes | Riboprobe or antibody | ||||||
|---|---|---|---|---|---|---|---|
| ISH
|
Immunostain
|
||||||
| Plp/Dm20 | Plp/Dm20 exon 5 | PLP-CT | PLP specific | ||||
| WT | null | WT+ | ND | WT+ | ND | ||
| null | rumpshaker | rumpshaker + | ND | rumpshaker + | ND | ||
| null | jimpy | jimpy + | Both negative | jimpy − | jimpy + | ||
| rumpshaker | jimpy | ND | ND | rumpshaker + | ND | ||
The genotype indicates the two alleles at the X-linked Plp locus. The riboprobe for Plp/Dm20 detects the majority of the coding region for the two transcripts. Exon 5 of the Plp/Dm20 transcript is deleted in jimpy. When used in conjunction with the Plp/Dm20 probe, it provides confirmation that jimpy cells are being detected. The PLP-CT antibody detects both PLP and DM20. The PLP-specific antibody is specific for that isoform and is used to detect jimpy cells.
Although the populations of competing oligodendrocytes are generated equally, their final pattern of distribution is clearly uneven, particularly in relation to the patches in the optic nerve. To determine whether the basis for this was established early in development or subsequently, we examined optic nerve and spinal cord from compound heterozygotes, expressing null and rumpshaker alleles, at various time points through the initial myelination process. In the optic nerve at P9 and subsequently, definite PLP+ patches were present in the heterozygotes. In contrast, the deposition of the myelinating PLP+ cells was uniform in male rumpshaker littermates, and the myelin basic protein (MBP) staining, which represents both mutant and null cells, was uniform in the heterozygotes (Fig. 9, A–D). In the heterozygote spinal cord at P1, an indication of smaller patches was present, although less consistent than in the optic nerve (Fig. 9, G–I).
Discussion
Female heterozygotes associated with most missense mutations of the X-linked Plp gene exhibit few, if any, clinical signs, suggesting that the majority of oligodendrocytes express the wild-type, rather than the mutant, allele. A progressive loss of oligodendrocytes expressing the mutant allele in jimpy heterozygotes has been proposed, based indirectly on total cell counts or levels of the jimpy transcript (Kagawa et al., 1994; Skoff and Ghandour, 1995). By generating compound heterozygotes, we can identify the mutant cells unequivocally. This approach demonstrates that oligodendrocytes expressing the jimpy allele are indeed lost over time and introduces the concept of a competitive hierarchy between cells expressing different alleles of the Plp gene.
The starting populations of mutant cells in the heterozygotes are not reduced
Our results show that in compound heterozygotes, oligodendrocytes expressing rumpshaker or jimpy alleles of the Plp gene are generated in approximately equal numbers as those expressing the null allele. Similarly, in heterozygotes expressing wild-type or null alleles, the number of wild-type cells is normal; so from this we infer that those expressing the null allele are not reduced. This finding is not unexpected, as studies of affected male animals with Plp mutations indicate that the generation and initial populations of oligodendrocytes are normal (Pringle et al., 1997; Thomson et al., 1999). Because the starting populations of mutant oligodendrocytes are not reduced, subsequent cell loss is likely to account for the diminished proportions of mutant cells in the older mice.
A hierarchy in survival of oligodendrocytes expressing different Plp alleles
Spontaneous missense mutations of the Plp gene are associated with increased numbers of dead oligodendrocytes in affected males. The magnitude of cell death varies between different mutations, being greater in those associated with severe phenotypes (Knapp et al., 1986; Schneider et al., 1992). Misfolded PLP is retained within the RER and has been implicated in the death of oligodendrocytes (Gow and Lazzarini, 1996; Gow et al., 1998), although formal proof is currently lacking. Abnormal oligodendrocyte death is not a feature of PLP-deficient mice, suggesting that this protein is not essential for cell survival (Klugmann et al., 1997; Yool et al., 2001) and cell death in the mutants is not due to a lack of functional PLP. However, in adult Plp +/− heterozygotes, wild-type oligodendrocytes always predominate when in competition with those expressing a null allele, suggesting that the presence of PLP/DM20 confers a definite advantage. In similar competitive situations, the cells expressing a null allele always predominate over those with a mutant allele, and rumpshaker cells fare better than jimpy oligodendrocytes. Using the amount of rumpshaker myelin as an indicator, <25% of cells expressing the rumpshaker allele survive when competing with PLP-deficient oligodendrocytes, whereas a majority survive when matched against jimpy cells. Thus, the survival of the cell appears to depend not only on the intrinsic severity of a particular mutation but also on the allele being expressed by competitor cells. This variability in cell survival strongly suggests that oligodendrocyte death must be influenced by factors additional to any intrinsic toxicity of the misfolded PLP or the absence of normal PLP.
Why do some mutant oligodendrocytes survive?
The proportion of jimpy oligodendrocytes decreased over time in the compound heterozygotes expressing jimpy and null alleles. The age at which a significant reduction occurred varied according to the region of CNS and correlated with the temporal pattern of myelination. A similar regional variation in cell death has been reported in male jimpy mice (Knapp et al., 1986), although such mice survive for only ∼4 wk. The use of the long-surviving compound heterozygotes demonstrated that a small number of jimpy cells was present at P100 or older. The low number and scattered distribution of such cells suggested that they had survived from the period of postnatal gliogenesis. We attempted to address this aspect by labeling compound heterozygotes with BrdU between embryonic day (E) 17 and P9, when gliogenesis is taking place, and then detecting the label in the mature mice. We were, however, unable to detect any BrdU-labeled oligodendrocyte nuclei after this lengthy period and formal proof of prolonged survival is still lacking. We did show by BrdU labeling that some jimpy oligodendrocytes are generated in mice older than 1 mo and that half of the BrdU-labeled cells were NG2+ and therefore potentially competent to give rise to oligodendrocytes. This finding is not unexpected, as the normal adult CNS contains a population of slowly proliferating oligodendrocyte progenitors (Levison et al., 1999; Nishiyama et al., 1999; Horner et al., 2000; Levine et al., 2001) and a marginal increase in proliferation rate occurs in older jimpy heterozygotes (Rosenfeld and Friedrich, 1986). The present study did not distinguish which Plp allele was activated in these progenitors, but presumably only 50% are potential jimpy oligodendrocytes.
Assuming that a small proportion of jimpy oligodendrocytes is capable of a prolonged survival raises the question as to what differentiates such cells from those that die. As surviving cells express readily detectable amounts of misfolded PLP, their longevity cannot be due to an absence of this potentially damaging product. One possible reason could relate to an association with axons. During normal myelinogenesis, in excess of 50% of oligodendrocytes generated may die, probably as a result of failure to associate with axons and secure necessary survival factors (Barres et al., 1992; Barres and Raff, 1994, 1999). Mature oligodendrocytes appear much less dependent on axonal contact (Ludwin, 1990; McPhilemy et al., 1990). We suggest that the surviving mutant oligodendrocytes may have established sufficient axonal support during the critical early period. This hypothesis would not require such cells to maintain axonal contact to ensure their survival indefinitely.
A Darwinian model explains the differential survival of oligodendrocytes and the optic nerve patches
Our data fit well with a “Darwinian” model in which developing oligodendrocytes expressing the various Plp alleles compete to survive. Those cells more adept at associating with axons gain the essential survival factors, whereas the less competent die. This model readily explains the differential survival of both the rumpshaker and null oligodendrocytes when set against disparate competitor cells and can account for the disproportionate patches of allotypes in the optic nerve. In the early neonatal period, there are clearly areas in the optic nerve, and to a lesser extent in the spinal cord, with clusters of mutant cells adjacent to zones where these cells are absent. Equally, there are areas in both regions where the two cell populations are intermingled. To generate a patch of mutant cells suggests prolonged clonal expansion from a common progenitor or the fortuitous contiguous settlement of progenitors expressing the mutant allele or a combination of both processes. If clonal expansion is the sole reason for a patch, it is difficult to envisage why the entire length of both optic nerves is not involved. To determine the extent of clonal expansion will require the recognition of progeny of individual progenitors, possibly through retroviral labeling before their migration into the nerve. Whatever process leads to a patch of cells with a single allotype, there appears to be a difference between the optic nerve and the remainder of the CNS in that the former region is more markedly affected. Once a patch of mutant oligodendrocytes has been laid down, we propose that there are two likely fates. Mutant cells capable of establishing an association with axons survive and generate thin myelin sheaths, as evidenced by the patches of rumpshaker myelin in the optic nerves of the compound heterozygotes. In contrast, mutant cells failing to establish this contact will be eliminated. If the two competing populations of cells are initially intermingled, as in the majority of the CNS, the more dominant cell will largely take over the territory of the “less fit” oligodendrocyte. Evidence for such a repair mechanism is found in the spinal cord and brain stem of heterozygotes with spontaneous mutations of the Plp gene (Knapp et al., 1986; Cuddon et al., 1998). If mutant cells originally forming a patch are eliminated, as in the optic nerves, there appears to be minimal repair and the end result is a zone of amyelinated axons, as found in heterozygotes with the jimpy and myelin-deficient mutations (Skoff and Montgomery, 1981; Duncan et al., 1993).
Implications for heterozygotes of spontaneous mutations of the Plp gene
Large amyelinated patches are a feature of the optic nerve of animals heterozygous for the jimpy, myelin-deficient, and shaking mutations of the Plp gene (Skoff and Montgomery, 1981; Duncan et al., 1987, 1993), whereas patches of naked axons are much rarer in spontaneous rumpshaker heterozygotes (Fanarraga et al., 1991). The patches appear random and show marked variation in number and size, even between the two nerves of a single heterozygote (Duncan et al., 1993). The basis of this phenotype is revealed by our studies, as discussed above. However, it is not clear why there is little repair of the amyelinated patches in the optic nerve in contrast to other regions of the CNS. Recent studies have emphasized the large number of oligodendrocyte progenitors that populate the CNS in normal adults (Levison et al., 1999; Nishiyama et al., 1999), and our study has demonstrated numerous proliferating NG2+ cells in the compound heterozygotes. One might anticipate that such progenitors expressing the wild-type (or null) allele would myelinate the bare axons. Why this does not occur is a topic we are currently pursuing and has relevance to remyelination in disorders, such as Multiple Sclerosis.
One prediction from the present study is that female patients heterozygous for a mutant allele associated with a “mild” Pelizaeus-Merzbacher disease phenotype in affected males may be more severely affected than those heterozygotes with an allele causing a “severe” phenotype in the respective male patients. The cells expressing the mild allele survive in competition with wild-type cells, whereas those with the severe allele die and are replaced by normal cells or myelin. There is indeed evidence for this in some cases of Pelizaeus-Merzbacher disease/Spastic Paraplegia type 2 (Garbern et al., 1999; Sivakumar et al., 1999). Similarly, in the animal mutants shaking pup and jimpy, there is evidence for progressive replacement of mutant oligodendrocytes in the spinal cord and brain stem by wild-type cells (Knapp et al., 1986; Cuddon et al., 1998).
Materials and methods
Generation of and characterization of mice
Female jimpy heterozygotes (Plp jp/+) were maintained on a C3H/101 background without the tabby linkage. Female rumpshaker heterozygotes (Plp jp-rsh/+ ) or homozygotes (Plp jp-rsh/jp-rsh) were on an identical background (the benign nature of the rumpshaker mutation allows generation of homozygous female mutants). The derivation and characterization of the Plp-null mutants, which are maintained on a C57BL6 background, has been previously described (Klugmann et al., 1997; Griffiths et al., 1998b). The Plp knockout mice do not express Plp mRNA, as detectable by in situ hybridization, nor PLP protein, as detectable by Western blotting and immunocytochemistry (Klugmann et al., 1997; Yool et al., 2001). Female mice carrying the jimpy or rumpshaker mutation were crossed with Plp − /Y to generate compound heterozygotes carrying null and mutant alleles (Plp jp/− and Plp jp-rsh/−). jimpy heterozygotes were mated with rumpshaker males to generate female mice expressing both mutant alleles (Plp jp/ jp-rsh).
Mice were genotyped by PCR, as described previously (Schneider et al., 1992; Klugmann et al., 1997; Thomson et al., 1999), using DNA derived from tail biopsies, or identified by immunostaining of cryosections of spinal cord with a PLP COOH-terminal antibody (see below).
BrdU labeling
BrdU (Sigma-Aldrich) was used to identify mitotic cells. Mice aged 30 d were injected intraperitoneally with 50 μg/g BrdU in 0.9% saline at midday on three consecutive days and tissues were sampled on the fifth day.
Tissue sampling
Animals were killed at various ages between P1 and 12 mo. Mice were perfused by intracardiac injection of paraformaldehyde–glutaraldehyde mixture (Griffiths et al., 1981), and cervical spinal cord and optic nerve were processed for resin embedding or perfused with buffered neutral formalin and processed for paraffin embedding. For double staining with BrdU and NG2, APC, or CD45, mice were perfusion fixed with periodate-lysine-paraformaldehyde fixative (McLean and Nakane, 1974) and cervical cord, brain, and optic nerves were transferred to 20% sucrose and then embedded in OCT compound (Sakura Fintek), snap frozen, and prepared for cryosectioning. Cervical and thoracic spinal cord, brain, and optic nerve from other mice were embedded, unfixed in OCT compound, snap frozen in isopentane cooled in liquid nitrogen, and prepared for cryosectioning.
In situ hybridization
Cryosections (15 μm) of transverse cervical spinal cord or saggital sections of the brain were hybridized with the 35S-labeled or DIG-labeled PLP-1 riboprobe detecting both Plp and Dm20 transcripts as previously described (Griffiths et al., 1989; Vouyiouklis et al., 2000). Exon 5 is deleted from the final transcript of jimpy Plp mRNA; a riboprobe specific to exon 5 was generated for studies involving these mutants. Autoradiograms were counterstained with haematoxylin and examined using darkfield and transmitted light. DIG was detected using an alkaline phosphatase conjugate and BCIP/NBT or HRP and diaminobenzidine. A summary of the riboprobes used and the alleles they detect is shown in Table I.
Immunostaining
Antibodies.
PLP/DM20 was detected using a polyclonal antibody to COOH-terminal residues 271–276, which are common to both isoforms (N.P. Groome, Oxford Brookes University, Oxford, UK). PLP was identified with an antibody raised against the PLP-specific region, residues 117–129 (E. Trifilieff, University of Strasbourg, Strasbourg, France). A summary of the PLP antibodies used and the alleles they detect is shown in Table I. MBP was detected using a rat monoclonal (clone 12; N.P. Groome). Myelin oligodendrocyte glycoprotein (MOG) was identified with a mouse monoclonal antibody (S. Piddlesden, University of Wales, Cardiff, Wales). Caspase 3 was detected with a rabbit polyclonal antibody (R&D Systems). BrdU was labeled with a mouse monoclonal antibody (Sigma-Aldrich). The APC antigen, which marks mature oligodendrocytes, was identified with a mouse monoclonal antibody (CC-1 clone; Oncogene Research Products). NG2 was detected with a rabbit polyclonal antibody (Chemicon International Ltd.) and CD45 with a rat polyclonal antibody (Serotec Ltd.).
Immunostaining.
Resin sections (1 μm) and paraffin wax sections (8 μm) were immunostained with the anti–PLP COOH-terminal antibody using the peroxidase antiperoxidase technique (Sternberger et al., 1970; Trapp et al., 1981). Cryosections (15 μm) were stained with antibodies to the PLP COOH-terminal and the PLP-specific region and with the caspase 3 and MOG antibodies, using indirect immunofluorescence. Sections were fixed in 4% paraformaldehyde in PBS for 20 min followed by 0.5% Triton X-100– PBS for 30 min at room temperature, and blocked in 0.1% Triton X-100, 0.2% pig skin gelatin in PBS for 30 min at room temperature. Primary antibodies were applied overnight at 4°C, and the secondary conjugates for 30 min at room temperature in the blocking buffer.
Sections labeled with BrdU were treated with 50% HCl/1% Triton X-100 for 10 min at room temperature, after the paraformaldehyde fixation. The anti-BrdU antibody was applied for 2 h at room temperature in 0.2% Triton X-100/PBS. After labeling with the secondary conjugate, the sections were immunostained for PLP or caspase 3. For NG2, APC, or CD45 double label with BrdU, cryostat sections were incubated overnight with appropriate antibodies, followed by the secondary conjugate. Sections were then fixed in 50% acetic acid/50% ethanol before continuing with the BrdU stain, as described.
Some cryosections were labeled with 2.2 μg/ml DAPI in H2O for 1 min at room temperature. Cryosections were mounted in Citifluor antifade medium.
Quantification of PLP+ myelin sheaths and cells
To determine the proportion of PLP+ and PLP− myelin sheaths in Plp jp-rsh/− mice, resin sections of thoracic spinal cord were immunostained for PLP/DM20. Two regions of the ventral columns on each side of the ventromedian fissure were photographed and printed at a final magnification of 2,800. A lined grid was placed on the prints and all fibers touching the lines were scored for their PLP status. Over 1,000 fibers were counted per animal and groups of four mice were analyzed at each age.
The numbers of PLP+ cells were determined in various regions of the CNS in Plp jp/− mice. The optic nerve and corpus callosum were sectioned longitudinally and the cervical cord cut transversely. Cryosections were immunostained with a PLP-specific antibody (to detect the oligodendrocytes expressing the jimpy allele) and with an anti-APC antibody to label mature oligodendrocytes. Nuclei were labeled with DAPI. Images of antibody and DAPI-stained cells were merged (Adobe Photoshop® 6.0; Adobe Systems) and all immunopositive cell bodies containing a nucleus, within a defined area, were counted (Image-Pro Plus; Media Cybernetics). As PLP+ cells occurred randomly, the fields for imaging were selected under phase optics (×20 objective) to avoid bias. Fields were selected along the lengths of the optic nerve and corpus callosum and in the ventral columns of the spinal cord. Groups of four to six animals were analyzed at various ages from P10 to P100.
Quantification of PLP+ cells in the spinal cord of compound heterozygotes aged P1 was performed as above, except that comparisons were made with affected male littermates. We found that the APC reaction was capricious at this age and could not be used to quantify the total oligodendrocyte count, which was estimated from the male littermates.
Statistical analysis
In compound heterozygotes expressing a null or a rumpshaker allele, each allele should be expressed in 50% of the oligodendrocytes and their myelin sheaths, if neither allele confers an advantage. To test the hypothesis that cells expressing the rumpshaker allele are at a disadvantage, we determined whether the percentage of PLP-positive rumpshaker myelin sheaths, as determined above, was less than the theoretical 50% value (represented by the total number of PLP-positive and PLP-negative sheaths × 0.5) using a one-tailed Mann-Whitney test. Comparison of the proportions of PLP-positive myelin sheaths or PLP-positive cells at different ages was performed using ANOVA with Bonferroni's Multiple Comparison Test as the post-test. Significance was P ≤ 0.5. Analyses were performed using the GraphPad Prism software (GraphPad Software).
Acknowledgments
We are grateful to Prof. N.P. Groome and Drs. E. Trifilieff and S. Piddlesden for the gift of antibodies.
This study was supported by Action Research and the Wellcome Trust.
P.J. Dickinson's present address is Department of Surgical and Radiological Sciences, School of Veterinary Medicine, University of California, Davis, Davis, CA 95616.
Footnotes
Abbreviations used in this paper: APC, adenomatous polyposis coli; E, embryonic day; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; P, postnatal day; PLP, proteolipid protein.
References
- Barres, B.A., and M.C. Raff. 1994. Control of oligodendrocyte number in the developing rat optic nerve. Neuron. 12:935–942. [DOI] [PubMed] [Google Scholar]
- Barres, B.A., and M.C. Raff. 1999. Axonal control of oligodendrocyte development. J. Cell Biol. 147:1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres, B.A., I.K. Hart, H.S.R. Coles, J.F. Burne, J.T. Voyvodic, W.D. Richardson, and M.C. Raff. 1992. Cell death in the oligodendrocyte lineage. J. Neurobiol. 23:1221–1230. [DOI] [PubMed] [Google Scholar]
- Bartlett, W.P., and R.P. Skoff. 1986. Expression of the jimpy gene in the spinal cords of heterozygous female mice. I. An early myelin deficit followed by compensation. J. Neurosci. 6:2802–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison, D., and W. Stoffel. 1994. Disruption of the compacted myelin sheath of axons of the central nervous system in proteolipid protein-deficient mice. Proc. Natl. Acad. Sci. USA. 91:11709–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil, P. 2000. Cell death in the oligodendrocyte lineage: a molecular perspective of life/death decisions in development and disease. Glia. 29:124–135. [DOI] [PubMed] [Google Scholar]
- Cuddon, P.A., D. Lipsitz, and I.D. Duncan. 1998. Myelin mosaicism and brain plasticity in heterozygous females of a canine X-linked trait. Ann. Neurol. 44:771–779. [DOI] [PubMed] [Google Scholar]
- Duncan, I.D., J.P. Hammang, and K.F. Jackson. 1987. Myelin mosaicism in female heterozygotes of canine shaking pup and myelin-deficient rat mutants. Brain Res. 402:168–172. [DOI] [PubMed] [Google Scholar]
- Duncan, I.D., K.F. Jackson, J.P. Hammang, D. Marren, and R. Hoffman. 1993. Development of myelin mosaicism in the optic nerve of heterozygotes of the X-linked myelin-deficient (md) rat mutant. Dev. Biol. 157:334–347. [DOI] [PubMed] [Google Scholar]
- Fanarraga, M.L., I.R. Griffiths, M.C. McCulloch, J.A. Barrie, B.M. Cattanach, P.J. Brophy, and P.G.E. Kennedy. 1991. Rumpshaker: an X-linked mutation affecting CNS myelination. A study of the female heterozygote. Neuropathol. Appl. Neurobiol. 17:323–334. [DOI] [PubMed] [Google Scholar]
- Fanarraga, M.L., I.R. Griffiths, M.C. McCulloch, J.A. Barrie, P.G.E. Kennedy, and P.J. Brophy. 1992. Rumpshaker: an X-linked mutation causing hypomyelination. Developmental differences in myelination and glial cells between the optic nerve and spinal cord. Glia. 5:161–170. [DOI] [PubMed] [Google Scholar]
- Fanarraga, M.L., I. Sommer, I.R. Griffiths, P. Montague, N.P. Groome, K.-A. Nave, A. Schneider, P.J. Brophy, and P.G.E. Kennedy. 1993. Oligodendrocyte development and differentiation in the rumpshaker mutation. Glia. 9:146–156. [DOI] [PubMed] [Google Scholar]
- Garbern, J., F. Cambi, M. Shy, and J. Kamholz. 1999. The molecular pathogenesis of Pelizaeus-Merzbacher disease. Arch. Neurol. 56:1210–1214. [DOI] [PubMed] [Google Scholar]
- Gow, A., and R.A. Lazzarini. 1996. A cellular mechanism governing the severity of Pelizaeus- Merzbacher disease. Nat. Genet. 13:422–428. [DOI] [PubMed] [Google Scholar]
- Gow, A., V.L. Friedrich, Jr., and R.A. Lazzarini. 1994. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J. Neurosci. Res. 37:574–583. [DOI] [PubMed] [Google Scholar]
- Gow, A., C.M. Southwood, and R.A. Lazzarini. 1998. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J. Cell Biol. 140:925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, I.R., I.D. Duncan, and M. McCulloch. 1981. Shaking pup: a disorder of central myelination in the spaniel dog. II. Ultrastructural observations on the white matter of cervical spinal cord. J. Neurocytol. 10:847–858. [DOI] [PubMed] [Google Scholar]
- Griffiths, I.R., L.S. Mitchell, K. McPhilemy, S. Morrison, E. Kyriakides, and J.A. Barrie. 1989. Expression of myelin protein genes in Schwann cells. J. Neurocytol. 18:345–352. [DOI] [PubMed] [Google Scholar]
- Griffiths, I.R., I. Scott, M.C. McCulloch, J.A. Barrie, K. McPhilemy, and B.M. Cattanach. 1990. Rumpshaker mouse: a new X-linked mutation affecting myelination: evidence for a defect in PLP expression. J. Neurocytol. 19:273–283. [DOI] [PubMed] [Google Scholar]
- Griffiths, I.R., M. Klugmann, T.J. Anderson, C.E. Thomson, D.A. Vouyiouklis, and K.-A. Nave. 1998. a. Current concepts of PLP and its role in the nervous system. Microsc. Res. Tech. 41:344–358. [DOI] [PubMed] [Google Scholar]
- Griffiths, I.R., M. Klugmann, T.J. Anderson, D. Yool, C.E. Thomson, M.H. Schwab, A. Schneider, F. Zimmermann, M.C. McCulloch, N.L. Nadon, and K.-A. Nave. 1998. b. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 280:1610–1613. [DOI] [PubMed] [Google Scholar]
- Horner, P.J., A.E. Power, G. Kempermann, H.G. Kuhn, T.D. Palmer, J. Winkler, L.J. Thal, and F.H. Gage. 2000. Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J. Neurosci. 20:2218–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, M., I. Sommer, M. Schachner, and K.-A. Nave. 1996. Monoclonal antibody O10 defines a conformationally sensitive cell-surface epitope of proteolipid protein (PLP): evidence that PLP misfolding underlies dysmyelination in mutant mice. J. Neurosci. 16:7920–7929. [DOI] [PMC free article] [PubMed]
- Kagawa, T., J. Nakao, M. Yamada, K. Shimizu, T. Hayakawa, K. Mikoshiba, and K. Ikenaka. 1994. Fate of jimpy-type oligodendrocytes in jimpy heterozygote. J. Neurochem. 62:1887–1893. [DOI] [PubMed] [Google Scholar]
- Klugmann, M., M.H. Schwab, A. Pühlhofer, A. Schneider, F. Zimmermann, I.R. Griffiths, and K.-A. Nave. 1997. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 18:59–70. [DOI] [PubMed] [Google Scholar]
- Knapp, P.E., R.P. Skoff, and D.W. Redstone. 1986. Oligodendroglial cell death in jimpy mice: an explanation for the myelin deficit. J. Neurosci. 6:2813–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, H., E.P. Hoffman, and H.G. Marks. 1994. The rumpshaker mutation in spastic paraplegia. Nat. Genet. 7:351–352. [DOI] [PubMed] [Google Scholar]
- Levine, J.M., R. Reynolds, and J.W. Fawcett. 2001. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 24:39–47. [DOI] [PubMed] [Google Scholar]
- Levison, S.W., G.M. Young, and J.E. Goldman. 1999. Cycling cells in the adult rat neocortex preferentially generate oligodendroglia. J. Neurosci. Res. 57:435–446. [PubMed] [Google Scholar]
- Ludwin, S.K. 1990. Oligodendrocyte survival in Wallerian degeneration. Acta Neuropathol. (Berl). 80:184–191. [DOI] [PubMed] [Google Scholar]
- McLean, I.W., and P.K. Nakane. 1974. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J. Histochem. Cytochem. 22:1077–1083. [DOI] [PubMed] [Google Scholar]
- McPhilemy, K., L.S. Mitchell, I.R. Griffiths, S. Morrison, A.W. Deary, I. Sommer, and P.G.E. Kennedy. 1990. Effect of optic nerve transection upon myelin protein gene expression by oligodendrocytes: evidence for axonal influences on gene expression. J. Neurocytol. 19:494–503. [DOI] [PubMed] [Google Scholar]
- Nave, K.-A., C. Lai, F.E. Bloom, and R.J. Milner. 1987. Splice site selection in the proteolipid protein (PLP) gene transcript and primary structure of the DM-20 protein of central nervous system myelin. Proc. Natl. Acad. Sci. USA. 84:5665–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama, A., A.S. Chang, and B.D. Trapp. 1999. NG2+ glial cells: a novel glial cell population in the adult brain. J. Neuropathol. Exp. Neurol. 58:1113–1124. [DOI] [PubMed] [Google Scholar]
- Pringle, N.P., N.L. Nadon, D.M. Rhode, W.D. Richardson, and I.D. Duncan. 1997. Normal temporal and spatial distribution of oligodendrocyte progenitors in the myelin-deficient (md) rat. J. Neurosci. Res. 47:264–270. [DOI] [PubMed] [Google Scholar]
- Rosenfeld, J., and V.L. Friedrich, Jr. 1986. Oligodendrocyte production and myelin recovery in heterozygous jimpy mice: an autoradiographic study. Int. J. Dev. Neurosci. 4:179–187. [DOI] [PubMed] [Google Scholar]
- Schneider, A., P. Montague, I.R. Griffiths, M.L. Fanarraga, P.G.E. Kennedy, P.J. Brophy, and K.-A. Nave. 1992. Uncoupling of hypomyelination and glial cell death by a mutation in the proteolipid protein gene. Nature. 358:758–761. [DOI] [PubMed] [Google Scholar]
- Sivakumar, K., N. Sambuughin, B. Selenge, J.W. Nagle, D. Baasanjav, L.D. Hudson, and L.G. Goldfarb. 1999. Novel exon 3B proteolipid protein gene mutation causing late-onset spastic paraplegia type 2 with variable penetrance in female family members. Ann. Neurol. 45:680–683. [PubMed] [Google Scholar]
- Skoff, R.P., and M.S. Ghandour. 1995. Oligodendrocytes in female carriers of the jimpy gene make more myelin than normal oligodendrocytes. J. Comp. Neurol. 355:124–133. [DOI] [PubMed] [Google Scholar]
- Skoff, R.P., and I.N. Montgomery. 1981. Expression of mosaicism in females heterozygous for jimpy. Brain Res. 212:175–181. [DOI] [PubMed] [Google Scholar]
- Sternberger, L.A., P.H. Hardy, Jr., J.J. Cuculis, and H.G. Meyer. 1970. The unlabelled antibody-enzyme method of immunocytochemistry. Preparation and properties of soluble antigen-antibody complex (horseradish peroxidase-antihorseradish peroxidase) and its use in identification of spirochetes. J. Histochem. Cytochem. 18:315–333. [DOI] [PubMed] [Google Scholar]
- Tan, S.-S., B. Faulkner-Jones, S.J. Breen, M. Walsh, J.F. Bertram, and B.E. Reese. 1995. Cell dispersion patterns in different cortical regions studied with an X-inactivated transgenic marker. Development. 121:1029–1039. [DOI] [PubMed] [Google Scholar]
- Thomson, C.E., T.J. Anderson, M.C. McCulloch, P.J. Dickinson, D.A. Vouyiouklis, and I.R. Griffiths. 1999. The early phenotype associated with the jimpy mutation of the proteolipid protein gene. J. Neurocytol. 28:207–221. [DOI] [PubMed] [Google Scholar]
- Trapp, B.D., Y. Itoyama, N.H. Sternberger, R.H. Quarles, and H. Webster. 1981. Immunocytochemical localization of Po protein in Golgi complex membranes and myelin of developing rat Schwann cells. J. Cell Biol. 90:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela, J.M., B. González, and B. Castellano. 1998. Understanding glial abnormalities associated with myelin deficiency in the jimpy mutant mouse. Brain Res. Brain Res. Rev. 26:29–42. [DOI] [PubMed] [Google Scholar]
- Vouyiouklis, D.A., J.A. Barrie, I.R. Griffiths, and C.E. Thomson. 2000. A proteolipid protein-(PLP)-specific pre-mRNA contains intron 3 and is upregulated during myelination in the CNS. J. Neurochem. 74:940–948. [DOI] [PubMed] [Google Scholar]
- Werner, H., M. Klugmann, M. Jung, M. Sereda, I.R. Griffiths, and K.-A. Nave. 1998. Mouse models of myelin diseases. Brain Pathol. 8:771–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yool, D.A., J.M. Edgar, P. Montague, and S. Malcolm. 2000. The proteolipid protein gene and myelin disorders in man and animal models. Hum. Mol. Genet. 9:987–992. [DOI] [PubMed] [Google Scholar]
- Yool, D.A., M. Klugmann, M. McLaughlin, D.A. Vouyiouklis, L. Dimou, J.A. Barrie, M.C. McCulloch, K.-A. Nave, and I.R. Griffiths. 2001. Myelin proteolipid proteins promote the interaction of oligodendrocytes and axons. J. Neurosci. Res. 63:151–164. [DOI] [PubMed] [Google Scholar]