

Abstract
A strategy is outlined for obtaining the free energy of a typical designed heteropolymer. The design procedure considers the probability that the target conformation is occupied in comparison with all the other conformations that could house the given sequence. Numerical calculations on lattice heteropolymer models are presented to illustrate the key physical principles.
Protein folding is an important example of the general class of problems involving conflicting constraints and, thence, a rugged energy landscape (1–6). The microscopic approaches of polymer theory and the results obtained in the study of spin glasses potentially add up to a powerful framework for the study of a variety of systems including proteins, polyampholytes, imprinted copolymers, and gels (1–13). Recently, many papers have been published that have attempted to use this framework for a sophisticated, analytic study of the phase diagram of designed heteropolymers (7, 14–19).
The functionality of a protein is mainly controlled by its structure in its native state (commonly assumed to be its ground state). An optimal tailoring of the structure of the native state of a protein by altering its amino acid sequence will enable the creation of proteins with desired functionality and will have applications in drug design. In general, a randomly chosen sequence will not have protein-like properties of a thermodynamically stable native state and rapid kinetic accessibility to it. The evolution of naturally occurring proteins with useful functionality and with native structures that are stable against mutations and small changes in solvent properties is the hallmark of a selection procedure or a design process.
The original idea for protein design (20–23) consists of running through sequences of amino acids to determine which sequence (or sequences) has the lowest energy in a target conformation. In this approach, a constraint is usually placed on the composition of amino acids in the test sequences, to avoid populating a sequence with just those amino acids that are most attractive to each other.
This idea has been formalized by noting that the probability for a particular sequence s to occur in a specific target conformation Γ is proportional to
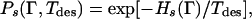 |
1 |
where Hs(Γ) is the energy of the sequence s in conformation Γ, and Tdes is a temperature at which the design is thought to occur. When Tdes = 0, the design is thought to be perfect and the sequence (or sequences) with the lowest energy in the target conformation are chosen, whereas for an infinite value of Tdes, there is no design at all and all sequences have the same Ps corresponding to what is known as the random heteropolymer case. In this problem, the annealed variables are the conformations, whereas the role of quenched random variables is played by the sequences.
We note that Eq. 1 is merely an approximation that requires modification to carry out the selection procedure for protein-like heteropolymers rigorously. Ps is affected by the probability of the sequence to be in other competing conformations and, thus, the design should maximize the relative probability of the sequence being in the target conformation. Such a correct design procedure thins out the competing low-lying energy states thereby inducing a funnel topography in the energy landscape (24, 25).
The free energy of a given sequence s at temperature T is given by
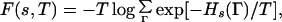 |
2 |
where the sum is over all the conformations of a self-avoiding walk.
The free energy of a typical random heteropolymer at a temperature T is obtained by averaging over the free energies of all sequences (with equal weight or corresponding to the infinite Tdes limit) (26):
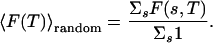 |
3 |
This equation was generalized (9, 14–19) to designed sequences by postulating that the ensemble-averaged free energy was obtained as a weighted average over all sequences and given by
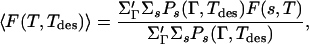 |
4 |
where the primed sum is over selected conformations that house the designed sequences. In refs. 14–19, these conformations were selected to be the compact ones.
To obtain the quenched average involving the logarithm in Eq. 2, one introduces n replicas. Further, because the sum over conformations also appears in Eq. 4, one needs yet another replica that couples to the previous n replicas after the summation over sequences which are the quenched variables.
In summary, the conventional analysis attempts to interpolate between two limits: a trivial one at infinite Tdes, in which all sequences have equal weight and no selection procedure is employed, and the second at Tdes = 0, in which only certain special sequences contribute. The goal is to have these special sequences be ones that are protein-like in the sense of having large thermodynamic stability. From Eq. 1, in the Tdes = 0 limit, nonzero weights are assigned only to those sequences whose ground-state energies have the lowest value among the ground-state energies of all sequences. Such sequences do not necessarily correspond to ones that are thermodynamically stable, but instead may very well be characterized by high degeneracies or low-lying excited states, thus making them physically uninteresting. As a consequence, the conventional analysis possibly may not interpolate between physically relevant limits.
We now proceed to the rigorous way of implementing this problem. Eqs. 2 and 4 are correct, whereas Eq. 1 will be replaced. It is still true that the free energy of a typical random heteropolymer at a temperature T is obtained by averaging over the free energies of all sequences (with equal weight or corresponding to the infinite Tdes limit) (25). The key error in the analysis is Eq. 1, which should be replaced by
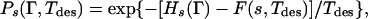 |
5 |
where F(s, Tdes) is defined in Eq. 2. Physically, Eq. 4 arises from the observation that the probability that a sequences is in the conformation Γ at a temperature Tdes, depends not only on the energy of the sequence in the conformation, but also involves the partition function in the denominator as a normalization (27–30). Thus, it does not suffice to merely consider the target conformation energy but the probability that the target conformation is occupied in comparison with all the other conformations that could house the given sequence. The correct procedure is clearly more cumbersome than the previous approaches and will entail the introduction of more replicas.
It is important to note that Eq. 1 is a special case of the correct Eq. 5, when the free energies of the protein-like sequences are essentially the same independent of sequence, i.e., self-averaging. It would be interesting to assess whether the free energies of protein-like sequences do become sequence-independent in the thermodynamic limit.
To illustrate the difference between the sequences selected by the two procedures, one based on Eq. 1 and the other on Eq. 5, we studied some representative quantities of an ensemble of sequences with the aid of numerical calculations with Tdes set equal to zero.
We begin by studying the behavior of an effective order parameter defined as
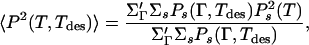 |
6 |
where
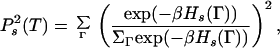 |
the unprimed sum is over all conformations and the primed sum is again over selected target conformations that house the designed sequences. The selected conformations in Eq. 1 are the maximally compact ones (conformations having the largest number of contacts), whereas those in Eq. 5 are the good conformations—conformations that are the unique native state of at least one sequence (some maximally compact conformations may be good as also ones that are not maximally compact).
Physically, as T → ∞, one expects that any sequence will have an equal probability to be in any of the numerous conformations available, thus making the order parameter small. In the T → 0 limit, if the selected sequences have a nondegenerate ground state, then the order parameter approaches 1. If the selected sequences have degenerate ground states with a degeneracy g(s), then 〈P2(0, 0)〉 is less than 1 and given by
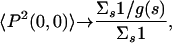 |
7 |
where the sum is over these sequences.
Let us now specialize to the HP (hydrophobic H and polar P) model introduced by Lau and Dill (31). It has been demonstrated (4, 31) that the properties of real proteins are mimicked reasonably well by those of chains of N beads made of only two types of amino acids (hydrophobic H and polar P) with the conformational space consisting of all self-avoiding walks on a two-dimensional square lattice. The advantage of the model is that, for moderate values of N, one may exactly enumerate both the sequences and the conformations. The interaction energies between the two types of amino acids are set to the values ɛHH = −1, ɛHP = 0, and ɛPP = 0.
Our numerical calculations using the HP model for chains with N = 9 and N = 12 are summarized in Fig. 1. The two sets of data represented by circles were determined using Eq. 5 for Ps(Γ, 0), whereas the two sets of data represented by triangles were determined using Eq. 1 for Ps(Γ, 0). Strikingly, the behavior at low temperature is qualitatively different in the two cases. For the HP model, the great majority of sequences with the ground state in maximally compact conformations with the lowest possible energy are degenerate with g(s) that increases with the length N of the chain. This accounts for the low value of the order parameter as measured from Eq. 1. Furthermore, the size dependence (from Fig. 1) is also different for the two cases, with Eq. 5 leading to the correct behavior.
Figure 1.

Plot of the order parameter 〈P2〉 versus T at Tdes = 0 for the HP model applied to chains of N = 9 and N = 12 beads. The number 1 or 5 in parentheses denotes the use of the corresponding equation in the text.
For the HP model, one may define protein-like sequences as those that have a unique ground-state conformation (and an associated energy gap between the ground state and the first excited state that is at least 1). Fig. 2 shows a histogram of the ground-state energies of the 1,569 such sequences for N = 16. The key point is that the selection procedure based on Eq. 1 would pick out all sequences (including the trivial HHHHHHHHHHHHHHHH sequence) that have a ground-state energy of −9 (corresponding to a maximally compact conformation with the maximum number of 9 HH possible contacts) irrespective of the ground-state degeneracy. The sequences thus selected would not be representative of the 1,569 protein-like sequences, which have a range of ground-state energies.
Figure 2.

The histogram of the ground-state energies of the 1,569 good sequences of 16 beads with two types of amino acids (HP model) on a square lattice.
To assess the role of the number of types of amino acids in possibly removing the degeneracy, we now proceed to consider the N = 16 model, but with 4 and then 8 types of amino acids. The first model consisted of an ensemble of chains with 16 beads made of 4 types of amino acids (H1, H2, P1, P2) mounted on all possible 802,075 two-dimensional conformations. Each location of the chain was assigned an amino acid selected at random with equal probability and the interaction energy matrix was taken to be
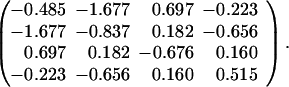 |
(The results are qualitatively the same for other sets of interaction parameters). The corresponding matrix for the 8 amino acid model (H1, H2, H3, H4, P1, P2, P3, P4) was
Approximately 60% of the former sequences have a unique ground state, whereas this number increases to 76% for the case with 8 amino acids.
Two measures (2, 6, 20, 33, 34) of the thermodynamic stability of a sequence in its native state are the energy gap, defined as the difference between the first excited state and the native state energies, and the z-score zs given by
 |
8 |
Here, 〈E〉 and σ are the average energy of a sequence s over all alternative conformations and the corresponding SD, respectively. E0 represents the ground-state energy of that sequence.
For each sequence with 16 beads the alternative conformations were taken to be all conformations with 6, 7, 8, and 9 contacts (a total of 30,169 conformations), but the native one. The graphs of the ground-state energies of these sequences versus the corresponding energy gaps (Fig. 3) indicate a broad distribution of ground-state energies. Notably, protein-like heteropolymers with a high thermodynamic stability characterized by large energy gaps do not necessarily have the lowest ground-state energy.
Figure 3.

(A) The plot of the ground-state energies of 1,146 randomly picked good sequences of 16 beads with 4 types of amino acids versus the energy gaps. (B) The plot of the ground-state energies of 1,517 randomly picked good sequences of 16 beads with 8 types of amino acids versus the energy gaps.
We turn now to a three-dimensional lattice model that has been considered standard for heteropolymer freezing studies (7). The sequences have 27 beads made up of all 20 types of amino acids and the space of conformations is restricted to the 103,346 maximally compact conformations that fit on a 3 × 3 × 3 lattice. Such a situation is expected to occur in this coarse-grained model of a protein when there is an overall attractive interaction between the amino acids. Each location of the chain was assigned an amino acid generated according to its frequency of occurrence in nature (35) and the 210 interaction energies between the amino acids were taken from table 3 of Miyazawa and Jernigan (36). For such a model, the great majority of sequences (approximately 90%) have nondegenerate ground states, so that a protein-like sequence might be defined as one having a thermodynamically stable ground state. Fig. 4A represents a plot of the ground-state energies of good sequences versus their energy gaps, and Fig. 4B is a graph of the ground-state energies versus zs. Here, as alternative conformations for each sequence, we took all maximally compact conformations but the native one. There are two notable features: first, the sequences having the lowest value of the ground-state energy (which would be the ones selected using Eq. 1) do not have the highest energy gap or zs; second, even if these could be considered protein-like sequences, there are many other equally good sequences that are not taken into account by Eq. 1 simply because their ground-state energies are not equal to the lowest one in the ensemble.
Figure 4.

(A) The plot of the ground-state energies of 2,012 randomly picked good sequences of 27 beads with 20 types of amino acids versus the energy gaps. (B) The plot of the ground-state energies of the same sequences as in A versus zs.
We conclude with some general observations. First, the rigorous approach does not require any constraints on the composition of amino acids. Second, an improper modification of Eq. 4 by allowing the sum over Γ to extend over all conformations would lead to a wrong result in which the average free energy 〈F〉 becomes trivially independent of Tdes because ΣΓ Ps(Γ, Tdes) = 1. Third, as pointed out before, it is useful to consider the Tdes = 0 case explicitly. There are two possible scenarios for the ground state of typical sequences. For simple models, such as the HP model of Lau and Dill (31), some sequences have a unique ground state, whereas most of them have degenerate ground states. The more generic situation is one in which, because of 20 kinds of amino acids and a more realistic interaction matrix, virtually each sequence has a unique ground state. However, the “well-designed” sequences have high thermodynamic stability with a small density of low-lying excited energy states. In such a scenario, for small values of Tdes, one obtains a sensible 〈F〉 as an average over predominantly those sequences that have a stability gap larger than or equal to Tdes. The order of taking limits of Tdes → 0 and the system size going to ∞ do not commute and the correct approach would be to allow Tdes → 0 after the thermodynamic limit is taken. Indeed, if one were to consider Tdes = 0 explicitly for finite systems, one would get the same result as for Tdes = ∞.
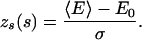
Acknowledgments
We are indebted to Sasha Grosberg and Vijay Pande for useful correspondence. This work was supported by Istituto Nazionale di Fisica Nucleare (Italy), Komitet Badan Naukowych Grant 2P03B-025-13, National Aeronautics and Space Administration, North Atlantic Treaty Organization, and the Petroleum Research Fund administered by the American Chemical Society.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Wolynes P G, Onuchic J N, Thirumalai D. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 2.Bryngelson J D, Onuchic J N, Socci J N, Wolynes P G. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 3.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dill K A, Bromberg S, Yue S, Fiebig K, Yee K M, Thomas D P, Chan H S. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camacho C J, Thirumalai D. Proc Natl Acad Sci USA. 1993;90:6369–6372. doi: 10.1073/pnas.90.13.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klimov D K, Thirumalai D. Phys Rev Lett. 1996;76:4070–4073. doi: 10.1103/PhysRevLett.76.4070. [DOI] [PubMed] [Google Scholar]
- 7.Pande, V. S., Grosberg, A. Y. & Tanaka, T. (1999) Rev. Mod. Phys., in press.
- 8.Wolynes P G. Proc Natl Acad Sci USA. 1997;94:6170–6175. doi: 10.1073/pnas.94.12.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plotkin S S, Wang J, Wolynes P G. J Chem Phys. 1997;106:2932–2948. [Google Scholar]
- 10.Wang J, Plotkin S S, Wolynes P G. J Phys France I. 1997;7:395–421. [Google Scholar]
- 11.Derrida B. Phys Rev Lett. 1980;45:79–82. [Google Scholar]
- 12.Pande V S, Grosberg A Y, Joerg C, Tanaka T. Phys Rev Lett. 1996;76:3987–3990. doi: 10.1103/PhysRevLett.76.3987. [DOI] [PubMed] [Google Scholar]
- 13.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 14.Ramanathan S, Shakhnovich E I. Phys Rev E. 1994;50:1303–1312. doi: 10.1103/physreve.50.1303. [DOI] [PubMed] [Google Scholar]
- 15.Pande V S, Grosberg A Y, Tanaka T. Proc Natl Acad Sci USA. 1994;91:12976–12979. doi: 10.1073/pnas.91.26.12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pande V S, Grosberg A Y, Tanaka T. Macromolecules. 1995;28:2218–2227. [Google Scholar]
- 17.Pande V S, Grosberg A Y, Tanaka T. J Chem Phys. 1995;103:9482–9491. [Google Scholar]
- 18.Pande V S, Grosberg A Y, Tanaka T. J Phys A. 1995;28:3657–3666. [Google Scholar]
- 19.Pande V S, Grosberg A Y, Tanaka T. Physica D. 1997;107:316–321. [Google Scholar]
- 20.Shakhnovich E I, Gutin A M. Proc Natl Acad Sci USA. 1993;90:7195–7199. doi: 10.1073/pnas.90.15.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pande V S, Grosberg A Y, Tanaka T. J Phys France II. 1994;4:1771–1784. [Google Scholar]
- 22.Pande V S, Grosberg A Y, Tanaka T. J Chem Phys. 1994;101:8246–8257. [Google Scholar]
- 23.Pande V S, Grosberg A Y, Tanaka T. Phys Rev E. 1995;51:3381–3392. doi: 10.1103/physreve.51.3381. [DOI] [PubMed] [Google Scholar]
- 24.Onuchic J N, Wolynes P G, Luthey-Schulten Z, Socci N D. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutin A M, Abkevich V I, Shakhnovich E I. Proc Natl Acad Sci USA. 1995;92:1282–1286. doi: 10.1073/pnas.92.5.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shakhnovich E I, Gutin A. Biophys Chem. 1989;34:187–199. doi: 10.1016/0301-4622(89)80058-4. [DOI] [PubMed] [Google Scholar]
- 27.Kurosky T, Deutsch J M. J Phys A. 1995;28:1387–1393. [Google Scholar]
- 28.Deutsch J M, Kurosky T. Phys Rev Lett. 1996;76:323–326. doi: 10.1103/PhysRevLett.76.323. [DOI] [PubMed] [Google Scholar]
- 29.Mirny L A, Shakhnovich E I. J Mol Biol. 1996;264:1164–1179. doi: 10.1006/jmbi.1996.0704. [DOI] [PubMed] [Google Scholar]
- 30.Seno F, Vendruscolo M, Maritan A, Banavar J R. Phys Rev Lett. 1996;77:1901–1904. doi: 10.1103/PhysRevLett.77.1901. [DOI] [PubMed] [Google Scholar]
- 31.Lau K F, Dill K A. Macromolecules. 1989;22:3986–3997. [Google Scholar]
- 32.Chan H S, Dill K A. Phys Today. 1993;(Feb.):24–32. [Google Scholar]
- 33.Goldstein R A, Luthey-Schulten Z A, Wolynes P G. Proc Natl Acad Sci USA. 1992;89:4918–4922. doi: 10.1073/pnas.89.11.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowie J U, Luthy R, Eisenberg D. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 35.Creighton T E. Proteins: Structures and Molecular Properties. New York: Freeman; 1993. p. 4. [Google Scholar]
- 36.Miyazawa S, Jernigan R. J Mol Biol. 1996;256:623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]