

Abstract
Calcineurin-dependent pathways have been implicated in the hypertrophic response of skeletal muscle to functional overload (OV) (Dunn, S.E., J.L. Burns, and R.N. Michel. 1999. J. Biol. Chem. 274:21908–21912). Here we show that skeletal muscles overexpressing an activated form of calcineurin (CnA*) exhibit a phenotype indistinguishable from wild-type counterparts under normal weightbearing conditions and respond to OV with a similar doubling in cell size and slow fiber number. These adaptations occurred despite the fact that CnA* muscles displayed threefold higher calcineurin activity and enhanced dephosphorylation of the calcineurin targets NFATc1, MEF2A, and MEF2D. Moreover, when calcineurin signaling is compromised with cyclosporin A, muscles from OV wild-type mice display a lower molecular weight form of CnA, originally detected in failing hearts, whereas CnA* muscles are spared this manifestation. We also show that OV-induced growth and type transformations are prevented in muscle fibers of transgenic mice overexpressing a peptide that inhibits calmodulin from signaling to target enzymes. Taken together, these findings provide evidence that both calcineurin and its activity-linked upstream signaling elements are crucial for muscle adaptations to OV and that, unless significantly compromised, endogenous levels of this enzyme can accommodate large fluctuations in upstream calcium-dependent signaling events.
Keywords: calmodulin, MEF2, NFAT, hypertrophy, calcium signaling
Introduction
Skeletal muscle fibers adapt to higher contractile loads by increasing their size and by expressing slower isoforms of muscle contractile proteins (Dunn and Michel 1997). Calcineurin, a Ca2+/calmodulin(CaM)-dependent phosphatase, appears crucial in the signaling of this adaptive response, since functional overload-induced fiber hypertrophy and fiber type transformations are prevented in vivo by administration of the specific calcineurin inhibitors cyclosporin A (CsA) and FK506 (Dunn et al. 1999). Calcineurin is likely activated in overloaded muscles via the chronic increases in intracellular calcium that occur under these conditions (Panchenko et al. 1974) as a result of a doubling of nerve-mediated muscle fiber activation (Gardiner et al. 1986) and load-related increases in insulin-like growth factor (Adams and Haddad 1996; Musaro et al. 1999; Semsarian et al. 1999). Once activated, calcineurin may signal downstream to genes involved in regulating muscle fiber size and myofibrillar protein phenotype via dephosphorylation of its substrate transcription factors, nuclear factor of activated T cells (NFAT) and myocyte enhancer factor 2 (MEF2) (Chin et al. 1998; Musaro et al. 1999; Semsarian et al. 1999; Wu et al. 2000).
Recently, a 10-fold overexpression of transcripts encoding a constitutively active form of the catalytic subunit of calcineurin (CnA*) has been shown to promote fast-to-slow fiber type transformations, but not fiber hypertrophy, in skeletal muscles of transgenic (Tg) mice under normal weightbearing conditions (Naya et al. 2000). Though these data corroborate original findings in cell culture with respect to the influence of calcineurin on slow fiber–specific genes (Chin et al. 1998), they suggest that upstream signaling effectors of this enzyme (i.e., nerve-mediated muscle activation and mechanical loading, insulin-like growth factor, etc.) are prerequisite to the initiation of muscle fiber growth. In support of the latter notion, retroviral-mediated gene transfer of CnA* has been shown to induce differentiation of skeletal myocytes only in the presence of extracellular calcium, the efficacy of which is dose dependent (Friday et al. 2000). This, together with reports that calcineurin acts in synergy with other calcium-dependent enzymes to activate the expression of gene targets in both muscle cells and T lymphocytes (O'Keefe et al. 1992; Wu et al. 2000), supports the contention that additional pathways may be involved in signaling muscle fiber growth (Murgia et al. 2000).
We thus tested the hypothesis that Tg mice expressing CnA* would display muscle fiber growth and more extensive fast-to-slow fiber type conversions when upstream signaling effectors of this enzyme are activated by functional overload (OV). We also investigated whether interference of Ca2+/CaM signaling would mimic the effects of calcineurin inhibitors (Dunn et al. 1999) and prevent these OV-induced fiber adaptations. To this end, Tg mice were generated that expressed either a transgene that encoded CnA* (O'Keefe et al. 1992), or a peptide (CaMBP) that binds Ca2+/CaM complexes and inhibits their signaling to CaM-dependent enzymes (Wang et al. 1995). We show that despite 3-fold higher calcineurin activity and enhanced dephosphorylation of its targets, NFATc1, MEF2A, and MEF2D, a 10-fold overexpression of CnA* mRNA did not influence fiber size nor fiber type proportions under normal contractile conditions, but matched the upstream functional requirements related to OV by displaying a similar doubling in size and slow fiber number as wild-type (WT) counterparts. Moreover, when calcineurin signaling was compromised with CsA, overexpression of CnA* spared overloaded muscles the appearance of a lower relative molecular weight form of this protein that was first identified in failing human hearts (Tsao et al. 2000). Finally, we show that OV-induced hypertrophy and fast-to-slow contractile protein type transitions were prevented in fibers expressing the CaMBP transgene. These results emphasize that calcineurin is a crucial signaling element in the induction of skeletal muscle fiber hypertrophy and fiber type conversions in response to increased contractile loading and show that this enzyme requires its upstream activators to effectively signal this adaptive response.
Materials and Methods
Tg Mice and Genotyping
CnA* Tg mice (C57/BL6 strain) were generated by overexpressing CnA* cDNA (O'Keefe et al. 1992) linked to either the muscle creatine kinase (MCK) promoter (Sternberg et al. 1988) or the myosin light chain (MLC)1f/3f promoter (Rosenthal et al. 1989). CaMBP Tg mice (ICR strain) were generated by overexpressing a CaMBP (Wang et al. 1995) linked to the human troponin I slow (TnIs) promoter (Corin et al. 1994). Transgenes were linearized and separated from bacterial vectors by electrophoresis and electroelution. Tg mice were generated by pronuclear injection of the transgene into fertilized embryos of mice using standard procedures (Hogan et al. 1994). Surrogate mothers gave birth to founder lines of Tg mice. Positive founders were identified and then crossed with WT mice of the same strain to generate F1 and F2 progeny. Stable lines of mice were generated from Tg offspring of F1 and F2 lines.
Founder Tg mice were identified by Southern blot analysis and their genotype confirmed using PCR screening. PCR was used to genotype subsequent generations of mice. Genomic DNA extracted from tail samples was used for these purposes. For PCR screening of CnA* Tg mice, genomic DNA was amplified using a sense primer specific for CnA* cDNA and an antisense primer specific for the SV-40 polyA of this transgene. A similar approach was used to screen CaMBP Tg mice. PCR reaction mixtures contained 0.5 μg of DNA, 10 mM dNTP, 200 ng of each primer, 1.5 mM MgCl2, PCR buffer, and 5 U of Taq polymerase (Qiagen Inc.). Cycling conditions were: 94°C for 5 min followed by 25 cycles of 30 s at 94°C, 1 min at 50°C, and 1 min at 72°C followed by a final 10-min extension at 72°C.
Northern Blot Analysis of CnA mRNA Expression
RNA was isolated from white gastrocnemius muscle using tripure isolation reagent (Boehringer) and resuspended in diethyl pyrocarbonate (DEPC)-H20. RNA (15 μg) was separated on a formaldehyde-2% agarose gel and transferred to a nylon membrane by capillarization. Membranes were probed with a 400-bp cDNA fragment from the CnA* transgene or a 340-bp cDNA fragment of the CaMBP transgene, labeled with [α32P] dATP and dCTP. Skeletal actin served as a loading control. Bands were detected using autoradiography.
RT-PCR and Calcineurin Phosphatase Assay
RNA was extracted from the distal portion of each plantaris muscle and 1 μg of total RNA was reverse-transcribed to cDNA (Dunn et al. 1999). cDNAs encoding type I myosin heavy chain (MHC), type IIb MHC, TnIs, troponin I fast (TnIf), and the 28 S rRNA subunit were PCR amplified using primers specific to these mRNA sequences (Dunn et al. 1999). CnAα cDNAs were amplified using primers designed from the mouse mRNA sequence (available at GenBank/EMBL/DDBJ under accession number ACJ05479): sense, 5′-GGTGGCTGGAGATGTCCGAGC-3′ (mRNA positions 65–85) and antisense, 5′-GGTGGTTCTTTGAATCGGTC-3′ (complementary to mRNA positions 721–740). GATA-2 cDNAs were amplified using primers designed from the mouse mRNA sequence (available at GenBank/EMBL/DDBJ under accession number AB000096): sense, 5′-CAACCATCTCGACTCGCAGG-3′ (mRNA positions 301–320) and antisense, 5′-GTGGGTGGGAAGCCGAAGAG-3′ (complimentary to mRNA positions 662–681). PCR reaction mixtures contained 2.5 μl of cDNA, 5 mM each dNTP, 10 pmol of each primer, 1.5 mM MgCl2, PCR buffer, and 0.65 U Taq (QIAGEN). Cycling conditions were: 1 min at 95°C, 1 min at 55°C, and 1 min at 72°C, followed by a final 5-min extension at 72°C. Cycle number was adjusted to permit comparison of PCR products across treatments within the linear phase of amplification. Products were run on 1% agarose–tris boric acid EDTA gels and stained with ethidium bromide.
Total calcineurin phosphatase activity was measured in plantaris homogenates by Isotechnika Inc. using a modification of a previously described assay (Fruman et al. 1992). In brief, tissue samples (∼25 mg) were mechanically disrupted by grinding in liquid nitrogen and then suspended in 200 μl lysis buffer (50 mM Tris, pH 8.0, 1 mM EDTA, 100 μM EGTA, 0.1% β-mercaptoethanol, 1 mM DTT, and 50 μg/ml of aprotinin). Samples were centrifuged and then the supernatant was diluted further with lysis buffer (1:4). An aliquot (30 μl) of each lysate was combined with 60 μl of assay buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 6 mM MgCl2, 500 mM DTT, 100 μg/ml BSA, 750 nM okadaic acid, 100 μM CaCl2, 100 nM CaM, and 15 μM of 32P-labeled–RII subunit of cAMP-dependent protein kinase) and samples were incubated at 30°C in the presence or absence of 1,500 ng/ml of CsA. After 15 min, reactions were stopped with 100 mM KH2PO4/5% trichloroacetic acid. The released inorganic phosphate was isolated using a Dowex cation–exchange column and measured via scintillation counting.
Protein Extraction and Western Blot Analysis
For extraction of whole cell protein, muscle samples (∼25 mg) were homogenized in 150 μl of RIPA buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM PMSF, 10 mM sodium fluoride, 1 mM sodium orthovanadate in PBS, pH 7.4). Homogenates were centrifuged at 20,000 g for 15 min at 4°C to obtain a clarified supernatant.
Plantaris extracts enriched in nuclear proteins were prepared according to the method of Blough et al. 1999. An equivalent amount of muscle tissue (∼25 mg) was homogenized in ice-cold lysis buffer (10 mM Hepes, pH 7.5, 10 mM MgCl2 5 mM KCl, 0.1 mM EDTA, pH 8.0, 0.1% Triton X-100, 1 mM DTT, 0.1 mM PMSF, 2 μg/ml each aprotinin and leupeptin) and centrifuged at 3,000 g for 5 min at 4°C to pellet nuclei and myofibrils. This pellet was then resuspended in 1 ml of ice-cold, high-salt buffer (20 mM Hepes, pH 7.9, 25% glycerol, 500 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, pH 8.0, 0.5 mM DTT, 0.2 mM PMSF, 2 μg/ml each leupeptin and aprotinin) for 30 min and then samples were centrifuged at 3,000 g for 5 min at 4°C. The supernatant, enriched in nuclear proteins, was then concentrated to 75 μl using a 5000 nominal molecular weight limit 4 ml Ultrafree® Filter Unit (Millipore). The protein concentration within each total cell and nuclear extract sample was determined with a Bradford assay using BSA as a standard. Whole cell and nuclear extracts were suspended in 1 vol of 2× electrophoresis sample buffer.
Each whole cell and nuclear sample (50–100 μg) was subjected to 6–12% SDS-PAGE electrophoresis and then transferred to PVDF membrane (Amersham Pharmacia Biotech). Equal loading of samples was verified via Coomassie blue staining of gel proteins and Ponceau S staining of membrane-bound proteins. For Western blotting, membranes were blocked in BLOTTO (5% milk, 0.1% Tween-20 in TBS, pH 8.0) for 1 h and then incubated for an additional hour with primary antibodies diluted in BLOTTO. Membranes were rinsed with 0.1% Tween-20 in TBS and then incubated for 1 h with horseradish peroxidase secondary antibody conjugates diluted in BLOTTO. Bound antibody complexes were developed using the ECL Plus kit and exposed to Hyperfilm ECL-Plus x-ray film (Amersham Pharmacia Biotech).
Antibodies used in this study were raised against: NFATc1 (Affinity Bioreagents, Inc. and Santa Cruz Biotechnology, Inc.), MEF2A (Santa Cruz Biotechnology, Inc. and gift from R. Prywes, Columbia University, New York, NY), MEF2D (gift from R. Prywes), CnAβ (StressGen Biotechnologies), and CnAα (Oxford Biomedical Research Inc. and Santa Cruz Biotechnology, Inc.). The MEF2A and MEF2D antibodies obtained from R. Prywes cross-react with MEF2C and were used to estimate the abundance of this protein in nuclear extracts. The CnA antibody (StressGen Biotechnologies) is raised against an epitope (FLQNNNLLSIIRAHEAQDAG, amino acids 264–283) in the catalytic region of human CnAβ (see Fig. 4 a), which is completely conserved in mouse CnAβ (available at GenBank/EMBL/DDBJ under accession number M81483). Anti-CnAα (Oxford) is raised against an epitope (RMPPRRDAMPSDA, amino acids 482–494) in the COOH terminus of human CnAα, which is fully conserved in mouse CnAα (available at GenBank/EMBL/DDBJ under accession number J05479), and partially conserved in and cross-reacting with mouse CnAβ (8 of 13 residues). Anti-CnAα (Santa Cruz Biotechnology, Inc.) is also raised against an epitope in the COOH terminus of this protein, but does not recognize other CnA isoforms.
Figure 4.
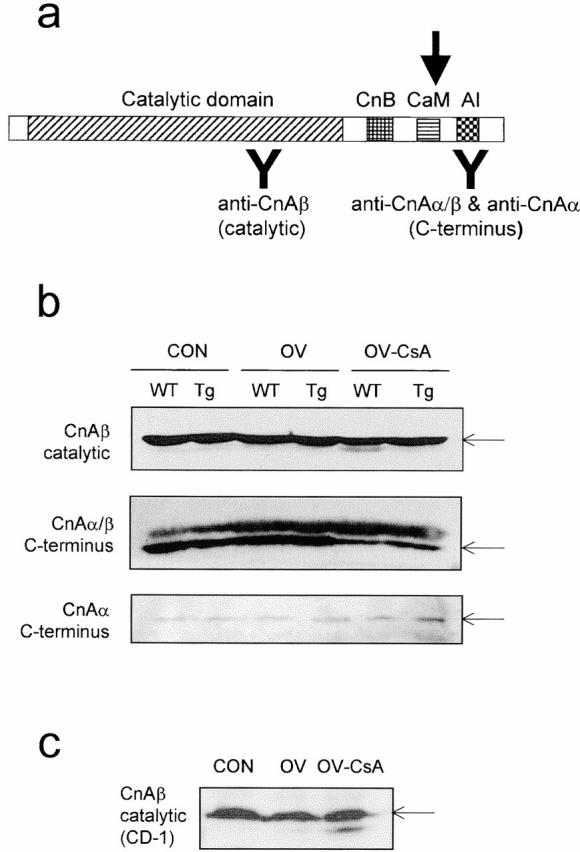
Overexpression of CnA* prevents the appearance of a lower relative molecular weight form of CnA in OV muscles when CnA signaling is compromised with CsA. (a) Cartoon depicting the catalytic, regulatory subunit-binding (CnB), CaM-binding (CaM), and auto-inhibitory (AI) domains of CnA and the location of epitopes recognized by the antibodies used in this study. The filled arrow shows the approximate location of where this protein is cleaved or prematurely terminated. (b) Western blots of plantaris protein extracts from WT or MCK-CnA* Tg mice after CON, OV, or OV plus CsA (OV-CsA) treatments probed with an antibody raised against an epitope in the catalytic region of CnAβ (top) or epitopes in the COOH terminus of CnAα or CnAβ (middle and bottom). The antibody used for Western blots in the middle panel cross-reacts with, and primarily detects CnAβ, the most abundant CnA isoform in skeletal muscle (see Materials and Methods), whereas the CnAα antibody used in the bottom panel recognizes only CnAα. Note that all three isoforms of CnA migrate to the same position on SDS-PAGE gels. The identity of the upper band in the middle panel is not known, but may represent a post-translational modification of CnA. c, Western blot analysis of plantaris extracts from WT mice of the CD-1 strain after the various treatment conditions probed with anti-CnAβ (catalytic). Arrows in b and c denote the size of full length CnAα and β (64 kD).
Functional Overload Surgery and CsA Administration
The plantaris muscle in each hindlimb of WT, CaMBP Tg, and CnA* Tg mice was overloaded by surgically removing the soleus and a major portion of the gastrocnemius muscle (Dunn et al. 1999). Some WT, MCK-, and MLC-CnA* Tg mice were also injected twice daily (12 h apart) with 25 mg/kg CsA (Dunn et al. 1999). This dose of CsA resulted in blood levels of this drug of 2,865 ± 517 ng/ml. Administration of this drug did not affect the health, growth nor noticeably alter the daily amount of locomotor activity displayed by these mice. After a 5-d or 4-wk treatment period, muscles were removed and quick frozen in melting isopentane. All surgical procedures were performed on mice anesthetized with a 100 mg/ml ketamine hydrochloride: 20 mg/ml xylazine cocktail in a volume ratio of (1.6:1). Treatment of animals was in accordance with the guidelines established by the Canadian Council on Animal Care.
Fiber Typing and Cross-sectional Size Measurements
Cryosections (10 μm) were cut from the same anatomical location in each muscle midbelly. Sections were blocked for 1 h in 5% goat serum in PBS and then probed with mouse antibodies specific for MHC type I (BA-F8), IIa (SC-71), IIb (BF-F3), or all MHCs but IIx (BF-35), followed by horseradish peroxidase–conjugated goat anti–mouse IgG or IgM (Dunn and Michel 1997). Antibody complexes were visualized using diaminobenzidine tetrahydrochloride. Fibers that expressed MHC type I, IIa, IIx only, or IIb were identified within three distinct areas of each muscle cross-section. Images of these areas were captured and fiber cross-sectional size measured using a microscope linked to a computer-based image analysis system (Dunn and Michel 1997). Fiber size values for each group were calculated as the mean (n = 3–4 muscles/group) of muscle fiber means (n = 20–60 fibers/muscle section).
Results
Generation of Mice Expressing Functional CnA*
Tg mice were generated that overexpressed CnA* either under the control of the MCK promoter which is active in all muscle fibers, or the fast MLC promoter which is operative only within fast fiber types and in the mouse plantaris represent ∼98% of the total fiber population. CnA* encodes a deletion mutant of the α isoform of murine CnA that lacks the COOH terminus autoinhibitory and CaM-binding domains, and displays constitutive activity in vitro (O'Keefe et al. 1992). Five independently derived founder mice were generated for each construct. For MCK-CnA*, four of five founders transmitted the transgene to the F1 generation and, of these, only one line (5237) showed CnA* expression at the mRNA level (Fig. 1 a). For the MLC-CnA* construct, three of five founders produced Tg offspring and only one line of mice (2276) expressed detectable levels of CnA* mRNA (Fig. 1 a). Tg mice from MCK-CnA* (5237) and MLC-CnA* (2276) lines were used in this study. CnA* mRNA was expressed at levels that were >10-fold higher than endogenous CnA in both 5237 and 2276 lines as assessed by Northern blot or RT-PCR analyses (Fig. 1, a and b). The expression of the transgene also resulted in threefold higher calcineurin phosphatase activity in the plantaris of MCK-CnA* Tg mice (Fig. 1 c), demonstrating that the CnA* transgene was functional in this muscle.
Figure 1.
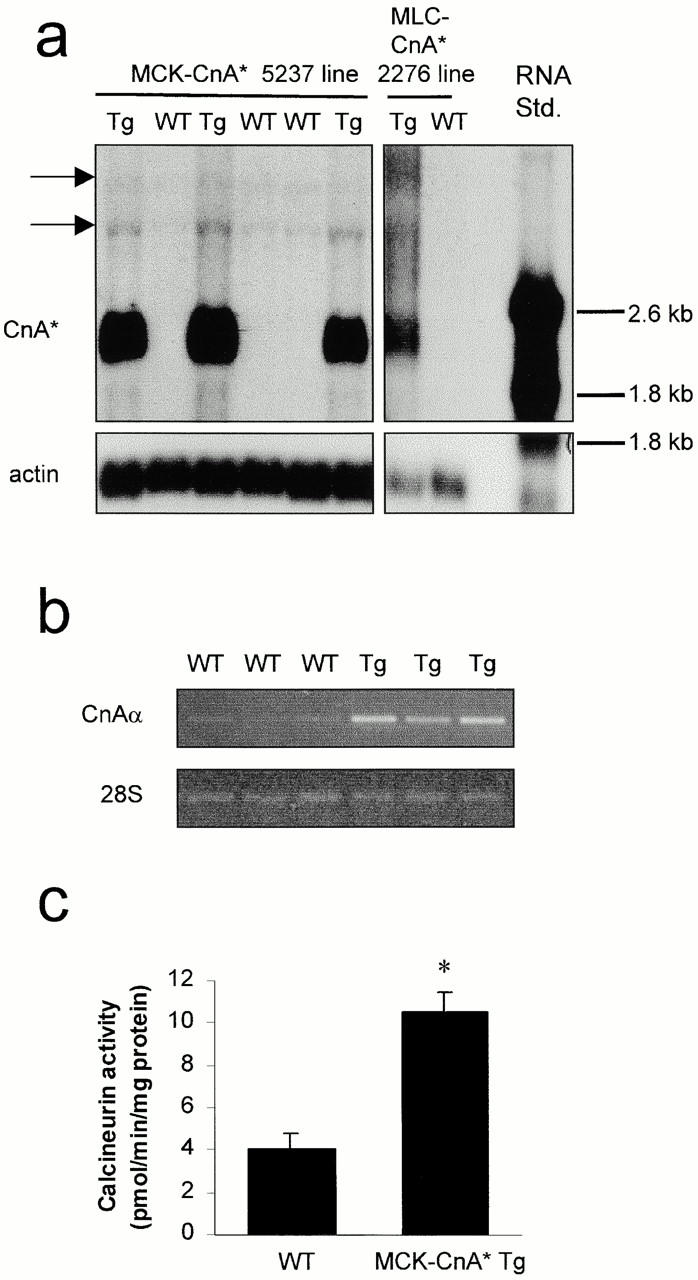
Overexpression of CnA* mRNA increases calcineurin activity in plantaris muscles. (a) Northern blots of CnA* mRNA expression in the plantaris of WT, MCK-, and MLC-CnA*-Tg mice. Loading control was α-skeletal actin. Arrows indicate the size of the 4.0 (upper) and 3.6-kB (lower) endogenous CnA transcripts. Note, the minor endogenous 1.6-kB CnA form is not shown. (b) RT-PCR analysis of total CnAα mRNA (endogenous plus transgene) expression in MCK-CnA* mice. Lanes are ethidium bromide-stained PCR products generated from individual muscle cDNA samples. The 28 S rRNA served as a loading control. (c) Mean ± SE of total calcineurin activity in WT and MCK-CnA* Tg plantaris muscles (n = three muscles/group). The asterisk denotes a difference (P < 0.05) from WT.
CnA* Expression and OV Increase CnA Signaling via NFATc1 and MEF2
Activation of calcineurin in skeletal myocytes has been shown to induce the dephosphorylation and nuclear localization of the NFATc1 transcription factor (Abbott et al. 1998; Chin et al. 1998; Musaro et al. 1999; Semsarian et al. 1999). Thus, to assess the relative influence of the CnA* transgene versus upstream activity-dependent effectors on calcineurin signaling, we examined the phosphorylation status and nuclear abundance of this calcineurin substrate in the plantaris of WT and MCK-CnA* Tg mice subjected to normal weightbearing or functional OV conditions. Consistent with previous findings in skeletal myocytes (Musaro et al. 1999; Semsarian et al. 1999), multiple NFATc1 bands ranging from 85–142 kD were detected on Western blots of plantaris whole cell and nuclear extracts (Fig. 2 a). Pretreatment of protein extracts with alkaline phosphatase produced only two lower molecular weight species of ∼85 and 90 kD, suggesting that the detected NFATc1 bands correspond to the hypophosphorylated (85 and 90 kD) and phosphorylated (>90 kD) forms of possibly two variants of this protein (see Rao et al. 1997). The sole difference in NFATc1 banding pattern between whole cell and nuclear isolates was the absence of the highest molecular weight 142-kD band in the nuclear fraction (Fig. 2 a), consistent with the notion that only a partial dephosphorylation of NFAT is required to uncover its nuclear localization signal and promote its nuclear import (Musaro et al. 1999). Expression of the CnA* transgene induced a subtle dephosphorylation of NFATc1 and an enrichment of this transcription factor in the nuclear fraction of the plantaris of normal weightbearing mice (Fig. 2 a, Con [control], compare lanes WT and Tg). Interestingly, OV alone induced a dephosphorylation of NFATc1 that was similar to that observed in CON MCK-CnA* Tg mice, indicating that the response was not potentiated by the CnA* transgene (Fig. 2 a, compare lanes OV and Con Tg). These effects of OV on NFATc1 in both treatment groups were blocked by administration of CsA (Fig. 2 a, compare lanes OV and OV-CSA). Taken together, these data suggest that CnA* expression and OV each activate calcineurin-dependent signaling in vivo and that the combined effect of these treatments on the dephosphorylation and nuclear translocation of NFATc1 is not additive.
Figure 2.

Effect of CnA* expression, OV, or OV plus CsA treatments on the phosphorylation status of NFATc1 and MEF2. (a) Western blots of NFATc1 in whole cell and nuclear extracts, and MEF2C in nuclear extracts, of WT and MCK-CnA* Tg plantaris muscles after 5 d of the various treatment conditions. Note the banding pattern for NFATc1 was obtained using two distinct antibodies raised against this protein (data not shown). MEF2C served as a loading control for nuclear extracts. (b) Western blot analysis of the total protein content of MEF2D and MEF2A in plantaris muscles of WT and MCK-CnA* Tg mice after 5 d of the various treatments. In a and b, filled arrows denote hypophosphorylated forms of each protein as determined by pretreatment of samples with alkaline phosphatase (+AP). Results in a and b represent the observations of three or more independent experiments.
A series of recent studies provide evidence that calcineurin may also target downstream genes through the dephosphorylation and activation of MEF2 transcription factors (Mao and Wiedmann 1999; Blaeser et al. 2000; Wu et al. 2000). Moreover, and of considerable significance, MEF2 cis-elements have been implicated in the regulation of a number of slow fiber–specific promoters (Chin et al. 1998; Esser et al. 1999). We thus investigated whether proteins of this family are dephosphorylated by calcineurin under MCK-CnA* Tg or OV conditions. We found that overexpression of CnA* alone induced a prominent dephosphorylation of MEF2D and a subtle dephosphorylation of MEF2A in normal weightbearing plantaris muscles as assessed by Western blots (Fig. 2 b). Like NFATc1, the prominent effect of the transgene on MEF2D dephosphorylation appeared to be matched by the effect of OV, with no observable amplification of this response in overloaded mice expressing CnA*. This dephosphorylation of MEF2D under these conditions appeared to be calcineurin-dependent since it was blocked by CsA administration (Fig. 2 b). By far the major influence on MEF2A was a pronounced dephosphorylation and increased abundance with OV, effects that were not potentiated by CnA* and only partly blocked by treatment with CsA (Fig. 2 b). The latter suggests that this transcription factor may act as both substrate and gene target of calcineurin. In contrast, expression of CnA* or OV did not produce any noticeable change in the mobility or abundance of MEF2C (Fig. 2 a), suggesting that this MEF2 isoform is not a target of this phosphatase. Taken together with NFATc1 findings, these data show that expression of CnA* promotes calcineurin signaling via NFATc1, MEF2A, and MEF2D in the plantaris under normal weightbearing conditions. Our data also show the effects of the CnA* transgene on these transcription factors to be largely matched, and in the case of MEF2A surpassed, by the effects of OV.
Calcineurin Requires Its Upstream Effectors to Promote Fiber Type Transitions and Growth
To assess the influence of upstream activity-dependent calcineurin effectors on the determination of the skeletal muscle phenotype, we examined plantaris fiber size and type proportions in WT, MCK-CnA* Tg, and MLC-CnA* Tg mice exposed to normal weightbearing or functional OV conditions. As expected, by 4 wk of OV we noted a doubling of plantaris relative mass (Fig. 3 a) and fiber cross-sectional size (Fig. 3 b) in WT mice. OV-WT plantaris muscles also displayed a characteristic increase in the number of fibers that expressed type I/slow MHC (Fig. 3 c), and, consistent with our previous findings (Dunn et al. 1999), a reciprocal elevation in transcript levels of slow fiber–specific genes (TnIs and I MHC) and decreased expression of fast (TnIf and IIb MHC) myofibrillar genes, compared with CON counterparts (Fig. 3 d).
Figure 3.

Overexpression of CnA* did not potentiate the hypertrophic response of plantaris muscles to OV or induce the expression of GATA-2. (a) Effect of 4 wk of OV on plantaris relative wet weight (MW) (mg/g body weight, BW) in WT and Tg mice. Values are means ± SE (n = 4–9/group). Asterisks denote a difference (P < 0.01) from respective sham control groups. (b) Effect of 4 wk of OV on the ratio of plantaris fiber cross-sectional area (CSA) relative to respective sham control values (broken line) for each fiber type in WT and Tg mice. Fiber CSA was measured in 20–60 fibers/muscle for 3–4 muscles/group. In this histogram, all bars are significantly different (P < 0.05) from respective sham controls which were not different from one another. (c) Effect of 4 wk of OV on the number of slow type fibers in the plantaris of WT and Tg mice. Values are means ± SE (n = 3–4/group). Asterisks denote a difference (P < 0.01) from the respective sham control. (d) RT-PCR analysis of transcript levels for TnIs, type I MHC, TnIf, IIb MHC, and 28 S rRNA in WT and MCK-CnA* Tg tissues after CON and 4 wk of OV. Lanes show ethidium bromide–stained PCR product bands generated from individual muscle cDNA samples. The 28 S rRNA served as a loading control. (e) Western blots of GATA-2 protein and RT-PCR analysis of GATA-2 mRNA expression in WT and MCK-CnA* Tg mice after 5 d of CON, OV, or OV plus CsA treatments. For PCR, lanes represent PCR product bands generated from pooled cDNA samples (n = 2/group). 28S rRNA expression was not different across treatments (not shown).
OV plantaris muscles that overexpressed CnA* (MCK-CnA* and MLC-CnA*) displayed a doubling in muscle mass (Fig. 3 a), fiber size (Fig. 3 b), and slow fiber number (Fig. 3 c), and increased expression of slow fiber–specific genes (Fig. 3 d) that was similar to WT counterparts. To our surprise, overexpression of CnA* did not influence slow fiber number (Fig. 3 c) or the expression of slow fiber–specific genes (Fig. 3 d) in the plantaris under normal contractile conditions, which is in sharp contrast to a recent study reporting fast to slow fiber transitions in mice that overexpressed this same transgene at comparable levels (Naya et al. 2000). Finally, expression of CnA* did not influence satellite cell differentiation, a process reported to be regulated by calcineurin (Friday et al. 2000), since the amount of central-nucleated fibers in histological sections was insignificant (range = 0–3 fibers/muscle section for all three groups) and not different between WT and Tg mice.
In light of the fact that none of the known targets of calcineurin measured above were up-regulated in response to expression of the CnA* transgene alone, we elected to assess the expression levels of GATA-2, another purported target of calcineurin implicated in skeletal myocyte hypertrophy (Musaro et al. 1999), in the plantaris of WT and MCK-CnA* Tg mice after CON and OV conditions. Surprisingly, muscles overexpressing CnA* did not display increased amounts of GATA-2 at the protein or mRNA levels (Fig. 3 e, Con, compare lanes WT and Tg). Moreover, although GATA-2 protein content was increased with OV, levels were even higher after CsA treatment (Fig. 3 e). In contrast to protein findings, GATA-2 mRNA levels were not significantly altered in response to the various treatment conditions (Fig. 3 e). These data suggest that this gene may be important for the OV response, but is not a major target of calcineurin in vivo. Taken together with muscle phenotype data, these findings suggest that calcineurin requires its upstream activity-dependent effectors to activate fast-to-slow fiber conversions and initiate fiber hypertrophy. These results also suggest that the level of nerve-mediated activity or mechanical loading is a better predictor of muscle fiber adaptations than the degree of calcineurin activation in these muscles.
CnA* Expression Prevents Appearance of a Shorter CnA Variant in OV-CsA Muscles
Recently, evidence has been presented that failing human hearts display a shorter, constitutively active, variant of CnA that is very similar to the deletion mutant CnA* harbored in our mice (Tsao et al. 2000). It is proposed that the manifestation of this form may serve as a molecular indicator of the inability of the endogenous calcineurin pool to meet the signaling requirements of contractile activity. Here we show that a lower relative molecular mass form of CnAβ, the most abundant skeletal muscle CnA isoform (Dunn, S., and R.N. Michel, unpublished observations), appears in skeletal muscle of WT mice under conditions of OV stress when calcineurin activity is severely compromised (decreased by 60%) by CsA. This phenomenon was not observed in OV MCK-CnA* Tg mice, suggesting that under these conditions the additional calcineurin activity is available to match upstream activity-related requirements. Using an antibody raised against an epitope in the catalytic domain of CnAβ (Fig. 4 a), we detected the full-length CnA form (64 kD) as well as a band of lower molecular weight on Western blots (Fig. 4 b, top, lane OV-CSA) corresponding to the reported size of a proteolysed form of CnA that displays Ca2+-independent, constitutive activity (Hubbard and Klee 1989). This smaller CnA form is likely the result of a COOH-terminal deletion, since it was not detected, even after long exposures to film, with an antibody that recognizes the COOH terminus of this protein (see Fig. 4, a and b, middle panel, lane OV-CSA WT). This lower relative molecular mass form was also detected in OV-CsA–treated plantaris muscles from another strain of mice (Fig. 4 c). Although the antibody used in the middle panel in Fig. 4 b recognizes both CnAα and CnAβ, the form detected in this Western blot likely corresponds to CnAβ, since the observed pattern of expression across treatments differs from that obtained using another antibody that only recognizes CnAα (Fig. 4 b, bottom, see also Materials and Methods).
CaMBP Expression Prevents OV-related Fiber Hypertrophy and Fast-to-Slow Fiber Conversions
To further explore the role of calcineurin in regulating fiber growth, we generated Tg mice that express a synthetic peptide that binds to, and inhibits the ability of Ca2+/CaM complexes to activate CaM-dependent enzymes (Wang et al. 1995). CaMBP was expressed in skeletal muscles under the control of the TnIs promoter, which restricts expression of this transgene to the slow/I MHC fiber population. Though these fibers constitute only a small contingent (∼2%) of plantaris cells in control mice, they are amongst the smallest and most highly recruited fibers in this muscle (Gardiner et al. 1986), and during OV are amongst the first to undergo a rapid doubling of size and a tripling in their numbers (Fig. 3 c; Dunn et al. 1999). Four independently derived founder mice were generated that expressed the CaMBP transgene and all founders transmitted the transgene to the F1 generation. Of these, only one line (6444) showed high level expression of CaMBP at the mRNA level (Fig. 5 a) and was used for this study. Here we show that in OV animals, expression of CaMBP completely blocked the growth of plantaris slow/type I fibers which harbored the transgene, but not fast/type II, fibers (Fig. 5b vs. c and d), and prevented the characteristic fast-to-slow fiber conversions (slow fiber No. = 24 ± 8, OV CaMBP vs. 59 ± 4, OV WT; P < 0.05). Consistent with our previous findings (Dunn et al. 1999), OV-induced hypertrophy was also prevented across all fiber types when mice were administered CsA (Fig. 5 d). Moreover, CaMBP only influenced the growth and transformation of slow/type I fibers under conditions of overload, since type I fiber size (582 ± 110 μm2) and slow fiber number (28 ± 6) in CON CaMBP plantaris muscles were not different from CON WT counterparts. In the predominantly slow soleus, type I fibers were also not significantly affected by the expression of the CaMBP transgene (data not shown), supporting the notion that calcineurin is of major importance only when contractile loading is increased above normal levels. Taken together, these results emphasize that calcineurin and other Ca+2/CaM-dependent pathways are critical for the signaling of muscle fiber growth and fast-to-slow conversions in response to increased loads.
Figure 5.
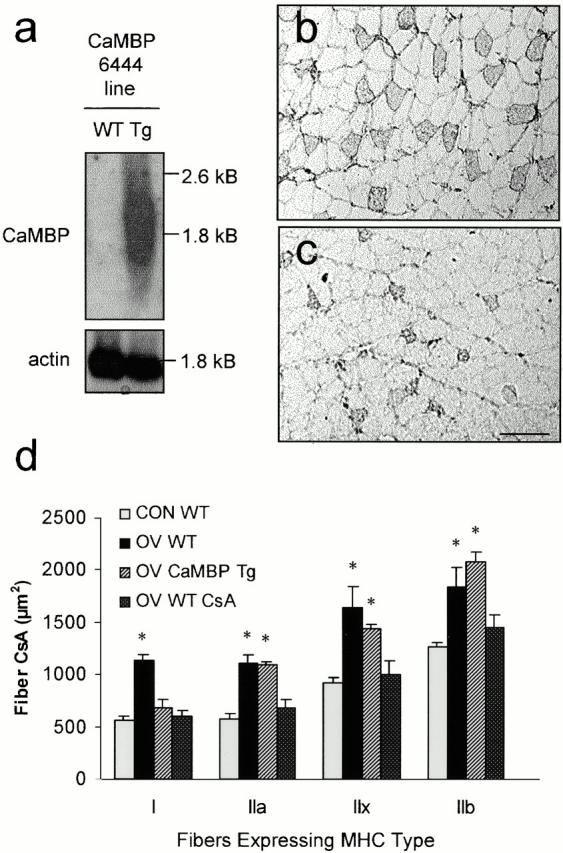
Overexpression of CaMBP in slow fibers prevented hypertrophy of these cells in response to 4 wk of OV. (a) Northern blot analysis of CaMBP mRNA expression in WT and Tg mice. α-skeletal actin served as a loading control. (b and c) Cross-sections from OV WT (b) or OV CaMBP Tg (c) plantaris muscles immunolabeled for type I MHC. Bar, 100 μm. (d) Mean ± SE cross-sectional size of plantaris fibers of CON WT, OV WT, OV CaMBP Tg, and OV WT CsA mice after 4 wk of treatment typed for their expression of the various MHCs. n = 3–4 muscles/group. The asterisks denote differences (P < 0.05) from CON WT.
Discussion
Calcineurin, a Ca2+/CaM-dependent phosphatase, has been implicated in the hypertrophic response of skeletal muscles to increased physiological loads (Dunn et al. 1999). The recent finding that expression of an activated form of Cna in skeletal muscles in vivo is not sufficient to induce fiber hypertrophy (Naya et al. 2000), suggests that the upstream effectors of this enzyme (i.e., activity-related increases in cytosolic Ca2+) are required for the initiation of skeletal muscle growth. Expanding upon this notion, we show that skeletal muscles overexpressing an activated CnA transgene, despite displaying threefold higher calcineurin activity and enhanced dephosphorylation of target substrates, exhibited fiber sizes and fiber type profiles indistinguishable from WT counterparts under normal weightbearing conditions. Moreover, these muscles did not display potentiated fiber growth or more extensive fast-to-slow fiber conversions in response to OV. On the other hand, expression of the CnA* transgene did spare OV-CsA muscles the manifestation of a shorter, constitutively active form of CnA, originally detected in failing human hearts (Tsao et al. 2000). We also show that expression of a peptide preventing CaM from signaling downstream to target enzymes (Wang et al. 1995), mimics the effects of calcineurin inhibitors in blocking OV-induced fiber growth and fast-to-slow fiber conversions. Taken together, these results emphasize that calcineurin, in conjunction with other activation-linked signal transduction events, is important for the initiation of fiber adaptations to increased contractile loads, and suggest that unless significantly compromised, the endogenous pool of this enzyme can accommodate the signaling requirements related to extremes in functional demand.
Transcription Factors Downstream of Calcineurin
Studies of skeletal myocytes have shown that NFAT and MEF2 cis-acting control elements are necessary for the calcineurin-dependent activation of TnIs and myoglobin promoter-linked reporter constructs (Chin et al. 1998). It appears that calcineurin may signal to these target genes via the direct dephosphorylation and transcriptional activation of NFATc1 and MEF2A (Chin et al. 1998; Mao and Wiedmann 1999; Wu et al. 2000). Consistent with this, we show for the first time that NFATc1, MEF2A, and MEF2D transcription factors are dephosphorylated, and MEF2A protein increased, in a calcineurin-dependent fashion in mature skeletal muscles in vivo in response to either CnA* expression or OV. Of these, MEF2D appears to be the preferred target of calcineurin in skeletal muscle since the dephosphorylation of this protein was more complete than either MEF2A or NFATc1 under these conditions. For the most part, the effect of OV on the dephosphorylation of these transcription factors was largely matched and never potentiated by the effect of the CnA* transgene. This combined with the finding that both dephosphorylation and abundance of MEF2A were markedly greater with OV, suggests that by far, muscle activity-linked signal transduction elements are major determinants of the signaling flux via calcineurin.
The fact that a gross change in phosphorylation status was observed for MEF2A and MEF2D, but not MEF2C, in response to CnA* expression or OV, suggests that only a subset of MEF2 isoforms (i.e., MEF2A and MEF2D) are substrates of calcineurin. This notion is supported by the finding that MEF2A and 2D are more effective than other MEF2 isoforms in enhancing the calcineurin-dependent activation of a slow-fiber-specific enhancer (Wu et al. 2000). Moreover, a MEF2 site implicated in the calcineurin-responsiveness of the Nur77 promoter in T lymphocytes is shown to bind only MEF2A and MEF2D proteins (Blaeser et al. 2000). Although the location of the calcineurin Ser/Thr dephosphorylation site(s) on MEF2A and MEF2D is not known, findings that the dephosphorylation of MEF2 by calcineurin in myocytes and T lymphocytes increases its transcriptional activity without affecting its DNA binding (Woronicz et al. 1995; Wu et al. 2000), suggest that such a site(s) may lie in a transactivation domain conserved in both of these proteins (Black and Olson 1998). Our MEF2 dephosphorylation findings thus concur with data showing increased MEF2-dependent transcription in the fast-twitch EDL under normal weight-bearing conditions in Tg mice harboring this same MCK-CnA* construct (Wu et al. 2000).
Matching of Calcineurin Activity to Upstream Contractile-dependent Signals
Despite the fact that overexpression of CnA* enhanced the dephosphorylation of target substrates in the plantaris, it did not influence slow fiber number or the expression of slow fiber-specific genes under normal contractile conditions. These results are in sharp contrast to a recent report of fast to slow fiber transitions in mice that overexpressed this same transgene at comparable levels (Naya et al. 2000). These previous data are difficult to reconcile with ours since fiber transformations were assessed in whole hindlimb preparations and calcineurin activities not reported. Nonetheless, the fact that a 10-fold overexpression of CnA* mRNA did not induce fiber hypertrophy in either study, reinforces the notion that in order to promote skeletal muscle fiber growth in vivo, increases in calcineurin activity must be matched to upstream activity-dependent effectors (i.e., contractile loading-related signaling events). Indeed, the finding that OV MCK-CnA* and OV MLC-CnA* Tg mice displayed a similar hypertrophic response and activation of the slow fiber program as WT counterparts, suggests that contractile activity is a major determinant of these calcineurin-mediated fiber adaptations. These data are therefore consistent with our previous notion (Dunn et al. 1999) that skeletal muscle cell signaling via calcineurin does not occur to a great extent under normal contractile conditions, and is only initiated when the nerve-mediated frequency of Ca2+ oscillations becomes higher than normal as seen under conditions of compensatory growth (Panchenko et al. 1974). Our findings in skeletal muscle contrast with those in heart in that expression of this same CnA* transgene induces cardiac hypertrophy when driven by the α-MHC promoter in Tg mice (Molkentin et al. 1998). The disparate effects of CnA* observed between cardiac and skeletal muscle may relate to differences in the strength of the MCK versus α-MHC promoter in these respective tissues. Alternatively, the possibility exists that because of the chronically active nature of this cell type, the threshold requirement of signaling via upstream activity-linked effectors (i.e., sustained levels of cytosolic Ca2+) is already met under normal cardiac contractile loading conditions.
Regarding the potential identity of these activity-dependent signal transduction events, there is mounting evidence that calcineurin must interact with parallel calcium-sensitive signaling pathways in order to fully activate downstream target genes (O'Keefe et al. 1992; Blaeser et al. 2000; De Windt et al. 2000; Friday et al. 2000; Wu et al. 2000). For instance, calcineurin synergizes with phorbol ester-dependent pathways to stimulate the IL-2 promoter in T lymphocytes and the expression of atrial natriuretic factor in cardiomyocytes (O'Keefe et al. 1992; De Windt et al. 2000). Similarly, calcineurin acts in conjunction with CaM-dependent kinase IV to fully activate the myoglobin promoter in cultured skeletal myocytes (Wu et al. 2000) and the Nur77 promoter in T lymphocytes (Blaeser et al. 2000). Moreover, retroviral-mediated gene transfer of CnA* induces skeletal myogenesis in vitro only in the presence of extracellular Ca2+ (Friday et al. 2000). Additionally, there is evidence that MAP kinase pathways are activated in response to increased contractile activity and play a role in regulation of the slow fiber phenotype (Murgia et al. 2000). In this context, MEF2 is an enticing candidate as an integrator of calcineurin and other activation-linked signal transduction pathways, since this transcription factor is both dephosphorylated by calcineurin and phosphorylated by various CaM kinases, ERK5, p38, and PKC (Black and Olson 1998; Wu et al. 2000).
An alternative possibility is that calcineurin signaling may converge with other activity-linked pathways via the association of GATA with NFAT (Molkentin et al. 1998; Musaro et al. 1999). Indeed, activation of calcineurin promotes the association of these two transcription factors via the dephosphorylation of NFATc1 and increased expression of GATA-2 under conditions of skeletal myocyte growth (Musaro et al. 1999). Consistent with findings from hypertrophic myocytes, we found this protein to be upregulated in the plantaris in response to OV, but not lowered by CsA treatment, suggesting that this transcription factor may be important for growth but not necessarily a gene target of calcineurin. Given that GATA is also known to associate with MEF2 (Morin et al. 2000), and that we observed fiber hypertrophy only when NFATc1 and MEF2 were dephosphorylated and GATA-2 increased, leads us to postulate that NFAT, MEF2, and GATA proteins act in synergy to transactivate target genes that lead to fiber growth in response to OV. Future studies should help identify the particular permutations of these transcription factors involved in the activation of slow fiber–specific genes versus those modulating adult fiber size.
Response of Skeletal Muscle to Limited CnA Availability
Although CnA* expression did not potentiate the fiber adaptations to OV, it did spare OV-CsA muscles the appearance of a shorter, COOH terminus deletion variant of CnA. This variant, resembling a form first identified in failing human hearts (Tsao et al. 2000), may harbor a deletion of the autoinhibitory domain at its COOH terminus (Fig. 5 a) thereby conferring constitutive activity to this enzyme (Hubbard and Klee 1989; O'Keefe et al. 1992). It is proposed that the manifestation of this shorter form of CnA may serve as a molecular indicator of injury related to excessive contractile stress or the inability of the endogenous calcineurin pool to meet the signaling requirements of contractile activity (Tsao et al. 2000). Our finding that the shorter form of CnA was detected in OV WT plantaris muscles treated with CsA, but was prevented by CnA* expression, suggests the latter scenario more likely since both tissues would have been subjected to the same contractile stress. Though it is not clear at present why the expression of the transgene blocked this OV- and CsA-sensitive appearance of the CnA variant, it may relate to the fact that the product of the transgene and this variant are probably functionally analogous (i.e., constitutively active). In this sense, increasing calcineurin activity may be a valid pharmacological strategy to counter muscle pathologies related to excessive contractile stress (e.g., postpolio syndrome) or clinical conditions in which signaling via this enzyme is compromised. Moreover, tissue detection of this shorter form of CnA may also prove to be a valuable diagnostic indicator of a stressed neuromuscular system.
The fact that the shorter form of CnA did not appear in the plantaris of WT mice in response to OV alone suggests that the endogenous calcineurin pool is sufficient to accommodate signaling under these conditions. In this respect, skeletal muscle appears to mimic the brain, a tissue displaying the highest levels of calcineurin, since overexpression of CnA* in neurons does not enhance calcineurin-dependent processes such as long-term depression (Winder et al. 1998). Thus, excitable cells displaying burst activation profiles such as muscle fibers and neurons may possess a functional reserve of this enzyme to ensure that calcineurin-dependent signaling pathways remain sensitive to the large fluctuations in calcium that may potentially occur in these cell types in response to activation.
CaM Dependency of Skeletal Muscle Adaptation
To further examine the role of calcineurin in the regulation of muscle fiber growth in vivo (Dunn et al. 1999), Tg mice were generated that overexpressed CaMBP in skeletal muscles under the control of the TnIs promoter. This strategy allowed us to restrict the expression and related effects of this transgene to slow/type I MHC fibers within the same muscle. CaMBP is a peptide that would have actions analogous to calcineurin inhibitors in preventing signaling of CaM to target enzymes. Consistent with our previous findings using specific calcineurin inhibitors (Dunn et al. 1999), overexpression of CaMBP completely blocked hypertrophy of plantaris slow/I MHC fibers which harbored the transgene. Interestingly, expression of CaMBP also prevented OV-related fast-to-slow fiber conversions, suggesting that the OV-sensitive initiation of the slow fiber program (i.e., TnIs expression) and consequent activation of the transgene within plantaris fast/II MHC fibers resulted in a reverberatory “on-off” circuit that prevented any further transformation of these cells. The fact that CaMBP expression did not affect plantaris slow fibers under normal weightbearing conditions, is consistent with the notion that calcineurin is of major importance only when contractile loading is increased above normal levels (Dunn et al. 1999). Although these results do not preclude a direct involvement of CaM kinases in these fiber adaptations, the fact that expression of CaMBP mimicked the effects of calcineurin inhibitors, emphasizes the critical role of this phosphatase in regulating fiber growth and fiber transformations. Finally, since the effects of the CaMBP transgene are limited to within mature skeletal muscle fibers, these findings support our previous conclusion (Dunn et al. 1999) that the modulating effect of calcineurin on fiber growth is likely restricted to events intrinsic to the cell rather than to extra-fiber phenomena related to satellite cell differentiation (Friday et al. 2000).
In summary, we provide evidence that both calcineurin and its upstream activity-linked effectors are required for the induction of fiber hypertrophy and fast-to-slow fiber conversions in response to increased contractile loads. The fact that calcineurin inhibitors and CaMBP influence fiber phenotype only under conditions of OV, taken together with the finding that OV is a major stimulus for calcineurin signaling via NFATc1 and MEF2 in vivo, reinforces our previous notion that calcineurin is a major modulator of skeletal fiber growth and fiber type transitions only when cytosolic Ca2+ is sustained at levels that are relatively higher than “normal” as seen under conditions of increased contractile loading (Panchenko et al. 1974). Future studies will aim to identify the target genes of calcineurin that regulate mature fiber growth and the accessory signaling events that coordinate with this phosphatase to initiate OV-induced fiber adaptations.
Acknowledgments
Thanks to Dr. R.S. Williams for assistance in the generation of Tg mice and C. Humphries for help in maintaining the Tg lines. We also thank Dr. R. Prywes for providing the MEF2D and 2A and Dr. D.J. Parry for providing MHC antibodies. S.E. Dunn and R.N. Michel are also affiliated with the Faculty of Health Sciences, University of Western Ontario, London, Ontario.
This work was funded by the Natural Sciences and Engineering Research Council of Canada to R.N. Michel.
Footnotes
E.R. Chin's present address is Department of Cardiovascular and Metabolic Diseases, Pfizer Global Research and Development, Groton, CT 06340.
Abbreviations used in this paper: CaM, calmodulin; CnA, catalytic subunit of calcineurin; CnA*, activated form of the catalytic subunit of calcineurin; CsA, cyclosporin A; MCK, muscle creatine kinase; MEF2, myocyte enhancer factor 2; MHC, myosin heavy chain; MLC, myosin light chain; NFAT, nuclear factor of activated T cells; OV, overload; Tg, transgenic; TnIf, troponin I fast; TnIs, troponin I slow.
References
- Abbott K.L., Friday B.B., Thaloor D., Murphy T.J., Pavlath G.K. Activation and cellular localization of the cyclosporine A-sensitive transcription factor NF-AT in skeletal muscle cells. Mol. Biol. Cell. 1998;9:2905–2916. doi: 10.1091/mbc.9.10.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams G.R., Haddad F. The relationships among IGF-1, DNA content, and protein accumulation during skelelal muscle hypertrophy. J. Appl. Physiol. 1996;81:2509–2516. doi: 10.1152/jappl.1996.81.6.2509. [DOI] [PubMed] [Google Scholar]
- Black B.L., Olson E.N. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- Blaeser F., Ho N., Prywes R., Chatila T.A. Ca2+-dependent gene expression mediated by MEF2 transcription factors. J. Biol. Chem. 2000;275:197–209. doi: 10.1074/jbc.275.1.197. [DOI] [PubMed] [Google Scholar]
- Blough E., Dineen B., Esser K. Extraction of nuclear proteins from striated muscle tissue. BioTechniques. 1999;26:202–206. doi: 10.2144/99262bm05. [DOI] [PubMed] [Google Scholar]
- Chin E.R., Olson E.N., Richardson J.A., Yang Q., Humphries C., Shelton J.M., Wu H., Zhu W., Bassel-Duby R., Williams R.S. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber phenotype. Genes & Development. 1998;12:2499–2509. doi: 10.1101/gad.12.16.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corin S.J., Juhasz O., Zhu L., Conley P., Kedes L., Wade R.. Structure and expression of the human slow twitch skeletal muscle troponin I gene. J. Biol. Chem. 1994;269:10651–10659. [PubMed] [Google Scholar]
- De Windt L.J., Lim H.W., Haq S., Force T., Molkentin J.D. Calcineurin promotes protein kinase C and c-jun NH2-terminal kinase activation in the heart. J. Biol. Chem. 2000;275:13571–13579. doi: 10.1074/jbc.275.18.13571. [DOI] [PubMed] [Google Scholar]
- Dunn S.E., Michel R.N. Coordinated expression of myosin heavy chain isoforms and metabolic enzymes within overloaded rat muscle fibers. Am. J. Physiol. 1997;273:C371–C383. doi: 10.1152/ajpcell.1997.273.2.C371. [DOI] [PubMed] [Google Scholar]
- Dunn S.E., Burns J.L., Michel R.N. Calcineurin is required for skeletal muscle hypertrophy. J. Biol. Chem. 1999;274:21908–21912. doi: 10.1074/jbc.274.31.21908. [DOI] [PubMed] [Google Scholar]
- Esser K., Nelson T., Lupa-Kimball V., Blough E. The CACC box and myocyte enhancer factor-2 sites within the myosin light chain 2 slow promoter cooperate in regulating nerve-specific transcription in skeletal muscle. J. Biol. Chem. 1999;274:12095–12102. doi: 10.1074/jbc.274.17.12095. [DOI] [PubMed] [Google Scholar]
- Friday B.B., Horsley V., Pavlath G.K. Calcineurin activity is required for the initiation of skeletal muscle differentiation. J. Cell Biol. 2000;149:657–665. doi: 10.1083/jcb.149.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D.A., Klee C.B., Bierer B.E., Burakoff S.J. Calcineurin phosphatase activity in T-lymphocytes is inhibited by FK506 and cyclosporin A. Proc. Natl. Acad. Sci. 1992;89:2685–3690. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner P., Michel R.N., Browman C., Noble E. Increased EMG of rat plantaris during locomotion following surgical removal of its synergists. Brain Res. 1986;380:114–121. doi: 10.1016/0006-8993(86)91435-6. [DOI] [PubMed] [Google Scholar]
- Hogan B.R., Beddington R., Constantini F., Lacy E. Manipulating the Mouse Embryo 1994. Cold Spring Harbor Laboratory, ; Cold Spring Harbor, NY: 79–203. pp. pp [Google Scholar]
- Hubbard M.J., Klee C.B. Functional domain structure of calcineurin Amapping by limited proteolysis. Biochemistry. 1989;28:1868–1874. doi: 10.1021/bi00430a066. [DOI] [PubMed] [Google Scholar]
- Mao Z., Wiedmann M. Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. J. Biol. Chem. 1999;274:31102–31107. doi: 10.1074/jbc.274.43.31102. [DOI] [PubMed] [Google Scholar]
- Molkentin J.D., Lu J.-R., Antos C.L., Markham B., Richardson J., Robbins J., Grant S.R., Olson E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin S., Charron F., Robitaille L., Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO (Eur. Mol. Biol. Organ.) J. 2000;19:2046–2055. doi: 10.1093/emboj/19.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia M., Serrano A.L., Calabria E., Pallafacchina G., Lomo T., Schiaffino S. Ras is involved in nerve-activity-dependent regulation of muscle genes. Nature Cell Biol. 2000;2:142–147. doi: 10.1038/35004013. [DOI] [PubMed] [Google Scholar]
- Musaro A., McCullagh K.J.A., Naya F.J., Olson E.N., Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999;400:581–585. doi: 10.1038/23060. [DOI] [PubMed] [Google Scholar]
- Naya F.J. , Mercer B., Shelton J., Richardson J.A., Williams R.S., Olson E.N. Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J. Biol. Chem. 2000;275:4545–4548. doi: 10.1074/jbc.275.7.4545. [DOI] [PubMed] [Google Scholar]
- O'Keefe S.J., Tamura J., Kincaid R.L., Tocci M.J., O'Neill E.A. FK-506- and CsA-sensitive activation of the interleuken-2 promotor by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- Panchenko L.F., Aliev M.K., Meerson F.Z. State of the calcium pump of the sarcoplasmic reticulum in compensatory hyperfunction and hypertrophy of skeletal muscle. Bull. Exp. Biol. Med. 1974;77:272–274. doi: 10.1007/BF00802477. [DOI] [PubMed] [Google Scholar]
- Rao A., Luo C., Hogan P.G. Transcription factors of the NFAT familyregulation and function. Annu. Rev. Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Rosenthal N., Kornhauser J.M., Donoghue M., Rosen K.M., Merlie J.P. Myosin light chain enhancer activates muscle-specific, developmentally regulated gene expression in transgenic mice. Proc. Natl. Acad. Sci. USA. 1989;86:7780–7784. doi: 10.1073/pnas.86.20.7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semsarian C., Wu M.J., Ju Y.K., Marciniec T., Yeoh T., Allen D.G., Harvey R.P., Graham R.M. Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signaling pathway. Nature. 1999;400:576–581. doi: 10.1038/23054. [DOI] [PubMed] [Google Scholar]
- Sternberg E.A., Spizz G., Perry W.M., Vizard D., Weil T., Olson E.N. Identification of upstream and intragenic regulatory elements that confer cell-type-restricted and differentiation-specific expression on the muscle creatine kinase gene. Mol. Cell. Biol. 1988;8:2896–2909. doi: 10.1128/mcb.8.7.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao L., Neville C., Musaro A., McCullagh K.J.A., Rosenthal N. Revisiting calcineurin and human heart failure. Nature Med. 2000;6:2–3. doi: 10.1038/71478. [DOI] [PubMed] [Google Scholar]
- Wang J., Campos B., Jamieson G.A., Jr., Kaetzel M.A., Dedman J.R. Functional elimination of calmodulin within the nucleus by targeted expression of an inhibitor peptide. J. Biol. Chem. 1995;270:30245–30248. doi: 10.1074/jbc.270.51.30245. [DOI] [PubMed] [Google Scholar]
- Winder D.G., Mansuy I.M., Osman M., Moallem T.M., Kandel E.R. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- Woronicz J.D., Lina A., Calnan B.J., Szychowski S., Cheng L., Winto A. Regulation of Nur77 orphan steroid receptor in activation-induced apoptosis. Mol. Cell. Biol. 1995;15:6364–6376. doi: 10.1128/mcb.15.11.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Naya F.J., McKinsey T.A., Mercer B., Shelton J.M., Chin E.R., Simard A.R., Michel R.N., Bassel-Duby R., Olson E.N., Williams R.S. MEF2 response to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO (Eur. Mol. Biol. Organ.) J. 2000;19:1963–1973. doi: 10.1093/emboj/19.9.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]