

Abstract
An approach that combines limited proteolysis and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) has been developed to probe protease-accessible sites of ribosomal proteins from intact ribosomes. Escherichia coli and Thermus thermophilus 70S ribosomes were subjected to limited proteolysis using different proteases under strictly controlled conditions. Intact ribosomal proteins and large proteolytic peptides were recovered and directly analyzed by MALDI-MS, which allows for the determination of proteins that are resistant to proteolytic digestion by accurate measurement of molecular weights. Larger proteolytic peptides can be directly identified by the combination of measured mass, enzyme specificity and protein database searching. Sucrose density gradient centrifugation revealed that the majority of the 70S ribosome dissociates into intact 30S and 50S subunits after 120 min of limited proteolysis. Thus, examination of ribosome populations within the first 30-60 min of incubation provide insight into 70S structural features. Results from E. coli and T. thermophilus revealed that a significantly larger fraction of 50S ribosomal proteins have similar limited proteolysis behavior than the 30S ribosomal proteins of these two organisms. The data obtained by this approach correlate with information available from the high resolution crystal structures of both organisms. This new approach will be applicable to investigations of other large ribonucleoprotein complexes, is readily extendable to ribosomes from other organisms, and can facilitate additional structural studies on ribosome assembly intermediates.
The ribosome, found in all organisms, is the subcellular organelle that performs the activity of protein synthesis. Prokaryotic ribosomes, which sediment at 70S, have a molecular mass of approximately 2.5 ×106 Da and consist of two subunits, the 50S and the 30S. Each subunit has a unique number of proteins and ribosomal RNAs (rRNAs). Eukaryotic ribosomes, which sediment at 80S, are substantially larger and more complex than their prokaryotic counterparts. Two subunits, the 60S and 40S, together contain at least 78 unique proteins and 4 rRNAs. The small subunit binds messenger RNA (mRNA) and mediates the interactions between mRNA and transfer RNAs (tRNAs). The larger subunit catalyzes peptide-bond formation. During the initiation phase of protein synthesis, the two subunits behave independently, assembling into complete ribosomes only when elongation is about to begin.
A fundamental prerequisite for understanding the biochemical interactions occurring within the ribosome during protein synthesis is a detailed knowledge of the structure of this ribonucleoprotein (RNP) complex. Some of the more successful approaches for characterizing ribosomes include crosslinking [1-4], cryo-electron microscopy (cryo-EM) [5-7], tritium bombardment [8], nuclear magnetic resonance (NMR) [9] and electrospray mass spectrometry [10-13]. One of the most successful approaches has been the development of X-ray crystallography within the past several years, wherein high-resolution crystal structures of ribosomal subunits or intact ribosomes have been obtained that provide detailed structural information at the atomic level [14-18]. However, as noted recently by Selmer et al [18], the presence of crystal structures alone does not translate into an accurate assignment of ribosomal proteins. Thus, analytical approaches that can provide information regarding ribosome components, organization and topology and that are readily extendable to new organisms will assist in structural assignments as ribosomes from new organisms are crystallized and studied.
Limited proteolysis has been used to investigate structure-function relationships in proteins and the interacting sites in protein-nucleic acid complexes [19-21]. Limited proteolysis can reveal details of the exposed sites of protein-nucleic acid complexes, which is useful for modeling three-dimensional structures. While limited proteolysis yields useful information regarding the topology of biomolecules, it has not been widely applied to studies of RNP complexes in general, or the ribosome in particular, due to the complexity of data interpretation as previous investigations of the ribosome by this approach relied upon 2-D gel electrophoresis as the separation/detection scheme [22, 23].
Mass spectrometry has been used as the readout step in a limited proteolysis approach for characterizing macromolecular structure [24-29]. In those previous reports, single proteins with nucleic acids or ligands [24, 26] or virus complexes [28] were investigated by limited proteolysis with mass spectrometry. Most limited proteolysis studies previously reported have been carried out for observing the conformational change of a protein with and without bound nucleic acid or ligands, and for probing the structural and dynamic differences between the holo and apo form of a protein [25]. Based on the interaction between a protein and a nucleic acid or ligand, the stable domain of a protein or the likely sites of binding within the protein could be directly determined by mass spectrometry without requiring sample isolation after limited proteolysis.
Here we expand the use of limited proteolysis with matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) to a much larger and more complex macromolecular assembly containing multiple proteins and RNAs. In the present study the protease-accessible sites of ribosomal proteins (r-proteins) within Escherichia coli and Thermus thermophilus intact 70S ribosomes have been identified by a combination of limited proteolysis and MALDI-MS. With this approach, undigested proteins can be unambiguously assigned based upon their accurately measured molecular weights. By comparative limited proteolysis using trypsin and Proteinase K, the r-proteins whose surface exposed regions are resistant to trypsin but not general proteolysis were identified. We identified six small subunit r-proteins and 22 large subunit r-proteins that are resistant to limited proteolysis using trypsin in both E. coli and T. thermophilus. In addition, some proteins were determined to be stable in their truncated form and are retained within the ribosomal subunit structures. As existing E. coli and T. thermophilus crystal structures were used to confirm the results found in this study, this new approach should be applicable to examining ribosomes whose topology or crystal structures are not yet known and can help inform future structural characterizations of ribosomal subunits or intact ribosomes.
EXPERIMENTAL
Materials
Tryptone and yeast extract were obtained from Difco Labs (Detroit, MI). Buffer reagents, Proteinase K, and the MALDI peptide calibration kit were obtained from Sigma (St. Louis, MO). Sinapinic acid (SA) and α-cyano-4-hydroxycinnamic acid (CHCA) were obtained from Fluka (Milwaukee, WI). Acids and organic solvents were HPLC grade or better. Trypsin (sequencing grade) and DNase were purchased from Promega (Madison, MI).
Preparation of 70S Ribosomes
Thermus thermophilus HB8 (ATCC 27642) was a gift from S. Gregory and A. Dahlberg (Brown University, Providence RI), and conditions for culturing T. thermophilus and isolating 70S ribosomes have been described elsewhere [30]. E. coli (MRE 600) tight coupled 70S ribosomes were obtained following the cell growth and harvesting procedures described previously [31]. Tight-coupled 70S ribosomes were isolated as follows. Fifteen g of frozen E. coli cell paste was resuspended in 22.5 ml of buffer A (20 mM Tris-HCl, pH 7.5,100 mM NH4Cl, 10.5 mM magnesium acetate, 0.5 mM EDTA). After the addition of DNAse I (8 μg per g of cell paste), the cells were disrupted in a French press. The disrupted cell slurry was then centrifuged for 1 hour at 30,000 ×g. The supernatant was carefully removed, and then layered over a sucrose cushion of buffer B (1.1 M sucrose, 20 mM Tris-HCl, pH 7.5, 0.5 M KCl, 10.5 mM magnesium acetate, 0.5 mM EDTA), and centrifuged at 45,000 rpm in a 45 Ti rotor for 17.5 hours. The ribosomal pellet was washed lightly in buffer C (1.5 M ammonium sulfate, 10 mM magnesium acetate, 400 mM KCl, 20 mM Tris-Cl, pH 7.5) to remove any “skin” of cell debris, and resuspended in the same buffer. The ribosomal peak was diluted in buffer E (10 mM Tris-HCl, pH 7.5, 50 mM KCl, 60 mM NH4Cl, 10.5 mM magnesium acetate, 0.25 mM EDTA), pelleted overnight by centrifugation at 43,000 rpm in a 45Ti rotor, and resuspended in buffer E. The tight coupled 70S ribosomes were then separated from excess 50S or 30S subunits by ultracentrifugation with a gradient of 10 - 40% sucrose in buffer E. The tight-coupled 70S ribosomes were diluted in buffer E without sucrose and pelleted as above.
Limited Proteolysis of 70S Ribosomes
Limited proteolysis using trypsin or Proteinase K was carried out in a buffer of 10 mM Tris-HCl (pH 7.7), 10 mM magnesium acetate, 60 mM NH4Cl, and 3 mM β-mercaptoethanol at 37 °C. The enzyme to ribosome ratio was adjusted to 1:500 (w/w) to achieve time-resolved cleavage with around 250 μg of ribosome present initially. The ribosome concentration was determined by UV absorbance (1 A260 unit = 26 pmole for 70S [32]). The total reaction mixture volume was 80 μL. Aliquots were withdrawn from the reaction mixture at specified time intervals for electrophoretic and mass spectrometric analysis. For electrophoretic analysis, 1 μL of reaction mixture was removed, combined with the electrophoresis loading buffer and then immediately heated in a boiling water bath for 10 min to halt proteolysis. For MALDI analysis, 9 μL of reaction mixture was removed and combined with 1 μL of 25% trifluoroacetic acid (TFA) to quench the reaction. As controls, r-proteins isolated from 70S ribosomes by acetic acid treatment were resuspended in buffer E and then incubated with trypsin, and 70S ribosomes were incubated without added protease. All control reactions were prepared for analysis by either electrophoresis or MALDI-MS as described above.
Sucrose Density Gradient Centrifugation
The integrity of ribosomal subunits was investigated by using a 0-45% sucrose gradient in a buffer containing 10 mM Tris-HCl, pH 7.5, 10.5 mM magnesium acetate, 60 mM NH4Cl and 3 mM 2-mercaptoethanol. A 0-45% gradient was made by adding 36 ml of 22.5% solution of sucrose to polyallomer centrifuge tubes. Tubes were frozen at -20 °C overnight and thawed at 4 °C. Samples after limited proteolysis were loaded on top of sucrose gradient and centrifuged at 19000 rpm for 17 hours in an SW28 rotor with a Beckman XL-80 ultracentrifuge (Beckman Coulter, Palo Alto, CA) at 4°C. Sucrose gradients were fractionated using a Density Gradient Fractionation System (ISCO Inc., Lincoln, NE). Fractions were collected and monitored at 260 nm.
SDS-PAGE and In-Gel Digestion
Sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out using 12.5% polyacrylamide gel of 1-mm thickness, run at 250 V for 4 - 5 hours. The SDS-PAGE was visualized by staining with Coomassie Brilliant Blue R250. As necessary, excised gel bands were digested with trypsin as described [33] and analyzed by MALDI-MS.
MALDI Analysis
All MALDI-MS experiments were done on a Bruker Reflex IV reflectron MALDI time-of-flight mass spectrometer (Bruker Daltonics, Billerica, MA, USA) equipped with a nitrogen laser as previously described [34]. MALDI samples were prepared by combining 1 μL of TFA-treated ribosomal solution with 9 μL of the SA matrix. Protein mass spectra were obtained in the positive ion mode at an acceleration voltage of 20 kV, extraction voltage of 17.1 kV and lens voltage of 10.1 kV by accumulating 300 laser shots. The laser power was adjusted to slightly above the threshold to obtain optimal resolution and signal-to-noise ratios. All samples were analyzed under identical instrumental parameters.
Data Analysis
Proteolytic digestion products were identified using the SequenceEditor software provided by the MALDI manufacturer and Protein Prospector [35]. Images of the 30S and 50S subunit structures were produced using MacPyMol [36] from the 3.5 Å crystal structure of E. coli (accession 2AVY and 2AW4) [17] and the 2.8 Å crystal structure of T. thermophilus (accession 2J00 and 2J01) [18].
RESULTS AND DISCUSSION
Several factors including protein-rRNA or protein-protein interactions, location of proteins within the ribosome, protease:ribosome ratio, time of incubation, pH, temperature, and protease specificity will influence the data obtained from limited proteolysis experiments. Thus, optimization of a limited proteolysis strategy for intact ribosomes composed of over 50 proteins and several large RNAs requires examination of several experimental variables.
Initially, E. coli (strain MRE 600) tight coupled 70S ribosomes were subjected to time-course measurements of proteolysis employing a site-specific protease, trypsin, and a non-specific protease, Proteinase K, as described in the Experimental section. Optimization of the limited proteolysis conditions was carried out by varying the enzyme to ribosome ratio from 1:100 (w/w) to 1:2000 (w/w). MALDI-MS was used to analyze the proteolytic fragments and intact proteins from ribosomes. MALDI-MS data were compared to that obtained by SDS-PAGE as well as to data reported by other techniques [7, 23, 37]. As necessary to confirm proteolytic fragment identities, in-gel digestion followed by MALDI peptide mass fingerprinting was also done. The results of these initial studies yielded an optimal enzyme to ribosome ratio of 1:500 (w/w) at pH 7.7 and 37 °C, and these conditions were then used for all subsequent investigations. While the optimal enzyme to ribosome mole ratio of 1:5 is lower than typically seen in other limited proteolysis studies, it is not inconsistent with prior literature reports [23, 24] and, as discussed below, was found to provide sufficient information on surface accessible proteins while maintaining the overall organization of the ribosome and ribosomal subunits.
Limited Proteolysis and Ribosome Integrity
To confirm that the limited proteolysis conditions are appropriate to reveal information on the organization of 70S ribosomes and ribosomal subunits, a series of sucrose density gradient (SDG) centrifugation studies were conducted. The results of these studies are shown in Figure 1. As seen here, limited proteolysis does lead to dissociation of 70S ribosomes into the 30S and 50S subunits and that dissociation is complete within 500 min of incubation for both E. coli and T. thermophilus under the conditions used in this study. Significantly, even up to 1500 min of incubation, there is no loss to subunit organization as reflected by the presence of 30S and 50S subunits in these data. These results do show, however, that conclusions specific to the 70S organization are limited to the first 60 min or less of incubation, and that data at 500 min and beyond are reporting on subunit organization only.
Figure 1.

Sucrose density gradient centrifugation studies of intact E. coli and T. thermophilus 70S ribosomes upon incubation with trypsin. For both organisms, the 70S ribosomes begin dissociating within 30 min of incubation and have become completely dissociated by the 500 min incubation period.
Limited Trypsin Proteolysis of E. coli 70S ribosomes
Figure 2 shows a series of mass spectra from the limited proteolysis of E. coli 70S ribosomes using trypsin. Intact proteins were assigned by accurate mass measurement of the ribosome mixture before proteolysis (T = 0 min) and by comparison to protein molecular weights predicted for E. coli from entries in the SwissProt database as before [34]. The r-proteins L2, L3, L6, L11, L13, L14, L15, L16, L17, L18, L20, L21, L22, L23, L24, L25, L28, L29, L30, L32, L33, L34, L35, and L36 of the large subunit, and r-proteins S4, S8, S9, S12, S13, S15, S16, S17, and S20 of the small subunit were still detected as intact proteins even after incubation with trypsin for 1500 min. After 1500 min, an additional 0.5 μg of trypsin was added to the reaction mixture and incubation was continued for 60 min. Even under these conditions, no difference in the mass spectral results was seen as compared to the data obtained at 1500 min (data not shown). The significantly greater number of large subunit as compared to small subunit r-proteins that were resistant to limited proteolysis using trypsin is a reflection of the different organization and r-protein:rRNA mass ratio of the two subunits [38, 39]. The mass spectral data limited to those proteins remaining intact after limited proteolysis with trypsin is summarized in Figure 3.
Figure 2.
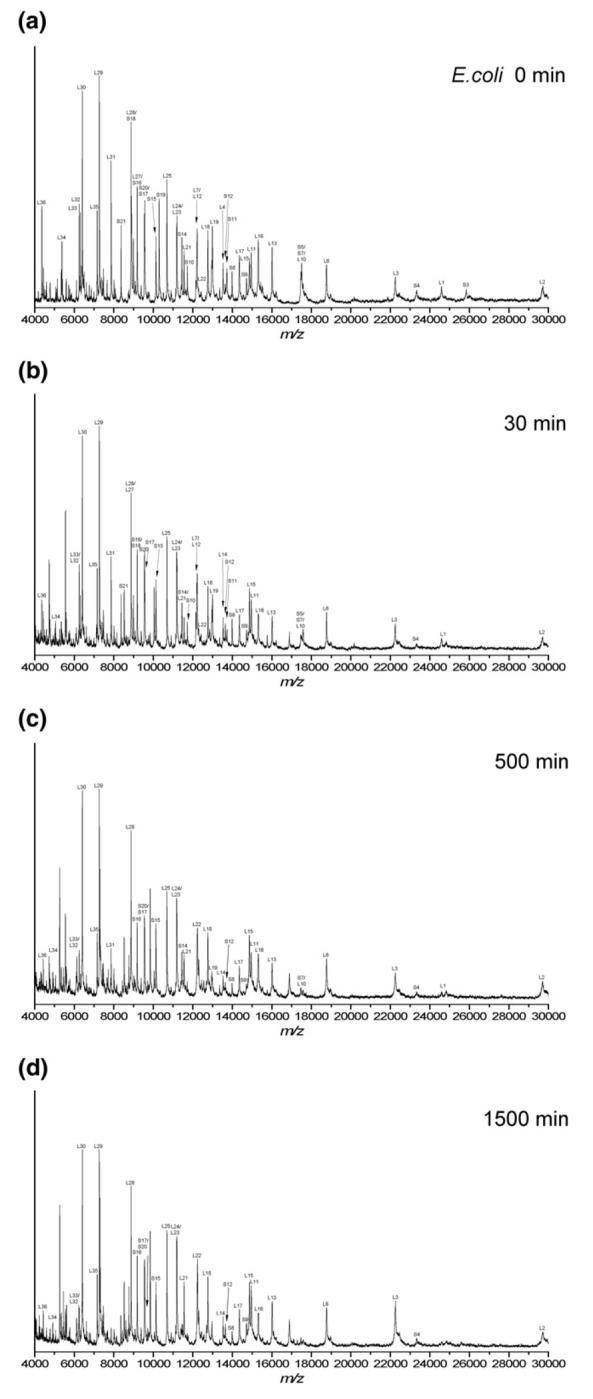
Representative MALDI mass spectral data after incubation of intact E. coli 70S ribosomes with trypsin under limited proteolysis conditions for the following time periods (a) 0 min, (b) 30 min, (c) 500 min, and (d) 1500 min. The intact ribosomal proteins detected after each incubation period are summarized in Figure 3. Table 1 summarizes the identification of new m/z values that arise during the limited proteolysis experiments.
Figure 3.

Intact E. coli ribosomal proteins from the (a) 30S subunit and (b) 50S subunit observed with MALDI-MS after incubation with protease for denoted time periods; (white bars) Trypsin and (black bars) Proteinase K.
In combination with the SDG data, there are three categories of r-proteins that do not remain intact throughout the entire incubation period examined in this study. The first category are r-proteins S2, S3, S6 and S19, which were digested within 30 minutes under the conditions employed here as determined by the disappearance of mass spectral peaks corresponding to the mass of the intact protein. As the SDG studies revealed that the majority of the sample contains intact 70S ribosomes at this incubation period, one can conclude that these ribosomal proteins are well-exposed on the ribosome surface or, at a minimum, have regions containing lysine(s) or arginine(s) that are sufficiently exposed for proteolysis. The second category contains r-proteins that are relatively instable to limited proteolysis with trypsin but whose original conformation, i.e., 70S vs. 30S/50S, cannot be determined by this experimental procedure. The r-proteins L7/L12, L27, S5, S11 and S18 were digested within 125 min under the conditions employed here. From the SDG studies, one can conclude that these proteins are susceptible to trypsin proteolysis either in their 70S or subunit conformations, if not both. The final category contains r-proteins which are digested after 125 minutes, here S7, S10, S14, S21, L1, L9, L10, L19 and L31. These data are likely reflecting subunit conformations as the SDG studies do not show intact 70S ribosomes at these longer periods.
In addition to the loss of mass spectral peaks from specific intact r-proteins, new ion signals at various m/z values were detected during these analyses. The mass spectral data of these new proteolytic fragments are summarized in Table 1. The new m/z values that arise during the limited proteolysis were identified, where possible, by combining knowledge of the specificity of trypsin with the mass values and original masses of the intact r-proteins. As seen in Table 1, the proteolytic fragments generally arise from cleavage of N- and/or C-terminal domains of the various r-proteins. Similar cleavages have been found during limited proteolysis studies of isolated r-proteins whose tertiary structure was maintained after extraction from ribosomes [40]. These data demonstrate that direct MALDI-MS analysis of limited proteolysis mixtures is compatible with both the identification of proteins susceptible to enzymatic proteolysis as well as defining the sequence location of proteolysis.
Table 1.
MALDI-MS analysis of E. coli proteolytic fragments generated by trypsin (Fig. 2). The identification of proteolytic fragments can occur even when m/z values are detected for intact proteins, as seen here for ribosomal protein L2. Protein fragments identified by asterisks, *, were still detected after 1500 min incubation with trypsin.
| Measured Mass [M+H]+ | Calculated Massa [M+H]+ | ΔM (Da) | Identity | Observed A.A. Residues | Loss of |
|---|---|---|---|---|---|
| 4738.2 | 4738.4 | -0.2 | L7/L12 | 75-121 | N-terminus |
| 4752.6 | 4752.4 | 0.2 | L7/L12 | 75-121 | N-terminus |
| 5270.9 | 5271.2 | -0.3 | L31* | 1-47 | C-terminus |
| 5461.6 | 5461.4 | 0.2 | L9* | 1-50 | C-terminus |
| 5555.1 | 5555.5 | -0.4 | L31* | 1-49 | C-terminus |
| 5617.3 | 5617.6 | -0.3 | L9* | 1-51 | C-terminus |
| 5761.3 | 5760.8 | 0.5 | S14 | 48-97 | N- and C-termini |
| 7128.4 | 7128.3 | 0.1 | L31 | 1-63 | C-terminus |
| 7315.7 | 7315.4 | 0.3 | S18* | 13-75 | N-terminus |
| 7339.1 | 7339.6 | -0.5 | L9 | 1-64 | C-terminus |
| 7721.5 | 7721.9 | -0.4 | S18 | 10-75 | N-terminus |
| 8162.6 | 8162.5 | 0.1 | S18 | 7-75 | N-terminus |
| 8357.9 | 8358.6 | -0.7 | L10* | 62-138 | N- and C-termini |
| 8529.9 | 8529.8 | 0.1 | L27* | 6-85 | N-terminus |
| 8629.1 | 8629.0 | 0.1 | S18 | 4-75 | N-terminus |
| 8658.1 | 8658.0 | 0.1 | L27 | 5-85 | N-terminus |
| 8768.8 | 8768.2 | 0.6 | S6 | 39-112 | N- and C-termini |
| 8870.7 | 8870.3 | 0.4 | S3 | 2-79 | C-terminus |
| 9715.9 | 9715.4 | 0.5 | S19* | 2-87 | C-terminus |
| 9844.9 | 9843.6 | 0.3 | S19* | 2-88 | C-terminus |
| 10043.6 | 10043.8 | -0.2 | S19 | 2-90 | C-terminus |
| 12310.6 | 12311.1 | -0.5 | S11* | 10-125/11-126/12-127/14-129 | N-and C-termini |
| 12248.5 | 12249.2 | 0.7 | L19* | 2-109 | C-terminus |
| 12566.1 | 12566.4 | -0.3 | S11 | 12-129 | N- and C-termini |
| 12861.2 | 12861.9 | -0.7 | S6 | 3-112 | N- and C-termini |
| 16225.1 | 16225.8 | -0.7 | S7 | 12-156 | N-terminus |
| 16895.1 | 16894.6 | 0.5 | S5* | 7-167 | N-terminus |
| 24842.3 | 24843.0 | -0.7 | S3* | 2-225 | C-terminus |
| 27597.6 | 27599.9 | -2.3 | L2* | 2-256 | C-terminus |
The average singly protonated mass of the tryptic products are shown as calculated using SequenceEditor ver. 1.0, provided by Bruker Daltonics and ProteinProspector.
To verify that the results obtained in these experiments reflected the organization of the ribosome, r-proteins were analyzed after incubating at 37 °C for 1500 min without trypsin. No difference in the mass spectral data obtained under these conditions as compared to the T = 0 min data in Figure 2 was found (data not shown). In addition, r-proteins that were isolated from the ribosome by acetic acid treatment were also resuspended in buffer and incubated with trypsin for 500 min. MALDI-MS analysis of this reaction mixture yielded no intact proteins (data not shown). Under these conditions, r-proteins are separated from rRNA and the organizational structure of the ribosome is destroyed.
To confirm that the MALDI-MS data accurately reflects the limited proteolysis of ribosomes, SDS-PAGE analysis was also done on the trypsin incubated ribosomes. In agreement with the MALDI-MS results, some protein bands do not decrease in intensity with time while other proteins are no longer detected on the gel (data not shown). Unlike the MALDI-MS results, specific lower molecular weight r-proteins are difficult to identify on the 1-D gel due to the poor resolution of this approach. However, the higher molecular weight proteins are easily monitored by 1-D PAGE and the response of these higher molecular weight proteins agrees with the response found by MALDI analysis.
Limited Proteinase K proteolysis of E. coli 70S ribosomes
In a manner similar to the above-described experiments with trypsin, E. coli 70S ribosomes were incubated with the non-specific protease, Proteinase K. In contrast to trypsin, which will only cleave at lysine or arginine residues, Proteinase K cleaves at any aromatic, aliphatic or hydrophobic amino acid residue. Thus, Proteinase K should provide a more general picture of exposed proteins within the ribosome as compared to trypsin. In addition, comparative proteolysis can be used to distinguish topological features that are specifically trypsin resistant, such as L7/L12, whose conformation is trypsin resistant in the absence of elongation factor G (EF-G) [22].
Figure S1 shows the mass spectrum of r-proteins remaining from the limited proteolysis of 70S ribosome using Proteinase K for 1500 min. Figure 3 summarizes the data from these MALDI experiments. The use of Proteinase K revealed a number of r-proteins that are rapidly digested upon limited proteolysis. Within the large subunit, r-proteins L1, L7/12, L9, and L17 are digested within the first 30 minutes of incubation. These results are in general agreement with those reported previously from limited proteolysis experiments on 50S subunits [23]. In addition, the small subunit r-proteins S2, S3, S6, S7, S10, S18 and S21 are digested within the first 30 minutes and S5 and S11 are digested within the first 60 minutes of incubation. The r-proteins L7/L12, L9, L17, S7, S18 and S21 were found to be well exposed and S2, S3 and S6 were found to be moderately exposed on intact ribosomes during hot tritium bombardment experiments [8]. Interestingly, S4, S20, L16 and L24 were also proposed to be well-exposed by tritium quantification yet are found to be significantly stable to proteolysis by Proteinase K suggesting differences in conformations between the two studies or differences in reactivity to proteolysis and tritium incorporation for these particular proteins.
As was done in the incubations with trypsin, a Coomassie Blue-stained PAGE analysis of several ribosome incubations was done for comparison (Figure S2). Similar trends are observed between the MALDI-MS data and the 1-D PAGE data: various proteins are observed to decrease in intensity with increased incubation with Proteinase K and new bands, presumed to be proteolytic fragments, are also detected. To identify several of the new bands generated during limited proteolysis that occurred at higher molecular weights, in-gel tryptic digestion and MALDI peptide mass fingerprinting for protein identification was done. As a representative example, a new band (circled in Fig. S2) appearing at a lower molecular weight than the band for intact ribosomal protein L2 (band B, Fig. S2) was detected in lane 8, which corresponds to a 1500 min incubation with Proteinase K. In-gel digestion and MALDI peptide mass fingerprinting yielded a match to ribosomal protein L2. Similar analyses were done on other bands, which are denoted in Figure S2. Table S1 summarizes the in-gel digestion and MALDI peptide mass fingerprinting results from the annotated bands from Figure S2. In addition to the lack of resolution at lower molecular weights for this 1-D gel, another obvious disadvantage of an electrophoretic-based approach is the need to perform additional identifications of new bands.
While these new bands could be identified by in-gel tryptic digestion and MALDI peptide mass fingerprinting, the use of a non-specific protease hinders determination of precise sites of proteolysis. Most significant, though, is the correlation between MALDI and PAGE results as well as the correlation between r-protein stability and limited proteolysis with the two enzymes. By comparison of the limited proteolysis results obtained between trypsin and Proteinase K, the r-proteins whose surface exposed regions are resistant to trypsin but not general proteolysis can be identified.
Comparison of E. coli MALDI-MS data obtained from different proteases
The E. coli r-proteins S12, S15, S16, S17, S20, L2, L3, L6, L13, L14, L15, L16, L18, L21, L22, L24, L25, L28, L30, L32, L34, and L36 were not completely digested by either trypsin or Proteinase K even at the longest periods of incubation (1500 min or 1500 + 60 min, respectively) examined in these studies. The r-proteins, L20, L29, L33, L35, S4, and S8, which were detected intact after the 1000 min incubation with Proteinase K but not trypsin, were eventually digested by Proteinase K after longer incubation periods. Thus, the trends for protein stability were similar for the two proteases used in this study with less r-proteins being stable when incubated with the non-specific protease. The use of the less specific protease, Proteinase K, resulted in 11 more completely digested proteins as compared to trypsin. Such results are to be expected given the lack of specificity for Proteinase K. However, because interactions between r-proteins and rRNA are usually through salt-bridges between positively charged residues on the proteins and phosphate oxygen atoms on the RNA [41], basic residues such as arginine and lysine that interact with rRNA through salt-bridges will be less susceptible to proteolysis. Thus, protease specificity may not account for all of the differences between trypsin and Proteinase K found in this study, although an examination of the data obtained in this study did not discern any relationship between the frequency of Arg/Lys residues within a protein and the rate of proteolysis with trypsin.
Limited Proteolysis of T. thermophilus 70S ribosomes
In a manner similar to that just described, limited proteolysis studies were also conducted on T. thermophilus (Figures 4 and S3). Intact proteins were assigned by accurate mass measurement of the ribosome mixture before proteolysis (T = 0 min) and by comparison to protein molecular weights predicted for T. thermophilus HB8 from entries in the SwissProt database as before [42]. The r-proteins L1, L2, L3, L6, L13, L14, L15, L16, L17, L18, L20, L21, L22, L23, L24, L28, L29, L30, L32, L33, L34, L35, and L36 of the large subunit, and r-proteins SThx, S6, S8, S9, S10, S14, S15 and S17 of the small subunit were still detected as intact proteins even after incubation with either trypsin or Proteinase K for 1500 min. The mass spectral data limited to those proteins remaining intact after limited proteolysis is summarized in Figures 5 and S4.
Figure 4.
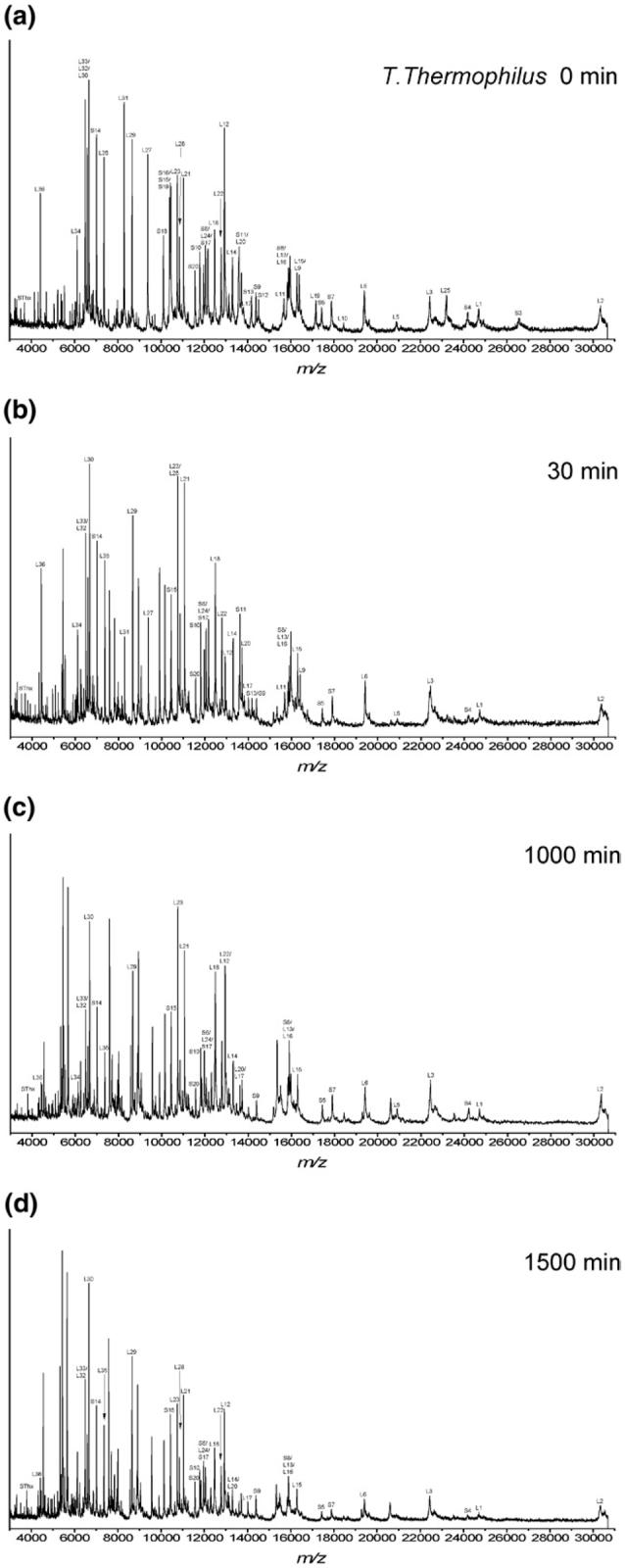
Representative MALDI mass spectral data after incubation of intact T. thermophilus 70S ribosomes with trypsin under limited proteolysis conditions for the following time periods (a) 0 min, (b) 30 min, (c) 500 min, and (d) 1500 min. The intact ribosomal proteins detected after each incubation period are summarized in Figures 5 and S4. Table 2 summarizes the identification of new m/z values that arise during the limited proteolysis experiments.
Figure 5.

Intact ribosomal proteins from the (a) 30S subunit and (b) 50S subunit observed with MALDI-MS after incubation with trypsin for denoted time periods; (white bars) T. thermophilus and (black bars) E. coli.
The r-proteins S3, S12, S16, S18, S19 and L19 were digested within 10 min under the conditions employed here as determined by the disappearance of mass spectral peaks corresponding to the mass of the intact protein. The SDG studies revealed that the majority of the sample contains intact 70S ribosomes at this incubation period, thus we conclude that these ribosomal proteins are well-exposed on the ribosome surface or, at a minimum, have regions containing lysine(s) or arginine(s) that are sufficiently exposed for proteolysis. Similarly, r-proteins L12, L19, L25, L31, S3, S5, S7, S12, S18 and S19 were digested within 60 minutes upon incubation with Proteinase K.
In addition to the loss of mass spectral peaks from these r-proteins, new ion signals at various m/z values were detected during these analyses. The mass spectral data of the tryptic fragments of T. thermophilus are summarized in Table 2. The new m/z values that arise during the limited proteolysis were identified, where possible, by combining knowledge of the specificity of trypsin with the mass values and original masses of the intact r-proteins. As seen in Table 2, the proteolytic fragments generally arise from cleavage of N- and/or C-terminal domains of the various r-proteins.
Table 2.
MALDI-MS analysis of T. thermophilus proteolytic fragments generated by trypsin (Fig. 4). Protein fragments identified by asterisks, *, were still detected after 1500 min incubation with trypsin.
| Measured Mass [M+H]+ | Calculated Massa [M+H]+ | ΔM (Da) | Identity | Observed A.A. Residues | Loss of |
|---|---|---|---|---|---|
| 4552.4 | 4552.3 | 0.1 | L19* | 1-39 | C-terminus |
| 5321.6 | 5321.3 | 0.3 | L14*,b,c | 1-49 | C-terminus |
| 5426.5 | 5426.4 | 0.1 | L31* | 1-48 | C-terminus |
| 5449.9 | 5449.5 | 0.4 | L9* | 1-50 | N-terminus |
| 5653.6 | 5653.5 | 0.1 | L12*,c | 2-57 | C-terminus |
| 6116.9 | 6117.3 | -0.4 | L9 | 1-56 | C-terminus |
| 6127.4 | 6127.2 | 0.2 | L25 | 20-72 | N- and C-terminus |
| 7582.5 | 7582.1 | 0.4 | S18* | 24-88 | N-terminus |
| 7809.6 | 7809.4 | 0.2 | S18 | 22-88 | N-terminus |
| 8880.0 | 8879.3 | 0.7 | S3* | 2-79 | C-terminus |
| 8913.8 | 8913.3 | 0.5 | L27* | 6-85 | N-terminus |
| 9042.0 | 9041.5 | 0.5 | L27 | 5-85 | N-terminus |
| 9565.9 | 9565.2 | 0.7 | S19* | 2-85 | C-terminus |
| 9674.5 | 9674.3 | 0.2 | L9 | 62-148 | N-terminus |
| 9893.9 | 9894.6 | -0.7 | S19 | 2-88 | C-terminus |
| 10143.4 | 10143.9 | -0.5 | L9 | 58-148 | N-terminus |
| 10299.6 | 10300.1 | -0.5 | L9 | 57-148 | N-terminus |
| 10130.0 | 10130.8 | -0.8 | S16* | 1-85 | C-terminus |
| 11144.2 | 11144.1 | 0.1 | S13* | 2-99 | C-terminus |
| 11852.6 | 11853.2 | -0.6 | S17*,c | 2-101 | C-terminus |
| 12303.7 | 12303.1 | 0.6 | S11* | 13-129 | N-terminus |
| 12459.6 | 12459.3 | 0.3 | S11 | 12-129 | N-terminus |
| 12686.5 | 12686.6 | -0.1 | S11 | 10-129 | N-terminus |
| 13092.7 | 13092.3 | 0.4 | L19* | 1-111 | C-terminus |
| 13465.2 | 13465.8 | -0.6 | S13* | 2-120 | C-terminus |
| 13593.1 | 13593.9 | -0.8 | S13 | 2-121 | C-terminus |
| 14015.3 | 14015.7 | -0.4 | S12* | 2-127 | C-terminus |
| 14272.2 | 14272.8 | -0.6 | L19 | 1-120 | C-terminus |
| 15336.0 | 15335.1 | 0.9 | L16*,c | 6-141 | N-terminus |
| 18442.5 | 18441.3 | 1.2 | L1*,c | 20-191 | N- and C-terminus |
| 20594.5 | 20595.9 | -1.4 | L25* | 1-182 | C-terminus |
The average singly protonated mass of the tryptic products are shown as calculated using SequenceEditor ver. 1.0, provided by Bruker Daltonics and ProteinProspector.
The ion at m/z 5321 can be generated from L2 (168-216, m/z 5321.1) and L14 (1-49, m/z 5321.3). Although these cannot be differentiated at the mass accuracy available here, it is noted that the ion abundance of intact L14 but not L2 decreases with increasing incubation with trypsin.
Intact proteins also detected.
Comparison of E. coli and T. thermophilis limited proteolysis data
Figure 5 summarizes the trypsin limited proteolysis mass spectral data obtained in this study. As noted in this figure, there is generally good agreement between the limited proteolysis results found for E. coli and T. thermophilus, thus the method developed here reports on the structure of (bacterial) ribosomes. As discussed above, most r-proteins for both organisms are detected intact even after long periods of incubation with trypsin or Proteinase K and likely demonstrate no conformational changes upon dissociation of intact 70S ribosomes into the small and large subunits.
A few r-proteins, S3, S11, S18, S19 and L27, were readily digested (within 30 minutes) for both organisms suggesting similarities in trypsin susceptibility within the 70S structure. The tryptic peptide S3 (2-79) was detected in both organisms. S3 has been found to contain basic residues which protrude into the upstream tunnel of the entry pore for the mRNA [43]. S11 and S18 are believed to have extensive contacts with S1 [43], which was lost in the ribosome preparations used in this work, possibly accounting for their limited stability here. The C-terminus of S19 was digested with trypsin in these studies. This terminus points toward the interface side of 30S and this region was found disordered in the T. thermophilus 30S structure [37]. L27 is found to interact within the P site and S11 within the E site [18], which may account for their limited stability in this work as the ribosomes investigated did not have occupied A, P or E sites.
Among the r-proteins that were found to be particularly susceptible to trypsin, a larger number were unique to the particular organism being studied. For E. coli, S5, S6, S10 and L12 were significantly less stable than their T. thermophilus counterparts. Similarly, for T. thermophilus, S12, S13, S16, L11, L19, L25 and L31 were significantly less stable than their E. coli counterparts. Within this group, all proteins except L31 have longer C-termini tails than found in E. coli. These extended regions tend to be basic and solvent-exposed likely leading to the difference in results seen between T. thermophilus and E. coli, and L31 has recently been remodeled on the 2.8 Å 70S bacterial crystal structure into a position that is consistent with the ease it dissociates from the ribosome [18].
Comparison with Other Biophysical Information
Those proteins readily hydrolyzed in both cases should be surface accessible thus providing information on the ribosome topography. The positions of the r-proteins from the 30S and 50S subunits found to be readily digested by both trypsin and Proteinase K are shown on the E. coli 70S 3.5 Å crystal structure [17] and on the T. thermophilus 2.8 Å crystal structure [18] in Figure 6. The crystal structures used here do not contain coordinates for r-proteins S1, L7/L12, L10, L11 (T. thermophilus only), L36 (T. thermophilus only) and L28 (E. coli only). Although coordinates for L10 and L12 are not available, these proteins were found to be susceptible to limited proteolysis in this study. As seen in Figure 6, the majority of unstable r-proteins are within the 30S subunit, specifically in the head and platform regions. Such results are not surprising as these regions were found to be more disordered in earlier X-ray structures, which hindered assignment of specific ribosomal proteins [7, 23, 37]. The 50S subunit and the body of the 30S subunit contain significantly fewer r-proteins susceptible to limited proteolysis. Similar information is presented in Figure S5, which places those r-proteins likely to be susceptible to proteolytic digestion within the 70S ribosome surface for clarity. The similarities between the proteolytic digestion of 30S head and platform proteins from E. coli and T. thermophilus are readily recognized.
Figure 6.

Surface presentation of the ribosomal proteins digested with both trypsin and Proteinase K. Color scheme: rRNA is represented in gray. Category one proteins (susceptible to digestion within 30 min) are represented in yellow. Category two proteins (susceptible to digestion within 125 min) are represented in green. Category three proteins (stable to digestion up to 1000 min) are represented in orange (see text). Proteins that were still detected intact even after 1500 min of incubation are represented in brown. (a-b) Views of the surfaces of the E. coli 70S ribosome with protease susceptible proteins identified. (a) to (b) are 180° rotations about the vertical axis. (a) View from the left-hand side with the 30S subunit on the right and the 50S subunit on the left. (b) View from the right-hand side with the 30S subunit on the left and the 50S subunit on the right. (c-d) Views of the surfaces of the T. thermophilus 70S ribosome with protease susceptible proteins identified. (c) to (d) are 180° rotations about the vertical axis with similar perspectives as for E. coli. Images of the 30S and 50S subunit structures were produced using MacPyMol [36] from the 3.5 Å crystal structure of E. coli (accession 2AVY and 2AW4) [17] and the 2.8 Å crystal structure of T. thermophilus (accession 2J00 and 2J01) [18].
Recently, Yamamoto and co-workers utilized MALDI-MS in combination with H/D exchange to probe the flexibility of E. coli 70S ribosomes [10]. They concluded that the extent of deuterium incorporation is closely related to the 30S and 50S assembly processes, with highly deuterated proteins being incorporated late in the assembly process. Because that H/D exchange approach, as well as the approach developed here, should probe ribosome topology, it is of interest to note that these limited proteolysis results are found to be only marginal related to assembly order. For example, r-proteins L6, L14, L16, L19, L25, L27, L28, L30, L31 and L32 are incorporated late in the assembly process of the 50S subunit [44, 45]. Within this list, only L19, L27 and L31 were found to be particularly susceptible to limited proteolysis with either protease. Moreover, each of these three r-proteins yielded stable tryptic fragments that were detectable even after 1500 min incubation with trypsin (Table 1). More relevant than the assembly process is the surface accessibility of these proteins as seen in Figure 6. Within the small subunit, the r-proteins incorporated late in the assembly process include S2, S3, S10, S14 and S21 [46, 47]. All except S14 are readily digested, but in addition it was also found that S5, S6, S11, S18 and S19 are also readily digested. As with the 50S r-proteins, limited proteolysis of 30S r-proteins reports surface accessibility of protease sites more than their stage in the assembly process.
Utility of the Present Method
There are several advantages to using MALDI-MS as the detection step for limited proteolysis of ribosomes. Because molecular weight is an intrinsic property, a detection method that reports molecular weights can be used to directly identify those proteins resistant to proteolysis. In addition, when a protease of high specificity, such as trypsin, is used, the identification of proteolytic fragments is also possible, especially when the hydrolyzed peptide is from the N- or C-terminus of the protein. In that case, a simple comparison of measured molecular weight values to those predicted based upon loss of an N- or C-terminus tryptic peptide is all that is needed to assign the identity of the proteolytic fragment and site of proteolysis (Figures S5 and S6). When internal peptide domains are cleaved, data analysis and interpretation becomes more difficult primarily due to the greater number of theoretical cleavage masses that must be calculated. One limitation of the approach described here is that some r-proteins, L4, L5, S1 and S2 (T. thermophilus only) are not detected in the control experiments, although S1 is known to be easily lost during most ribosome preparations. Furthermore, some proteins may be difficult to detect when all r-proteins are analyzed due to MALDI suppression effects as noted previously [48]. There are additional limitations of this approach given the time resolution available to study ribosome conformations. For example, tracking conformational rearrangements during translation via the specific experimental procedure presented in this work would not provide sufficient time resolution to reveal any meaningful information on that process. However, this approach should be applicable to studies that investigate binding/release of external ribosome factors, providing such interactions are sufficiently long-lived.
With the advent of high resolution crystal structures [17, 18], a significant advancement in our understanding of ribosome structure and function has resulted. However, as has been noted [49, 50], high resolution crystal structures are limited to a very small number of organisms and information from one organism can inform but not directly replace information from another organism. Thus, methods such as that developed here which reveal low resolution information regarding ribosome organization are still necessary, and can be especially useful for organisms for which no high resolution crystal structures are yet available. As noted in this work, information obtained by MALDI-MS can provide specific identification of r-proteins relatively resistant to proteolytic digestion, can denote r-proteins readily digested, and can identify stable r-protein proteolytic fragments. The approach presented does not destroy the overall structure of ribosomal subunits, thus the MALDI data must be reporting on surface accessible r-proteins. One of the drawbacks of the direct MALDI-MS readout of ribosomal proteins and proteolytic fragments is that, in the absence of any detected proteolytic fragment, one cannot determine whether a particular r-protein is being digested by the protease. Although this drawback is not inherent to MALDI-MS, it does preclude obtaining quantitative data on protein stability in the absence of isotopic labeling or internal standards.
CONCLUSIONS
The use of MALDI-MS for the direct determination of r-proteins resistant to limited proteolysis has been demonstrated. This approach can be used at the level of intact ribosomes and can provide information on sites of proteolysis when proteases of high specificity are used. The approach yielded information of ribosome topology that is comparable to that previously obtained by other approaches. The speed and sensitivity of MALDI-MS, combined with the measurement of molecular mass, an intrinsic property of proteins, will allow this approach to be extended to ribosomes from additional organisms. Moreover, this approach provides a general route to obtaining information on the topology, organization and conformational changes associated with RNP complexes.
The data obtained using E. coli and T. thermophilus ribosomes agrees well with existing crystal structures suggesting that this method is applicable to additional organisms, including eukaryotic systems that are significantly larger and more complex. In addition to providing information on intact ribosomes, this approach should be applicable to examining ribosome dissociation processes, subunit associations and conformational changes associated with the binding of external ribosomal factors. Another particular useful extension of this method would be the investigation of other RNP complexes for which little high-resolution structural data exists, such as the ribosomal subunit assembly intermediates [45, 46, 51]. Thus, while this present study does not advance directly our understanding of the E. coli or T. thermophilus ribosomes, it does demonstrate the feasibility of a relatively straight-forward and rapid mass spectrometric approach for limited proteolysis of large RNP complexes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Steven T. Gregory and Albert E. Dahlberg (Brown University) for the gift of T. thermophilus and Drs. S. Gregory and Yolanda Sanchez for helpful discussions. Financial support of this work was provided by the National Institutes of Health (GM 58843) and the University of Cincinnati.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Urlaub H, Kruft V, Bischof O, Muller E, Wittmann-Liebold B. Protein-rRNA binding features and their structural and functional implications in ribosomes as determined by cross-linking studies. EMBO J. 1995;14:4578–4588. doi: 10.1002/j.1460-2075.1995.tb00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiede B, Urlaub H, Neubauer H, Grelle G, Wittmann-Liebold B. Precise determination of RNA-protein contact sites in the 50S ribosomal subunit of Escherichia coli. Biochem. J. 1998;334:39–42. doi: 10.1042/bj3340039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambert JM, Boileau G, Cover JA, Traut RR. Cross-links between ribosomal proteins of 30S subunits in 70S tight couples and in 30S subunits. Biochemistry. 1983;22:3913–3920. doi: 10.1021/bi00285a029. [DOI] [PubMed] [Google Scholar]
- 4.Pohl T, Wittmann-Liebold B. Identification of a cross-link in the Escherichia coli ribosomal protein pair S13-S19 at the amino acid level. J. Biol. Chem. 1988;263:4293–4301. [PubMed] [Google Scholar]
- 5.Spahn CMT, Beckmann R, Eswar N, Penczek PA, Sali A, Blobel G, Frank J. Structure of the 80S Ribosome from Saccharomyces cerevisiae—tRNA-Ribosome and Subunit-Subunit Interactions. Cell. 2001;107:373–386. doi: 10.1016/s0092-8674(01)00539-6. [DOI] [PubMed] [Google Scholar]
- 6.Gao H, Sengupta J, Valle M, Korostelev A, Eswar N, Stagg SM, Van Roey P, Agrawal RK, Harvey SC, Sali A, Chapman MS, Frank J. Study of the structural dynamics of the E. coli 70S ribosome using real-space refinement. Cell. 2003;113:789–801. doi: 10.1016/s0092-8674(03)00427-6. [DOI] [PubMed] [Google Scholar]
- 7.Walleczek J, Schuler D, Stoffler-Meilicke M, Brimacombe R, Stoffler G. A model for the spatial arrangement of the proteins in the large subunit of the Escherichia coli ribosome. EMBO J. 1988;7:3571–3576. doi: 10.1002/j.1460-2075.1988.tb03234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agafonov DE, Kolb VA, Spirin AS. Proteins on ribosome surface: Measurements of protein exposure by hot tritium bombardment technique. Proc. Natl. Acad. Sci. U.S.A. 1997;94:12892–12897. doi: 10.1073/pnas.94.24.12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christodoulou J, Larsson G, Fucini P, Connell SR, Pertinhez TA, Hanson CL, Redfield C, Nierhaus KH, Robinson CV, Schleucher J, Dobson CM. Heteronuclear NMR investigations of dynamic regions of intact Escherichia coli ribosomes. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10949–10954. doi: 10.1073/pnas.0400928101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto T, Izumi S, Gekko K. Mass Spectrometry of Hydrogen/Deuterium Exchange in 70S Ribosomal Proteins from E. coli. FEBS Lett. 2006;580:3638–3642. doi: 10.1016/j.febslet.2006.05.049. [DOI] [PubMed] [Google Scholar]
- 11.Ilag L, Videler H, McKay A, Sobott F, Fucini P, Nierhaus K, Robinson C. Heptameric (L12)6/L10 rather than canonical pentameric complexes are found by tandem MS of intact ribosomes from thermophilic bacteria. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8192–8197. doi: 10.1073/pnas.0502193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rostom A, Fucini P, Benjamin D, Juenemann R, Nierhaus K, Hartl F, Dobson C, Robinson C. Detection and selective dissociation of intact ribosomes in a mass spectrometer. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5185–5190. doi: 10.1073/pnas.97.10.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benjamin D, Robinson C, Hendrick J, Hartl F, Dobson C. Mass spectrometry of ribosomes and ribosomal subunits. Proc. Natl. Acad. Sci. USA. 1998;95:7391–7395. doi: 10.1073/pnas.95.13.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 15.Wimberly BT, Broderson DE, Clemons WM, Morgan-Warrent RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 16.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JHD, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 17.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JHD. Structures of the Bacterial Ribosome at 3.5 A Resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 18.Selmer M, Dunham CM, Murphy FV, IV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S Ribosome Complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 19.Peng B-H, Lee JC, Campbell GA. In Vitro Protein Complex Formation with Cytoskeleton-anchoring Domain of Occludin Identified by Limited Proteolysis. J. Biol. Chem. 2003;278:49644–49651. doi: 10.1074/jbc.M302782200. [DOI] [PubMed] [Google Scholar]
- 20.Meyer EL, Strutz N, Gahring LC, Rogers SW. Glutamate Receptor Subunit 3 Is Modified by Site-specific Limited Proteolysis Including Cleavage by γ-Secretase. J. Biol. Chem. 2003;278:23786–23796. doi: 10.1074/jbc.M301360200. [DOI] [PubMed] [Google Scholar]
- 21.Hijarrubia MJ, Aparicio JF, Martin JF. Domain Structure Characterization of the Multifunctional alpha -Aminoadipate Reductase from Penicillium chrysogenum by Limited Proteolysis. Activation of alpha -Aminoadipate does not require the Peptidyl Carrier Protein or the Reduction Domain. J. Biol. Chem. 2003;278:8250–8256. doi: 10.1074/jbc.M211235200. [DOI] [PubMed] [Google Scholar]
- 22.Gudkov AT, Gongadze GM. The L7/L12 proteins change their conformation upon interaction of EF-G with ribosomes. FEBS Letter. 1984;176:32–36. doi: 10.1016/0014-5793(84)80906-0. [DOI] [PubMed] [Google Scholar]
- 23.Kruft V, Wittmann-Liebold B. Determination of Peptide Regions on the Surface of the Eubacterial and Archaebacterial Ribosome by Limit Proteolytic Digestion. Biochemistry. 1991;30:11781–11787. doi: 10.1021/bi00115a007. [DOI] [PubMed] [Google Scholar]
- 24.Cohen SL, Ferre-D’amare AR, Burley SK, Chait BT. Probing the solution structure of the DNA-binding protein Max by a combination of proteolysis and mass spectrometry. Protein Sci. 1995;4:1088–1099. doi: 10.1002/pro.5560040607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fontana A, Zambonin M, de Laureto PP, Filippis VD, Clementi A, Scaramella E. Probing The Conformational State of Apomyoglobin by Limited Proteolysis. J. Mol. Biol. 1997;266:223–230. doi: 10.1006/jmbi.1996.0787. [DOI] [PubMed] [Google Scholar]
- 26.Yang F, Cheng Y, Peng J, Zhou J, Jing G. Probing the conformational state of a truncated staphylococcal nuclease R using time of flight mass spectrometry with limited proteolysis. Eur. J. Biochem. 2001;268:4227–4232. doi: 10.1046/j.1432-1327.2001.02337.x. [DOI] [PubMed] [Google Scholar]
- 27.Leite JF, Amoscato AA, Cascio M. Coupled proteolytic and mass spectrometry studies indicate a novel topology for the glycine receptor. J. Biol. Chem. 2000;275:13683–13689. doi: 10.1074/jbc.275.18.13683. [DOI] [PubMed] [Google Scholar]
- 28.Bothner B, Dong XF, Bibbs L, Johnson JE, Siuzdak G. Evidence of viral capsid dynamics using limited proteolysis and mass spectrometry. J. Biol. Chem. 1998;273:673–676. doi: 10.1074/jbc.273.2.673. [DOI] [PubMed] [Google Scholar]
- 29.Gervasoni P, Staudenmann W, James P, Plueckthun A. Identification of the binding surface on β-lactamase for GroEL by limited proteolysis and MALDI-mass spectrometry. Biochemistry. 1998;37:11660–11669. doi: 10.1021/bi980258q. [DOI] [PubMed] [Google Scholar]
- 30.Cameron DM, Gregory ST, Thompson J, Suh M-J, Limbach PA, Dahlberg AE. Thermus thermophilus L11 Methyltransferase, PrmA, Is Dispensable for Growth and Preferentially Modifies Free Ribosomal Protein L11 Prior to Ribosome Assembly. J. Bacteriol. 2004;186:5819–5825. doi: 10.1128/JB.186.17.5819-5825.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spedding G. Ribosomes and Protein Synthesis. A Practical Approach Oxford University Press; 1990. Ribosomes and Protein Synthesis: A Practical Approach; pp. 1–27. [Google Scholar]
- 32.Stanley WM, Jr., Bock RM. Isolation and Physical Properties of the Ribosomal Ribonucleic Acid of Escherichia coli. Biochemistry. 1965;4:1302–1311. doi: 10.1021/bi00883a014. [DOI] [PubMed] [Google Scholar]
- 33.Rowley A, Choudhary JS, Marzioch M, Ward MA, Weir M, Solari RCE, Black WP. Application of protein mass spectrometry in cell biology. Methods. 2000;20:383–397. doi: 10.1006/meth.2000.0951. [DOI] [PubMed] [Google Scholar]
- 34.Suh M-J, Limbach PA. Investigation of protein isolation methods suitable for the Matrix-Assisted Laser Desorption/Ionization mass spectrometric analysis of ribonucleoprotein complexes. Eur. J. Mass Spectrum. 2004;10:89–99. doi: 10.1255/ejms.626. [DOI] [PubMed] [Google Scholar]
- 35.Clauser KR, Baker P, Burlingame AL. Role of Accurate Mass Measurement (±10 ppm) in Protein Identification Strategies Employing MS or MS/MS and Database Searching. Anal. Chem. 1999;71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- 36.DeLano WL. The PyMol Molecular Graphics System. DeLano Scientific; Palo Alta, CA, USA: 2002. http://www.pymol.org [Google Scholar]
- 37.Brodersen DE, Clemons WM, Carter AP, Wimberly BT, Ramakrishnan V. Crystal structure of the 30 S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16 S RNA. J. Mol. Biol. 2002;316:725–768. doi: 10.1006/jmbi.2001.5359. [DOI] [PubMed] [Google Scholar]
- 38.Noller HF, Hoffarth V, Zimniak L. Unusual resistance of peptidyl transferase to protein extraction procedures. Science. 1992;256:1416–1419. doi: 10.1126/science.1604315. [DOI] [PubMed] [Google Scholar]
- 39.Khaitovich P, Mankin AS, Green R, Lancaster L, Noller HF. Characterization of functionally active subribosomal particles from Thermus aquaticus. Proc. Natl. Acad. Sci. U.S.A. 1999;96:85–90. doi: 10.1073/pnas.96.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Littlechild J, Malcolm A, Paterakis K, Ackermann I, Dijk J. The tertiary structure of salt-extracted ribosomal proteins from Escherichia coli as studied by proton magnetic resonance spectroscopy and limited proteolysis experiments. Biochim. Biophys. Acta. 1987;913:245–255. doi: 10.1016/0167-4838(87)90336-0. [DOI] [PubMed] [Google Scholar]
- 41.Allers J, Shamoo Y. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J. Mol. Biol. 2001;311:75–86. doi: 10.1006/jmbi.2001.4857. [DOI] [PubMed] [Google Scholar]
- 42.Suh M-J, Hamburg D-M, Gregory ST, Dahlberg AE, Limbach PA. Extending Ribosomal Protein Identifications to Unsequenced Bacterial Strains Using Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Proteomics. 2005;5:4818–4831. doi: 10.1002/pmic.200402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson DN, Nierhaus KH. Ribosomal Proteins in the Spotlight. Critical Rev Biochem Mol Biol. 2005;40:243–267. doi: 10.1080/10409230500256523. [DOI] [PubMed] [Google Scholar]
- 44.Dohme F, Nierhaus KH. Total Reconstitution and Assembly of 50S subunits from Escherichia coli Ribosomes in Vitro. J. Mol. Biol. 1976;107:585–599. doi: 10.1016/s0022-2836(76)80085-x. [DOI] [PubMed] [Google Scholar]
- 45.Herold M, Nierhaus K. Incorporation of six additional proteins to complete the assembly map of the 50 S subunit from Escherichia coli ribosomes. J. Biol. Chem. 1987;262:8826–8833. [PubMed] [Google Scholar]
- 46.Held WA, Ballou B, Mizushima S, Nomura M. Assembly mapping of 30S ribosomal proteins from Escherichia coli. Further studies. J. Biol. Chem. 1974;249:3103–3111. [PubMed] [Google Scholar]
- 47.Mizushima S, Nomura M. Assembly mapping of 30S ribosomal proteins from E. coli. Nature. 1970;226:1214–1218. doi: 10.1038/2261214a0. [DOI] [PubMed] [Google Scholar]
- 48.Arnold RJ, Reilly JP. Observation of E. coli ribosomal proteins and their posttranslational modification by mass spectrometry. Anal. Biochem. 1999;269:105–112. doi: 10.1006/abio.1998.3077. [DOI] [PubMed] [Google Scholar]
- 49.Sergiev P, Leonov A, Dokudovskaya S, Shpanchenko O, Dontsova O, Bogdanov A, Rinke-Appel J, Mueller F, Osswald M, von Knoblauch K, Brimacombe R. The Ribosome. Cold Spring Harbor Laboratory Press, Cold Spring Harbor; NR: 2001. “Correlating the X-ray structures for halo- and thermophilic ribosomal subunits with biochemical data for the Escherichia coli ribosome”. [DOI] [PubMed] [Google Scholar]
- 50.Sergiev PV, Dontsova OA, Bogdanov AA. Chemical methods for the structural study of the ribosome: Judgment day. Mol. Biol. 2001;35:472–496. [PubMed] [Google Scholar]
- 51.Holmes KL, Culver GM. Mapping structural differences between 30S ribosomal subunit assembly intermediates. Nat. Struct. Biol. 2004;11:179–186. doi: 10.1038/nsmb719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.