

Abstract
A hallmark of systemic lupus erythematosus and the MRL murine model for lupus is the presence of anti–double-stranded (ds)DNA antibodies (Abs). To identify the steps leading to the production of these Abs in autoimmune mice, we have compared the phenotype and localization of anti-dsDNA B cells in autoimmune (MRL+/+ and lpr/lpr) mice with that in nonautoimmune (BALB/c) mice. Anti-dsDNA B cells are actively regulated in BALB/c mice as indicated by their developmental arrest and accumulation at the T–B interface of the splenic follicle. In the MRL genetic background, anti-dsDNA B cells are no longer developmentally arrested, suggesting an intrinsic B cell defect conferred by MRL background genes. With intact Fas, they continue to exhibit follicular exclusion; however, in the presence of the lpr/lpr mutation, anti-dsDNA B cells are now present in the follicle. Coincident with the altered localization of anti-dsDNA B cells is a follicular infiltration of CD4 T cells. Together, these data suggest that MRL mice are defective in maintaining the developmental arrest of autoreactive B cells and indicate a role for Fas in restricting entry into the follicle.
Keywords: tolerance, Fas, autoimmunity, antinuclear antibody, splenic architecture
Adefining feature of most autoimmune diseases is the presence of autoantibodies. The factors that account for the production of autoantibodies in diseased individuals and their absence in healthy individuals are not clear. Many studies have examined the characteristics of autoantibodies and disease pathology after the disease process is well underway; however, few studies have followed the fate of the autoreactive B cells themselves both before and after initiation of disease. To understand how tolerance is maintained in the B cell compartment, we and others have used Ig transgene (Tg)1 models that increase the frequency of autoreactive B cells and allow their fate to be tracked (1–5). Studies using Ig Tgs directed toward the model Ags hen egg lysozyme (HEL) and MHC class I have described several fates for autoreactive B cells in nonautoimmune mice, including clonal deletion, anergy, and receptor editing. The conclusion from these studies is that the fate of an autoreactive B cell depends on many factors, including the presence of T cell help and the form and location of the Ag (1–3, 6). Similar manifestations of B cell tolerance have also been described using Ig Tg models for Ags that are targeted in autoimmune diseases such as SLE (5, 7–13). Given that the nature of the in vivo Ag for SLE-associated specificities has not been identified, the role that different forms of Ag, as well as affinity, play in dictating a deleting versus anergizing phenotype cannot be assigned.
The MRL-lpr/lpr mouse is a much-studied model of systemic autoimmunity. MRL-lpr/lpr mice develop a similar autoantibody profile to SLE patients, with serum Abs directed against many nuclear Ags, such as DNA and histones (14). MRL+/+ mice, which do not carry the lpr mutation in Fas (15, 16), also develop autoantibodies, although with delayed kinetics (17). For both MRL-lpr/lpr and MRL+/+ mice, serum autoantibodies are not present at birth; instead the mice seroconvert as adults (14). What causes this breakdown in tolerance is unclear; potential candidates include dysregulation of autoreactive B cells, T cells, and/or a difference in the nature of the Ag(s) being recognized.
Using model Ags, many studies have addressed whether lpr/lpr mice are globally defective in B and/or T cell tolerance. Although one study suggests that negative selection to high doses of Ag is affected by the lpr/lpr mutation (18), the overwhelming conclusion from numerous reports is that, overall, both B and T cell tolerance is intact in lpr/lpr mice (19–23). However, all of the examples cited were investigating tolerance to Ags that are not spontaneously targeted in disease. MRL-lpr/lpr mice do produce autoantibodies to nuclear Ags, implying that tolerance to specific self-Ags is no longer intact. Importantly, the steps leading to this breakdown in tolerance have not been identified.
To understand how anti-DNA B cells are regulated, we have used the VH3H9 Tg, which was repeatedly isolated from anti-DNA Abs produced by diseased MRL-lpr/lpr mice (24). The VH3H9 H chain can pair with a variety of endogenous L chains to generate anti-DNA Abs as well as non-DNA Abs (25). Therefore, VH3H9 Tg mice give us the ability to study anti-DNA B cells, in the context of B cells with other specificities, in both nonautoimmune and autoimmune environments. Using the VH3H9 Tg on a nonautoimmune (BALB/c) background, we have identified a population of anti–double-stranded (ds)DNA B cells that persist in the periphery (10). These cells are developmentally arrested and have a shortened life span. Additionally, they localize to the T–B interface of the splenic follicle. These features of anergy are similar to those of anergic anti-HEL B cells in a diverse repertoire, with the exception that anti-HEL B cells are reported as not being developmentally arrested (3, 26, 27).
The presence of anti-DNA Abs in MRL-lpr/lpr mice suggests that the regulation of anti-DNA B cells is no longer intact; however, it does not tell us how either MRL genes or the lpr/lpr mutation influence the loss of tolerance. Although it is clear that there are distinct MRL and lpr contributions to autoimmunity, their precise consequences for the fate of autoreactive B cells have not been defined. To determine more specifically how the regulation of anti-dsDNA B cells breaks down in the autoimmune environment, the VH3H9 Tg was bred onto the MRL-lpr/lpr and MRL+/+ backgrounds and the fate of anti-dsDNA B cells monitored. Comparing the phenotype of anti-dsDNA B cells in MRL mice to that in BALB/c mice allows us to identify differences that may account for the production of anti- dsDNA Abs in autoimmune mice. Additionally, by examining VH3H9 MRL-lpr/lpr mice before and after anti- dsDNA Ab is detected in the serum, we may be able to correlate phenotypic changes with the onset of seroconversion. Finally, by comparing the fate of anti-dsDNA B cells in MRL-lpr/lpr with MRL+/+ mice, the contribution of Fas in the elimination of autoreactive B cells may be identified.
In this study, we document several changes in the status of anti-dsDNA B cells in both MRL+/+ and MRL-lpr/lpr mice that precede the expression of serum autoantibodies. MRL+/+ and MRL-lpr/lpr mice exhibit a defect in maintaining the developmental arrest of anti-dsDNA B cells. In MRL-lpr/lpr mice, two additional changes have been identified: the anti-dsDNA B cells are now able to enter the B cell follicle, and the T/B lymphoid architecture is disrupted before seroconversion. Thus, this study reveals potential mechanisms by which the MRL genetic background and the lpr mutation in Fas contribute to autoantibody production.
Materials and Methods
Mice
BALB/c mice were purchased from Harlan Sprague-Dawley, Inc. MRL-lpr/lpr and MRL+/+ mice were purchased from The Jackson Laboratory. VH3H9 Tg mice have been described previously (4). The VH3H9 Tg mice have been backcrossed onto the BALB/c and MRL backgrounds for at least 9 and 17 generations, respectively, and have been bred and maintained in the animal facility at The Wistar Institute. VH3H9 MRL-lpr/lpr mice deficient in the JH locus were obtained from M. Shlomchik (Yale University, New Haven, CT [28]). In all cases, age-matched BALB/c mice and/or Tg− littermates were used as controls. The presence of the VH3H9 Tg was determined by PCR amplification of tail DNA with primers specific for VH3H9 (4).
Antinuclear Antibody Assay
The presence of antinuclear Abs (ANAs) in serum was detected using permeabilized HEP-2 cells as the substrate (Antibodies Incorporated). Manufacturer's instructions were followed with serum samples tested at a 1:100 dilution. Anti-homogeneous nuclear (HN) ANA binding and ANA titers were detected with a combination, defined as total Ig, of FITC-conjugated goat anti–mouse Ig(H+L) and anti–mouse IgM reagents (Southern Biotechnology Associates). To test for the presence of λ ANAs, an FITC-conjugated goat anti–mouse λ reagent (Southern Biotechnology Associates) or LS136-biotin (anti-λ1; gift of Garnett Kelsoe, Duke University, Durham, NC) followed by streptavidin (SA)-FITC (Fisher Biotech) was used. The samples were then visualized under a fluorescent microscope and scored blind, without knowledge of age or genotype of the mice. The results using the monoclonal LS136 and polyclonal anti-λ reagents were comparable.
ANA Detection in Seroconversion.
Sera were tested from six litters of mice. These mice were bled once a week between 6 and 16 wk of age. These serial samples were tested, in the protocol described above, for the presence of total Ig anti-HN and Igλ ANAs. The first positive bleed was considered the seroconversion time point.
ANA Titers.
The anti-HN seroconversion time points of a subset of Tg− and VH3H9 MRL-lpr/lpr mice were examined for total Ig ANA titers. In each case, the time point was 10 wk of age. These samples were tested at an initial 1:100 dilution and at serial 10-fold dilutions. The serum titer was defined as the reciprocal of the last dilution at which positive staining was seen.
Cell Preparations
Bone marrow (BM), spleen, and LN cells were removed from VH3H9 Tg and Tg− mice. Single-cell suspensions were prepared and, where necessary, erythrocytes were removed by hypotonic lysis.
Identification of VH3H9/Vλ1 B Cells
Because the VH3H9 H chain Tg has been shown to be a good excluder of endogenous H chain rearrangement in the BALB/c background, we have followed the fate of anti-dsDNA B cells in VH3H9 Tg mice using anti-λ–specific reagents (4). Additionally, the restricted use of VH3H9 by λ+ cells was confirmed using JH−/− MRL-lpr/lpr mice (28). Several different reagents were used to track λ+ and λ1+ B cells (LS136, R11-153, JC5, and R26-46). Using these reagents and flow cytometry we have shown that the majority of λ+ B cells in VH3H9 Tg mice are λ1 (10). Therefore, we are able to follow VH3H9/Vλ1 B cells in MRL mice using anti-pan λ reagents.
Flow Cytometry Analysis
Cells (5 × 105) were surface stained according to standard protocols (29). The following Abs were used: RA3-6B2–FITC, –PE, or –biotin (anti-B220), R11-153–FITC (anti-Vλ1), R26-46–FITC (anti-Vλ total), R8-140–PE (anti-Igκ), 1D3-FITC or -biotin (anti-CD19), 7G6-FITC (anti-CD21/35), Cy34.1-FITC (anti-CD22), B3B4-FITC (anti-CD23), 3/23-FITC (anti-CD40), Mel-14–FITC (anti-CD62L, L-selectin), M1/69-FITC (anti–heat-stable antigen [HSA]), and IM7-FITC (anti-CD44) (PharMingen); 7E9-FITC (anti-CD21/35; gift of A. Naji, University of Pennsylvania, Philadelphia, PA); LS136-biotin (anti-Vλ1; gift of G. Kelsoe) and JC5.1-PE (anti-Vλ total; gift of J. Kearney, University of Alabama, Birmingham, AL); polyclonal anti-IgM–PE and SBA-1–PE (anti-IgD; Southern Biotechnology Associates); SA-Red670 (GIBCO BRL); SA-FITC (Fisher Biotech); and SA-PE (Vector Laboratories).
All samples were analyzed on a FACScan™ flow cytometer (Becton Dickinson) using CellQuest software. 15,000–40,000 events were collected for each sample and gated for live lymphocytes based on forward and side scatter.
In analyzing the cell surface phenotype, it became apparent that SA-Red670 binds to B cells from MRL-lpr/lpr mice. The reagent does not bind to conventional T cells in MRL-lpr/lpr mice, but does bind to ∼10% of the B220+CD3+CD4−CD8− (double-negative [DN]) T cells that accumulate. This binding segregates with the mouse strain: BALB/c and C57BL/6 B cells do not bind to SA-Red670, whereas MRL-lpr/lpr and MRL+/+ B cells do bind (data not shown). SA-Red670 binding is not due to the SA in the conjugate because neither SA-FITC nor SA-PE binds to MRL-derived cells (data not shown). Because Red670 is composed of PE plus a Cy5 residue (30), this suggests that Cy5 mediates the binding. It is unclear what Red670 binds on the B cell, but it does not appear to be mediated through Ig since the Iglow cells have the same amount of Red670 binding as the Ighigh cells (data not shown). Because all B cells in MRL mice and only a small fraction of DN T cells bind to SA-Red670, in some experiments we have used SA-Red670 as a marker to enrich for B cells. To identify B cells specifically, a second B cell marker that is absent on DN T cells, such as CD19 or Ig, was used to identify the B cells.
Immunohistochemistry
Spleens were suspended in OCT, frozen in 2-methyl-butane cooled with liquid nitrogen, sectioned, and fixed with acetone. The spleen sections were stored at −20°C and then stained according to the protocol described (31). In brief, the sections were blocked using PBS/5% normal goat serum (Sigma Chemical Co.)/0.1% Tween-20 and then stained with GK1.5-biotin (anti-CD4), 53-6.7–biotin (anti-CD8), 30H12-FITC (anti–Thy-1.2) (grown as supernatants), Cy34.1-FITC (anti-CD22), 281-2 (anti–syndecan-1) (PharMingen), MOMA-1 (anti–marginal metallophilic macrophages; Bachem), and/or anti-Igλ–alkaline phosphatase (AP) (Southern Biotechnology Associates). Streptavidin– horseradish peroxidase (HRP) or -AP (Southern Biotechnology Associates), polyclonal anti–rat-HRP (Jackson ImmunoResearch Laboratories), MAR18.5-biotin (anti–rat Ig, grown as supernatant), and anti-FITC–AP (Sigma Chemical Co.) or anti-FITC–HRP (Chemicon) were used as secondary Abs. HRP and AP were developed using the substrates 3-amino-9-ethyl-carbazole (3-AEC) and Fast Blue BB base (Sigma Chemical Co.), respectively.
Enzyme-linked Immunospot Assay
Splenic B cells were plated at 4 × 105 cells/well and diluted serially 1:4 in Multiscreen HA mixed cellulose ester membrane plates (Millipore) coated with unlabeled goat anti–mouse total Ig (Southern Biotechnology Associates). The Ig secreted by the plated cells was detected by AP-conjugated goat anti–mouse total Ig or Igλ as secondary Abs (Southern Biotechnology Associates) and visualized using NBT/BCIP substrate (nitroblue tetrazolium/ 5-bromo-4-chloro-3-indolyl phosphate; Sigma Chemical Co.).
Statistical Analysis
Statistical significance was determined using an unpaired nonparametric test and Instat software.
Results
In this study, we have used the VH3H9 Tg to track a specific population of anti-dsDNA B cells, in the context of a diverse repertoire, and compare their fate in nonautoimmune and autoimmune environments. Transfection and hybridoma analyses have shown that the germline Vλ1 L chain pairs with the VH3H9 H chain to generate an anti-dsDNA ANA+ Ab (25, 32). Using anti-λ–specific reagents, we have followed B cells of this specificity in terms of serum Ab expression, surface phenotype, splenic localization, and ability to differentiate into antibody-forming cells (AFCs).
The Kinetics of Seroconversion in MRL-lpr/lpr Mice.
As a first step in studying VH3H9 MRL-lpr/lpr mice, we examined the overall rate at which ANAs appeared in the serum. An age-matched cohort of Tg− and VH3H9 MRL-lpr/lpr mice was bled weekly from age 6 to 16 wk. The sera were tested for the presence of ANAs using reagents to detect total Ig, and scored as positive when they showed an HN staining pattern. This ANA pattern is found in a high frequency of SLE serum and correlates with the presence of anti-dsDNA, antihistone, and antichromatin Abs (33). Fig. 1 A shows that Tg− MRL-lpr/lpr mice became serum HN ANA+ at approximately the same time (10.1 ± 3.0 wk) as VH3H9 MRL-lpr/lpr mice (9.1 ± 2.0 wk; P = 0.2). To test if the VH3H9 Tg alters the amount of ANA in the serum, ANA titers were determined for a subset of the mice (Fig. 1 B). Again, no difference was detected between Tg− and VH3H9 MRL-lpr/lpr mice. Likewise, Fig. 1 C shows that the VH3H9 MRL-lpr/lpr mice seroconvert to Igλ ANA+ at an average of 9.2 ± 1.4 wk, the same time when total HN ANAs appear in the serum (9.1 ± 2.0 wk). Within the time-frame analyzed (ages 6.0–43.5 wk), the VH3H9/λ1 specificity was not detected in serum from MRL+/+ mice (data not shown). The fact that most Tg− MRL-lpr/lpr mice failed to seroconvert to Igλ ANA+ is consistent with data showing that the overwhelming majority of ANAs in Tg− MRL-lpr/lpr mice use the more abundant Igκ L chains (34). Given that VH3H9/Vλ1 ANAs arise with the same kinetics as other ANAs in both Tg− and Tg+ MRL-lpr/lpr mice, VH3H9/Vλ1 B cells appear to be a good model for following the fate of ANA B cells in general.
Figure 1.

The VH3H9 Tg does not change the seroconversion time point or titer of ANAs but does increase serum λ ANA frequency. An age-matched cohort of Tg− (○) and VH3H9 (•) MRL-lpr/lpr mice was bled weekly from the age of 6 to 16 wk. Sera were tested for the presence of ANAs by immunofluorescence. (A) Symbols represent individual mice at the age when anti-HN ANAs first appear. Tg− MRL-lpr/lpr mice became serum ANA+ at approximately the same time (10.1 ± 3.0 wk) as VH3H9 MRL-lpr/lpr mice (9.1 ± 2.0 wk; P = 0.2). n = 25 mice of each genotype. (B) ANA titers were determined for the seroconversion time point (10 wk) of a subset of mice (n = 6 mice of each genotype). The serum titer is defined as the reciprocal of the last dilution at which positive staining was seen. No difference was detected between Tg− and VH3H9 MRL-lpr/lpr mice. (C) Serum samples were tested for the appearance of λ ANAs. Symbols placed above the dotted line represent mice at the age when λ ANAs were first detected. Below the dotted line are symbols representing mice that failed to show λ ANAs by 15 wk of age. n = 17 Tg− MRL-lpr/lpr, n = 18 VH3H9 MRL-lpr/lpr mice.
Anti-dsDNA B Cells in VH3H9 Mice Show Evidence of Ag Encounter in the BM.
Having established the kinetics of seroconversion, we proceeded to compare the phenotype of anti-dsDNA B cells in mice that are serum ANA+ (>9 wk MRL-lpr/lpr) with mice that are ANA− (<9 wk MRL-lpr/lpr, MRL+/+, and BALB/c). Using λ-specific reagents and flow cytometry, we have previously shown that anti-dsDNA B cells are present in the periphery of nonautoimmune (BALB/c) mice with a decreased level (four- to fivefold) of surface Ig relative to Tg− B cells (10). Likewise, Igλ+ B cells are also present in the spleen and LNs of VH3H9 MRL-lpr/lpr and VH3H9 MRL+/+ mice, with the same four- to fivefold decreased level of surface Ig compared with Igλs in Tg− mice (Fig. 2 B, and data not shown). Interestingly, when Ig levels are compared for Igλ+ cells in the BM, the Ig level is also reduced, and to a similar extent, in VH3H9 MRL-lpr/lpr, VH3H9 MRL+/+ (Fig. 2 A), and VH3H9 BALB/c mice (10). We and others have interpreted a low level of Ig to be an indication of Ag encounter (3, 10–12, 35–38). Additionally, the extent of receptor downmodulation has been correlated with the available concentration of self-Ag (39, 40). Thus, the VH3H9/Igλ B cells in all backgrounds tested encounter their Ag, and this interaction first takes place in the BM.
Figure 2.

VH3H9/λ anti-dsDNA B cells are present with a reduced Ig density: (A) BM and (B) spleen in (left) MRL-lpr/lpr and (right) MRL+/+ mice. Dot plots are graphed on a log scale, and mean fluorescence intensity (MFI) is given for the λ+ cells in the boxed region. n = 7 mice of each genotype.
Cell Surface Phenotype of MRL+/+ and MRL-lpr/lpr B Cells Differs from BALB/c B Cells.
We next were interested in determining how encounter with Ag would affect the phenotype of the autoreactive cells from MRL+/+, MRL-lpr/lpr, and BALB/c mice. However, before the analysis of VH3H9 Tg anti-dsDNA B cells, we noted differences among the three background strains of mice in terms of B cell expression levels of several cell surface markers. Although a similar pattern of expression for all sets of mice was observed for the majority of markers, including B220 (Fig. 3 A), HSA, CD22, and CD44 (data not shown), there were notable exceptions in L-selectin, CD23, and CD21/35. The proportion of L-selectinlow B cells is increased in MRL+/+ and MRL-lpr/lpr mice compared with BALB/c mice (Fig. 3 A). Decreased levels of L-selectin can indicate activation, suggesting that activated B cells are accumulating in MRL+/+ and MRL-lpr/lpr mice (41, 42). Additionally, as has been previously reported, CD23 levels are also decreased on MRL-lpr/lpr B cells and this decrease is more apparent as the mice age (43; Fig. 3 A). A decrease in CD21/35 expression has been reported in MRL-lpr/lpr mice (44) as well as SLE patients (45–47). We also observe a twofold decrease in CD21/35 expression on a portion of the B cells in MRL-lpr/lpr mice compared with BALB/c and MRL+/+ mice. However, in our analysis, the most striking difference in the MRL mice both with and without the lpr mutation when compared with the BALB/c mice is the dramatic increase in CD21/35high cells (Fig. 3 A). This peak of CD21/35high cells, which are also IgMhigh (data not shown), accumulates with age in the MRL-lpr/lpr mice. Because CD23lowCD21/35highIgMhigh is reminiscent of the phenotype assigned to marginal zone (MZ) B cells (48), we are in the process of quantitating whether MRL mice have an exaggerated MZ area.
Figure 3.




Phenotypic analysis of Tg− and VH3H9 (A–C) splenic and (D) BM B cells in age-matched BALB/c, MRL+/+, and MRL-lpr/lpr mice. (A) Tg− mice. Histograms show staining of total B cell population (gated on B220+Ig+ cells). Percentages are given for the indicated gates on representative plots. There is an exaggerated population of CD21high cells (MFI > 200) present in MRL+/+ (32.9 ± 11.5%) and MRL- lpr/lpr (40.1 ± 9.9%) compared with BALB/c (7.9 ± 3.1%) mice (P < 0.0001). CD23 levels decrease on MRL-lpr/lpr B cells as the mice age (percentage of CD23low B cells at 4–7 wk, 48 ± 10.6% vs. at 8–14 wk, 70.4 ± 8.7%; P = 0.03). There is also an increased population of CD23low B cells in MRL+/+ mice (MRL+/+, 48 ± 11.3% vs. BALB/c, 16.4 ± 3.4%; P = 0.016), although not to the extent in 8–14 wk MRL- lpr/lpr mice (70.4 ± 8.7%; P = 0.0003). The proportion of L-selectinlow B cells is increased in MRL-lpr/lpr (38.8 ± 13.6%) and MRL+/+ (38.4 ± 13.9%) compared with BALB/c (23.5 ± 4.8%) mice (P = 0.009). VH3H9/λ B cells in (B) MRL+/+, BALB/c, and (C) MRL-lpr/lpr spleen. Histograms are gated on the total B cell population (thin line) and Igλ+ population (bold line). The underlaid histograms (thin line) were scaled down to allow for comparison to the λ+ B cells (which comprise ∼10% of the total B cell population). The majority of markers show that VH3H9/λ B cells in MRL+/+ and MRL-lpr/lpr mice, unlike in BALB/c mice, are no longer developmentally arrested; however, CD21/35 levels are decreased. VH3H9 BALB/c, VH3H9 MRL+/+, and VH3H9 MRL-lpr/lpr mice at 4–7 wk are λ ANA−, whereas VH3H9 MRL-lpr/lpr mice at 8–14 wk are λ ANA+. VH3H9/λ B cells in MRL-lpr/lpr and MRL+/+ spleens are developmentally mature regardless of the mouse's ANA status. (D) BM B cells were stained with CD22 and Igλ. VH3H9/λ B cells are Iglow at the CD22low stage (VH3H9/ Igλ MFI, 50 vs. Tg− Igλ MFI, 200) and do not decrease as the B cells mature (VH3H9/Igλ MFI, 68 vs. Tg− Igλ MFI, 480), suggesting an encounter with Ag at an early developmental stage. Note that there is a decrease in the CD22high population (23 to 6%) in MRL-lpr/lpr mice, with and without the VH3H9 Tg. This is consistent with a previous report that documents a decrease in the frequency of mature B cells in the BM of MRL-lpr/lpr mice (reference 87). All graphs are plotted on a log scale. n = 7 mice at each age range for each genotype.
Anti-dsDNA B Cells Are No Longer Developmentally Arrested in MRL+/+ and MRL-lpr/lpr Mice.
With differences among the three background strains noted, the developmental status of the anti-dsDNA B cells in VH3H9 MRL+/+, VH3H9 MRL-lpr/lpr, and VH3H9 BALB/c mice was compared using flow cytometry and a panel of cell surface markers. Previously, we have shown that anti-dsDNA B cells in VH3H9 BALB/c mice are developmentally arrested and show signs of activation (10). To examine the cell surface phenotype of splenic anti-dsDNA B cells, the relative expression levels of B220, HSA, CD21/35, CD22, CD23, and CD44 (29, 49–53) on VH3H9/λ B cells were compared with those on Tg− B cells in BALB/c (Fig. 3 B), MRL+/+ (Fig. 3 B), and MRL-lpr/lpr (Fig. 3 C) mice. The λ+ B cells in Tg− MRL+/+ (Fig. 3 B), MRL- lpr/lpr (Fig. 3 C), and BALB/c (10) mice have equivalent levels of all surface markers tested, suggesting that there is nothing inherently different about Igλ+ B cells. In contrast and as previously reported, anti-dsDNA B cells from VH3H9 Tg BALB/c mice appear developmentally arrested in that they express a low level, relative to mature splenic B cells, of B220 (decreased twofold), CD21/35 (decreased threefold), CD22 (decreased twofold), and CD23 (decreased twofold), an intermediate level of HSA (increased twofold), and an elevated level of CD44 (increased fourfold) (10; Fig. 3 B). Importantly, VH3H9/λ B cells on the MRL+/+ and MRL-lpr/lpr background are no longer developmentally arrested: they express mature levels of B220, HSA, CD22, and CD44 (Fig. 3, B and C). Additionally, VH3H9/λ MRL+/+ B cells express mature levels of CD23 (Fig. 3 B); however, in the MRL-lpr/lpr mice, CD23 is not a useful marker for maturation, given that the level of CD23 is dramatically reduced on all MRL-lpr/lpr B cells as the mice age (43; Fig. 3 A). The surface phenotype of VH3H9/λ B cells was also compared in MRL-lpr/lpr mice both before and after seroconversion to determine if autoantibody production is marked by a change in cell surface phenotype. Igλ+ B cells from both ANA− (age 4–7 wk) and ANA+ (age 8–14 wk) VH3H9 MRL-lpr/lpr mice are phenotypically mature (Fig. 3 C). One drawback of this analysis is that most of the markers tested are lost on B cells that have differentiated into AFCs. Thus, although we may be missing AFCs in the flow cytometry analysis, the majority of the VH3H9/λ B cell population is indistinguishable between ANA− and ANA+ MRL-lpr/lpr mice.
While several cell surface markers suggest that anti- dsDNA B cells in MRL+/+ and MRL-lpr/lpr mice are no longer developmentally arrested, CD21/35 levels are decreased on VH3H9/λ+ B cells, although not to the extent they are in the BALB/c background (decreased one- to twofold in MRL mice compared with threefold in BALB/c mice; Fig. 3, B and C). The reduced level of CD21/35 is not due to complement binding that in turn could block the Ab binding site, because staining with two different anti-CD21/35 Abs (7G6, which binds to the complement-binding site, and 7E9, which does not [54, 55]) gave similar results (data not shown). Why CD21/35 levels remain decreased on VH3H9 MRL+/+ and MRL-lpr/lpr λ+ B cells, despite normal levels of other developmental markers (B220, HSA, and CD22) is not clear. One possibility is that only those B cells expressing low levels of CD21/35 persist after negative selection, given that CD21/35 is a coreceptor for signals through the Ig receptor (56). A similar explanation has been described for the reduced levels of the CD8 coreceptor seen on self-reactive anti-HY TCR Tg T cells present in the periphery of male mice (57). Another possibility is that reduced CD21/35 levels reflect activation due to Ag encounter (44). Ig Tg B cells specific for model Ags, where Ag exposure can be controlled, may help to determine if Ig downmodulation in the presence of Ag is accompanied by a decrease in CD21/35.
One scenario that could account for the more advanced maturation of VH3H9/λ+ B cells in MRL as opposed to BALB/c mice is that they encounter Ag at a later developmental stage in the BM and thus their maturation goes unimpeded. As an indication of Ag encounter, the level of surface Ig on the λ+ B cells in the BM of VH3H9 and Tg− BALB/c, MRL+/+, and MRL-lpr/lpr mice was compared. As is shown in Fig. 3 D, the level of Ig on Tg− B cells increases as the B cells mature from a CD22low to a CD22high stage. If the VH3H9/λ+ B cells in the different genetic backgrounds are encountering Ag at a later stage, we would have predicted that the level of Ig would be higher on the less mature (CD22low) B cells in the VH3H9 Tg MRL mice and then decrease once they have encountered Ag. However, the level of Ig on VH3H9/λ+ B cells from all three backgrounds is low at the CD22low stage and remains low at the CD22high stage, suggesting that these B cells have all encountered Ag at an immature stage. This encounter with Ag does not halt the maturation of the B cells: in BALB/c mice, the VH3H9/λ+ B cells continue to develop to a slightly more mature stage as indicated by intermediate levels of HSA, B220, CD22, and CD23 (10; and Fig. 3 B), whereas in MRL mice (+/+ and lpr/lpr), the splenic B cells appear fully mature (Fig. 3, B and C).
To confirm that the cells we are following are using the VH3H9 Tg, we have repeated our experiments using VH3H9 MRL-lpr/lpr mice deficient at the JH locus (JH−/− [28]). As B cells in VH3H9 JH−/− mice are unable to rearrange endogenous H chain loci, the only H chain they express is the VH3H9 Tg. The results from VH3H9 MRL- lpr/lpr mice with and without an intact JH locus were indistinguishable in terms of phenotypic analysis of BM and splenic VH3H9/Vλ B cells (data not shown).
Altered Localization of Anti-dsDNA B Cells in MRL-lpr/lpr Mice.
Given the changes in developmental maturity of anti-dsDNA B cells in BALB/c versus MRL mice, we next looked to see what effect these changes have on the B cells' splenic localization. For the most part, B and T cells are located in discrete areas in the spleen, called the B cell follicle and periarteriolar lymphoid sheath (PALS), respectively. Previously, using Ig Tg mice, we and others have shown that anergic B cells localize to the T–B interface of the splenic follicle (10, 27). One hypothesis to account for this localization is that anergic B cells continually encountering Ag are at a competitive disadvantage for follicular space (27, 58). Another hypothesis is that the location of the B cells is dependent on the amount of Ag exposure, not competition with other naive B cells (39, 40). In both models, B cells encountering Ag move to the T–B interface, where they can interact with T cells. Immature B cells are also reported to first appear at the T–B interface in the spleen (59).
Given that the VH3H9/λ BALB/c B cells that localize to the T–B interface are both immature and have encountered Ag, we have been unable to distinguish which of these features is responsible for their localization (10; and Fig. 4 A). However, on the MRL+/+ and MRL-lpr/lpr backgrounds, VH3H9/λ B cells are no longer developmentally arrested, providing an opportunity to separate maturation status from Ag encounter. Igλ+ B cells in Tg− mice from autoimmune and nonautoimmune backgrounds are scattered throughout the splenic follicle (10; Fig. 4, A and B). However, similar to their counterparts in the BALB/c background, VH3H9/λ B cells from MRL+/+ mice localize to the T–B interface (Fig. 4 A). Therefore, follicular localization is not dependent on the developmental status of the autoreactive B cells, at least in MRL+/+ mice; rather, we suggest it is a consequence of Ag encounter.
Figure 4.
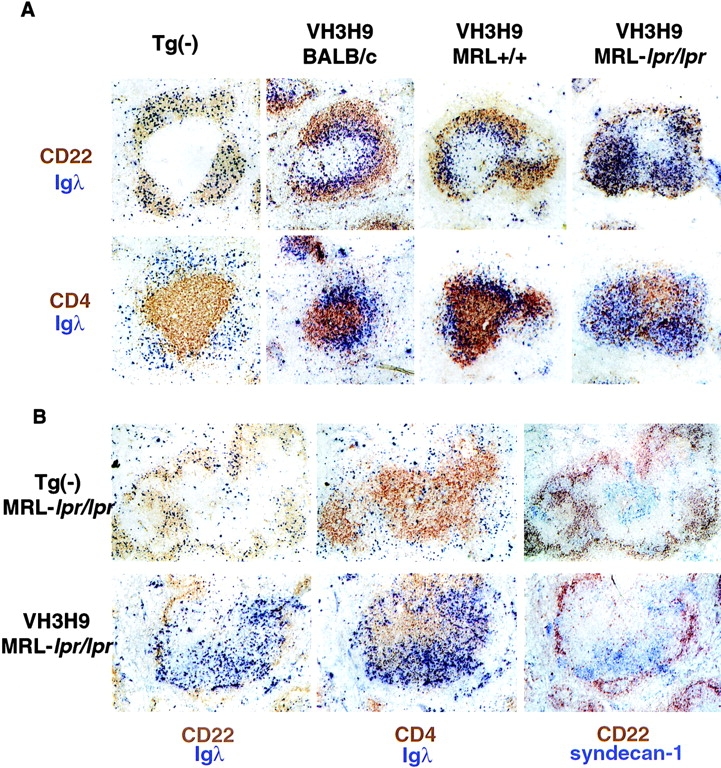
Localization of Igλ+ B cells. (A) Spleen sections from Tg−, VH3H9 BALB/c, VH3H9 MRL+/+, and VH3H9 MRL-lpr/lpr mice were stained with Abs against (top) CD22 and Igλ or (bottom) CD4 and Igλ. In Tg− mice of all three backgrounds, the Igλ+ cells are scattered throughout the B cell follicle. The follicle shown in the figure is from a Tg− MRL+/+ spleen. Igλ+ cells in VH3H9 MRL+/+ mice, like those in VH3H9 BALB/c, accumulate at the T–B interface. However, the VH3H9/λ B cells in MRL-lpr/lpr mice are found in the B cell follicle. Interestingly, CD4+ cells are also scattered in the B cell area with the λ+ B cells. Mice shown are 5 wk of age. (B) Spleen sections from (top) Tg− MRL-lpr/lpr and (bottom) VH3H9 MRL-lpr/lpr mice stained with Abs against (left) CD22 and Igλ, (middle) CD4 and Igλ, or (right) CD22 and syndecan-1 (AFCs). In the majority of follicles, VH3H9/Igλ AFCs are found in the PALS, although some follicles did not have syndecan-1+ cells. AFCs in Tg− MRL-lpr/lpr mice are also found in the PALS. AFCs are first detectable at 8 wk, and mice shown are 10 wk of age. Original magnification: ×100 (n = 10 mice of each genotype).
In striking contrast to the VH3H9/λ MRL+/+ B cells, VH3H9/λ+ B cells in the MRL-lpr/lpr background are not localized to the T–B interface; instead, they are located within the B cell follicle (Fig. 4 A). This pattern of localization is seen in mice as early as 4 wk of age, before their specificity is detectable in the serum. Identical results for localization of anti-dsDNA B cells were obtained using VH3H9 MRL-lpr/lpr mice with and without an intact JH locus, confirming that the cells we are following use the VH3H9 Tg (data not shown). Thus, while indistinguishable by developmental status, anti-dsDNA B cells in VH3H9 MRL+/+ and VH3H9 MRL-lpr/lpr mice exhibit different splenic localizations.
Disrupted Architecture in MRL-lpr/lpr Mice.
Disruptions in the splenic architecture of MRL-lpr/lpr mice have been known for some time and have been attributed to the accumulation of CD3+B220+CD4−CD8− (DN) T cells. It has been reported that these DN T cells are located between the T and B cell areas in the follicles of older MRL- lpr/lpr mice (60, 61). To confirm this, MRL-lpr/lpr spleen sections were stained with anti-B220 and anti-CD3, and coexpressing cells were identified by immunofluorescence. In agreement with previous studies, the B220+CD3+ cells were located in discrete areas between the PALS and what remained of the B cell follicle (data not shown).
In addition to the disruption in splenic architecture caused by DN T cells, we have identified a novel histological feature of MRL-lpr/lpr mice that is evident even before the emergence of DN T cells. In contrast to the usual segregation of B and T cells, CD4 T cells in VH3H9 MRL- lpr/lpr mice do not localize exclusively to the T cell area: a subpopulation of CD4 T cells are in the B cell follicle (Fig. 4 A). To examine this altered CD4 localization more closely, spleen sections were stained with MOMA-1, a marker for MZ macrophages, and either CD22, CD4, or CD8 (Fig. 5, A and B). In both BALB/c and MRL+/+ spleens, with and without the VH3H9 Tg, distinct B and T cell areas are present with few to no CD4 or CD8 T cells in the B cell area (Fig. 5 A). However, in VH3H9 MRL-lpr/lpr spleens, although distinct B and T cell areas are present, there is also a subpopulation of CD4 T cells scattered throughout the follicle (Fig. 5 B). CD4 T cells have been found in the B cell follicle in the context of germinal centers (GCs [62]). However, the CD4 T cells in the follicles of MRL-lpr/lpr mice are not like those described in GCs as they express Thy-1 (data not shown), a marker absent on GC T cells (62). CD8 T cells remain tightly compacted around the central arteriole, and neither CD4 nor CD8 T cells are present in the MZ of VH3H9 MRL-lpr/lpr mice (Fig. 5 B).
Figure 5.

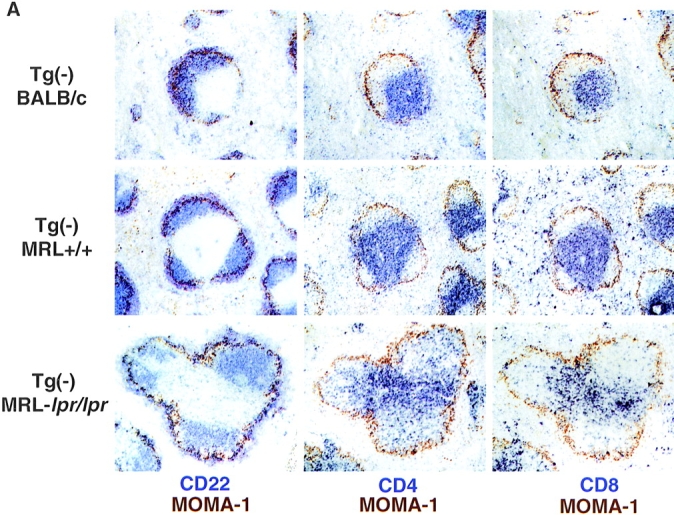
Disrupted architecture in MRL- lpr/lpr mice. (A) Spleen sections from (top) Tg− BALB/c, (middle) Tg− MRL+/+, and (bottom) Tg− MRL-lpr/lpr mice were stained with Abs against MOMA-1 (MZ metallophilic macrophages) and (left) CD22 (B cells), (middle) CD4 (T cells), or (right) CD8. In MRL-lpr/lpr mice, CD4 T cells are present scattered in the B cell area. CD8 T cells remain in the PALS. Mice shown are 12 wk old. (B) Spleen sections from age-matched (top) Tg− MRL-lpr/lpr and (bottom) VH3H9 MRL-lpr/lpr mice. VH3H9 Tg accelerates the appearance of disrupted architecture in MRL-lpr/lpr mice. Mice shown are 6 wk old. Original magnification: ×100 (n = 15 mice of each genotype).
To determine if the altered localization of CD4 T cells is a general feature of MRL-lpr/lpr mice, or is unique to the VH3H9 Tg, spleen sections from Tg− MRL-lpr/lpr mice were examined (Fig. 5, A and B). Unlike BALB/c and MRL+/+ spleens, the Tg− MRL-lpr/lpr spleens have CD4 T cells scattered throughout the B cell follicle (Fig. 5 A), showing that this disrupted architecture is not limited to VH3H9 mice. The fact that MRL-lpr/lpr and not MRL+/+ mice show disrupted architecture suggests that this alteration is attributable to a block in the Fas/FasL pathway. At the time points examined, CD4 infiltrates were not found in other organs such as the liver or kidney (data not shown).
Although the presence of the VH3H9 Tg is not necessary for the appearance of CD4 T cells in the B cell follicle, it does accelerate the process. Age-matched VH3H9 and Tg− MRL-lpr/lpr mice were analyzed for CD4 T cell infiltrates by immunohistochemistry (Fig. 5 B). Alterations in T/B architecture are seen as early as 4–5 wk in VH3H9 MRL-lpr/lpr spleens (Fig. 4 A, and data not shown); however, Tg− MRL-lpr/lpr spleens show no disruption until 6–8 wk (Fig. 5 B) and it becomes more pronounced at 10–12 wk (Fig. 5 A). VH3H9 BALB/c and VH3H9 MRL+/+ mice have normal splenic architecture, suggesting that the lpr mutation, not the VH3H9 Tg, is necessary for the disruption (Fig. 4 A, and data not shown). Therefore, in VH3H9 MRL-lpr/lpr mice, although increasing the frequency of autoreactive B cells does not accelerate the kinetics or titers of serum ANAs, it does accelerate the kinetics of T–B mixing in MRL-lpr/lpr mice.
Alterations in VH3H9 MRL-lpr/lpr Mice after Seroconversion.
In addition to noting differences in anti-dsDNA B cells in nonautoimmune versus autoimmune mice, we also examined VH3H9 MRL-lpr/lpr mice before and after seroconversion to determine if any architectural alterations correlate with the emergence of serum Ab. Although no difference by flow cytometry was detected between ANA+ and ANA− mice (Fig. 3 C), a difference was observed by immunohistochemistry (Fig. 4, A and B). In ANA+ mice, many darkly staining Igλ+ cells are present in the PALS as well as in the bridging channels to the red pulp (Fig. 4 B). These cells are not obvious in ANA− mice (data not shown). To determine if these darkly staining cells are AFCs, serial sections were stained for syndecan-1, a cell surface proteoglycan that is expressed on B cells that have differentiated into AFCs (63). The dark Igλ staining cells colocalize with syndecan-1 staining, indicating that they are AFCs. Importantly, many Igλ+/syndecan-1− cells remain in the B cell follicles even in ANA+ animals, suggesting that not all dsDNA B cells have differentiated into AFCs (data not shown). Syndecan-1+ cells are also found in the PALS in Tg− MRL-lpr/lpr mice, although in this case the vast majority are not Igλ+ (Fig. 4 B), consistent with Igκ+Igλ− ANAs in the serum (Fig. 1). This localization of AFCs to the inner T cell area, as opposed to the bridging channels to the red pulp (also referred to by others as the red pulp area adjacent to the T cell zone [31, 64, 65]), is similar to that reported by Marshak-Rothstein and colleagues for rheumatoid factors in MRL-lpr/lpr mice (61, 66).
As an alternate approach to quantitate the number of AFCs in the various mice, ex vivo enzyme-linked immunospot assays (ELISpots) were performed (Fig. 6, A and B). Using reagents to detect all AFCs, few to no AFCs were detectable in spleens from young (4–6 wk) Tg− and VH3H9 MRL-lpr/lpr, MRL+/+, and BALB/c mice, but became more pronounced in aged (10–12 wk) MRL-lpr/lpr mice (Fig. 6 A and data not shown). To follow anti-dsDNA AFCs in particular in VH3H9 MRL-lpr/lpr mice, Igλ AFCs were quantitated (Fig. 6 B). Igλ AFCs were readily detectable in older VH3H9 MRL-lpr/lpr mice. By ELISpot, only 5–10% of the Igλ+ cells in VH3H9 MRL-lpr/lpr mice are generating AFCs. The fact that not all Igλ+ cells are secreting Ab as shown by histology and ELISpots supports the idea that it is a breakthrough of a fraction of the anti-dsDNA B cells and not an overall breakdown of tolerance that leads to serum ANAs in MRL-lpr/lpr mice.
Figure 6.


AFCs in the spleen from ANA− (4–6 wk) and ANA+ (10–12 wk) Tg− MRL-lpr/lpr, VH3H9 MRL-lpr/lpr, and BALB/c mice. The number of (A) total Ig and (B) Igλ AFCs were determined by ex vivo ELISpot. The data are presented as the mean number of AFCs per 104 cells ± the SD of triplicate wells. Total Ig+ and Igλ+ AFC numbers were calculated by normalizing for total B220+Ig+ and B220+Igλ+ numbers as determined by flow cytometry, respectively. Presented data are from a representative mouse of each genotype. Total number of mice from five experiments: n = 5 Tg− BALB/c; n = 4 VH3H9 BALB/c; n = 4 ANA+ Tg− and VH3H9 MRL-lpr/lpr; and n = 2 ANA− Tg− and VH3H9 MRL-lpr/lpr mice. Note that a higher number of Igλ+ AFCs than total Ig+ AFCs were detected for the ANA+ VH3H9 MRL-lpr/lpr mice in this assay. This difference is attributed to a sensitivity difference in the reagents used to detect total Ig versus Igλ. Therefore, although comparisons can be made for the relative number of AFCs of a given type (i.e., between Igλ+ numbers), they cannot be used to compare absolute frequencies between types (i.e., total Ig vs. Igλ).
Discussion
Several studies have documented alterations in total B and T cell populations in autoimmune mice and SLE patients (43, 44, 61, 67). The unique aspect of the data presented here is that specific autoreactive B cells have been followed, both in nonautoimmune mice and in autoimmune-prone mice, as they develop autoantibodies. To do this we made use of the VH3H9 H chain Tg, which increases the frequency of anti-dsDNA B cells and allows their fate, in the presence of nonautoreactive B cells, to be compared in BALB/c, MRL+/+, and MRL-lpr/lpr mice. This model has allowed for the analysis not only of endpoints such as serum autoantibodies, but also of the B cells themselves and the factors that influence them during the process of seroconversion. In this study, we have described two novel observations regarding the breakdown of tolerance in MRL- lpr/lpr mice. The first is that MRL mice exhibit a defect in maintaining the developmental arrest of anti-dsDNA B cells. The second is that a deficiency in Fas allows these autoreactive cells to enter the B cell follicles from which they are normally restricted. Combining these observations, we propose that the transition of anti-dsDNA B cells from tolerized B cells on the BALB/c background to autoantibody-producing cells in the MRL-lpr/lpr is a multistep process that includes overcoming both developmental arrest and follicular exclusion.
The MRL Background Allows Maturation of Autoreactive Cells.
It has been clearly established that autoimmunity in MRL mice is polygenic, yet the mechanisms by which these different loci influence autoantibody production have not been defined (68–71). Although anti-dsDNA B cells are tolerized in BALB/c mice as manifested by their developmental arrest and accumulation at the T–B interface of the splenic follicle (10), anti-dsDNA B cells on the MRL+/+ and MRL-lpr/lpr backgrounds are developmentally mature. One possibility that could account for this difference is timing of initial Ag exposure. For example, if anti-dsDNA B cells in MRL mice are not exposed to the tolerizing Ag during their development, maturational arrest would not occur. However, the data presented are inconsistent with this scenario: for anti-dsDNA B cells in both MRL and BALB/c mice, the level of surface Ig is low at the same developmental stage in the BM. A second possibility is that although anti-dsDNA B cells in both BALB/c and MRL mice initially encounter their Ag at the same time in development, an absence of continual Ag stimulation in the MRL mice allows the B cells to subsequently progress to maturity. In the HEL B cell tolerance model in the context of bcl-2, the removal of Ag allows the development of the anergic B cells to progress (38). However, in all the mice studied here, anti-dsDNA B cells are uniformly Iglow in the spleen as well as in the BM, suggesting that constant Ag exposure is occurring in all cases. A third possibility, and one we favor, is that a defect in MRL mice allows autoreactive B cells to survive a selection checkpoint that would normally lead to developmental arrest. This is important in that the maturation state of a B cell may translate to significant changes in function in terms of B cell receptor–associated intracellular signaling capabilities and subsequent B cell responsiveness (72). Mapping studies by various groups have identified several autoimmunity-associated loci in MRL mice (68–71). It will be important to determine if any of these loci, together or in isolation, control the developmental maturation of autoreactive B cells.
The Role of Fas in Follicular Exclusion.
We observe in this study a previously undefined connection between Fas and the entry of cells to the B cell follicle. Comparing MRL mice with and without the lpr defect in Fas, we find that despite their mature status, anti-dsDNA B cells in MRL+/+ mice remain at the T–B interface, whereas in MRL-lpr/lpr mice, they are in the B cell follicle. Exclusion from the B cell follicle has been correlated with the decreased survival of B cells (58). However, studies using bcl-2 Tgs have shown that this correlation is not absolute, as simply increasing a B cell's life span does not alone alter the localization of anti-HEL B cells from the T–B interface (27). The data from the present study indicate that Fas can play an important role in determining follicular entry; what remains to be discovered is the mechanism by which it exerts this effect. A deficiency in Fas could act directly on the survival of the anti-dsDNA B cells. Alternatively, lack of Fas could have an indirect effect through the shaping of the peripheral T and B cell repertoires.
The lack of Fas could translate into an alteration in the availability of T cell help. It has been clearly established that T cells are required for the generation of autoantibodies in MRL-lpr/lpr mice (73–75). Additionally, it has been suggested that Ag-experienced B cells, such as the anti-dsDNA B cells followed here, are held at the T–B interface in the absence of T cell help (39, 58). In light of this idea, one possible interpretation of our results is that Fas normally plays a crucial role in eliminating the autoreactive T cell population. In its absence, a fundamental shift occurs in the T cell repertoire such that autoreactive T cell help is now available, and autoreactive B cells are given the signals they need to proceed past the T–B interface. The data presented here argue against this possibility. The anti-dsDNA B cells are found peppered throughout the B cell follicle and appear not to be proliferating (data not shown). If they had received T cell help, we would have expected to observe either (a) the compaction of B cells in follicular GCs, or (b) the formation of AFCs (31). Although at later time points (mice >8 wk) we do see evidence of AFCs, at the early time points (4–7 wk) dsDNA B cells appear exclusively in the follicles. Thus, the effect of Fas on follicular entry seems unlikely to be mediated solely through an effect on T cell help.
Alternatively, Fas could regulate follicular entry through its functions on the B cell itself. For example, Rathmell and colleagues have shown that anergic B cells, far from being rescued by T cell help, are killed in a Fas-dependent manner (76, 77). In the absence of Fas, then, it is possible that anti-dsDNA B cells would not die and thus would be able to proceed into the B cell follicle. One prediction from this is that anti-dsDNA B cells in Fas wild-type animals would have a reduced life span. Experiments are currently underway to determine if the life span of anti-dsDNA B cells is reduced in MRL+/+ compared with MRL-lpr/lpr mice. Again, however, if T cell help were present in the absence of Fas, we would expect to see evidence of further differentiation of the anti-dsDNA B cells in young MRL-lpr/lpr mice. Interestingly, experiments using TCR−/− mice, in the context of the HEL B cell tolerance model, have shown that T cells are not required to maintain follicular exclusion (78). Therefore, if a signal through Fas on the B cell is what restricts it to the T–B interface, other FasL-expressing cells would be required to deliver that signal.
A scenario that is consistent with our data is that the deficiency in Fas is affecting the proportion of autoreactive B cells in the peripheral B cell repertoire and, as such, alters B cell localization. Studies in the HEL system indicate that changes in the B cell repertoire can affect the localization of autoreactive B cells. In mice where anergic anti-HEL B cells make up the vast majority of the B cell population, they are present in the B cell follicle; however, when nonautoreactive B cells are also present, the anergic B cells are excluded from the B cell follicle (27, 58). If Fas plays a role in eliminating autoreactive B cells, then the lpr mutation could affect the proportion of autoreactive B cells in the periphery such that, instead of the majority of competing B cells being nonautoreactive, the anti-dsDNA B cells are in an environment with mostly other autoreactive B cells. Therefore, anti-dsDNA B cells in MRL-lpr/lpr mice would be present in the B cell follicle due to lack of competition and not because they are missing a signal through Fas to restrict their entry. Altering the composition of the B cell repertoire by transfer experiments of dsDNA B cells from VH3H9 MRL-lpr/lpr mice into MRL-lpr/lpr mice where the B cells express a nonautoreactive Ig Tg (23) should allow us to discern if the addition of competitive B cells now results in follicular exclusion of the anti-dsDNA B cells.
An additional component that must be considered is the role of Fas in Ag composition. The role of Ag in MRL-lpr/lpr autoantibody production remains controversial. BM chimera and allophenic experiments, mixing lpr/lpr and Fas wild-type cells, have suggested that intrinsic B and T cell defects in lpr/lpr mice, and not alterations in Ag milieu, are responsible for autoantibody production (79–82). However, the interpretation of these results is complicated. lpr/lpr T cells are known to express elevated levels of FasL (83, 84), and it is distinctly possible that the autoreactive non- lpr/lpr B cells in the chimeras, susceptible to Fas-mediated death upon activation, are simply triggered to die before they can contribute to autoantibody levels. Therefore, it remains a possibility that eliminating Fas-mediated death alters the antigenic make-up of MRL-lpr/lpr mice. Consistent with this, studies by Rosen and colleagues have shown that some of the nuclear Ags targeted in SLE are packaged into discrete blebs at the surface of apoptotic cells (85). They have further demonstrated that triggering apoptotic pathways by different stimuli generates unique substrate fragments that can then be clustered in these blebs (86). Preferential use of one pathway over another, as could occur in Fas-deficient MRL mice, would potentially alter the ability to produce and display tolerogenic or immunogenic epitopes. The fact that dsDNA B cells from both MRL+/+ and MRL-lpr/lpr mice are uniformly Iglow suggests that both are encountering a similar amount of Ag. However, it may be that a difference in antigenic context determines whether a B cell enters the follicle, as in MRL-lpr/lpr mice, or is held at the T–B interface, as in MRL+/+ mice. Because of the potential significance of this issue, experiments are currently underway to explore the nature of the in vivo Ag recognized by these anti-dsDNA B cells.
The Role of Fas in Regulating Entry of T Cells to the B Cell Follicle.
In examining the localization of anti-dsDNA B cells, we have identified a unique histological feature of CD4 T cells in both Tg− and VH3H9 MRL-lpr/lpr mice. A subpopulation of CD4 T cells, which normally are located in a discrete area around the central arteriole, have infiltrated the B cell follicle. The lack of this infiltrate in Tg− and VH3H9 MRL+/+ mice suggests that the absence of a Fas–FasL interaction is the determining factor. In VH3H9 MRL-lpr/lpr mice, the T cell splenic architecture is disrupted as early as 4 wk of age (before we see evidence of DN T cells) and becomes more apparent as the mice age. Interestingly, increasing the frequency of autoreactive B cells by the presence of the VH3H9 Tg, while not accelerating seroconversion, does accelerate the appearance of T–B mixing. Why a subpopulation of T cells resides in the B cell follicle in VH3H9 MRL-lpr/lpr mice is unclear, but the follicular localization of CD4 T cell infiltrates occurs in mice that also have follicular anti-dsDNA B cells. Our working model is that the increased frequency of autoreactive B cells in the VH3H9 Tg mouse plays a role in accelerating the influx of CD4 T cells. There is precedent for B cells, in general, affecting the activation status of T cells in MRL-lpr/lpr mice: B cell–deficient MRL-lpr/lpr mice do not have as many activated/memory T cells as B cell– sufficient MRL-lpr/lpr mice (28). To explore this idea further, we are examining the spleens of MRL-lpr/lpr mice carrying Ig Tgs in the absence of their cognate Ag (23). We would predict that the infiltration of T cells would occur at a decreased rate in mice with a B cell repertoire skewed away from autoreactivity. Alternatively, it may be the infiltrating CD4 T cells that allow the anti-dsDNA B cells to enter the follicle. To independently regulate the follicular entry of CD4 T cells and dsDNA B cells, we are breeding the VH3H9 Tg onto MHC class II–deficient MRL-lpr/lpr mice (74). These mice will allow us to determine if the altered localization of VH3H9/λ B cells is dependent on the presence of CD4 T cells.
Autoantibody Production.
This study has allowed us to compare the phenotype of the anti-dsDNA B cells in MRL-lpr/lpr mice before and after seroconversion. Even before seroconversion, the anti-dsDNA B cells are developmentally mature and in the B cell follicle. Furthermore, using a decreased level of surface Ig to indicate Ag encounter (3, 10–12, 35–38), anti-dsDNA B cells are Iglow in the BM, spleen, and LNs, suggesting continual Ag exposure, beginning in the BM. The consistent histological difference between VH3H9 MRL-lpr/lpr mice before and after seroconversion is the presence of Igλ AFCs in serum Igλ ANA+ animals. We note two striking characteristics of this AFC formation. The first is that, while mice immunized with model Ags, such as (4-hydroxy-3-nitrophenyl)acetyl (NP), form AFCs in the bridging channels to the red pulp (31, 64, 65), the VH3H9/Igλ AFCs are, in addition to being in the bridging channels, also located in the PALS. This altered localization of AFCs is consistent with that previously reported by Marshak-Rothstein and colleagues for rheumatoid factor AFCs in MRL-lpr/lpr mice (61, 66). Whether this localization of AFCs to the PALS is restricted to autoreactive B cells or extends to AFCs in response to foreign Ags is currently under study. The second difference is that even in serum Igλ ANA+ animals, not all of the Igλ B cells become AFCs, suggesting a breakthrough of only some autoreactive B cells and not a global breakdown that would affect the population as a whole. Therefore, we have segregated initial exposure of Ag and entry into the follicle from terminal differentiation into AFCs.
Conclusions.
In summary, the factors that are required to transform an autoreactive B cell population from a tolerant one to one producing Ab are clearly complex. This study has compared the phenotype and splenic localization of a population of anti-dsDNA B cells in nonautoimmune mice with anti-dsDNA B cells in autoimmune MRL+/+ and MRL-lpr/lpr mice. BALB/c mice actively regulate anti-dsDNA B cells as manifested by their developmental arrest, retention at the T–B interface, and lack of anti- dsDNA Ab in the serum. In MRL-lpr/lpr mice where anti-dsDNA Ab is present in the serum, the anti-dsDNA B cells are no longer developmentally arrested or excluded from the B cell follicle. Anti-dsDNA B cells in MRL+/+ mice are also not developmentally arrested; however, they remain at the T–B interface. This allows us to assign a defect in maturational arrest to the MRL genetic background. The maturation state of B cells may have profound effects on their life span and ability to be reactivated. Additionally, in this model, a defect in Fas is needed for the B cells to enter the follicle. However, even Fas-deficient B cells require an additional factor to become AFCs, as young VH3H9 MRL-lpr/lpr mice, where the λ+ B cells are mature and in the follicle, are serum−. Changes in T cell help or Ag presentation may be required for seroconversion. Interestingly, a defect in Fas also leads to an infiltration of CD4 T cells into the B cell follicle. What role this infiltrate plays in the production of autoantibodies is unknown. Given the requirement for CD4 T cells in the production of autoantibodies in MRL-lpr/lpr mice (73–75), it will be important to determine the specificity of these infiltrating CD4 T cells.
Acknowledgments
We thank Ashlyn Eaton, Michele Lutz, and Drs. Andrew Caton and Jason Cyster for critical reading of the manuscript, Dr. Clayton Buck for use of his cryostat and microscope, and Deepa Kurian for technical assistance. In addition we acknowledge the generous contribution of Dr. Mark Shlomchik for VH3H9 JH−/− MRL-lpr/lpr mice.
Abbreviations used in this paper
- AFC
antibody-forming cell
- ANA
antinuclear antibody
- AP
alkaline phosphatase
- BM
bone marrow
- DN
double-negative
- ds
double-stranded
- ELISpot
enzyme-linked immunospot assay
- GC
germinal center
- HEL
hen egg lysozyme
- HN
homogeneous nuclear
- HRP
horseradish peroxidase
- HSA
heat-stable antigen
- MFI
mean fluorescence intensity
- MZ
marginal zone
- PALS
periarteriolar lymphoid sheath
- ss
single-stranded
- SA
streptavidin
- Tg
transgene
Footnotes
Services provided by The Wistar Institute staff were supported by Core grant CA10815 and by grants from the National Institutes of Health (5R01 AI32137-06), the Arthritis Foundation, and the Pew Charitable Trust to J. Erikson. L. Mandik-Nayak is supported by Wistar Training grant CA-09171. S. Seo is supported by Medical Scientist Training Program grant 5T-32GM-07170.
References
- 1.Nemazee DA, Bürki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 2.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–769. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- 3.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 4.Erikson J, Radic MZ, Camper SA, Hardy RR, Carmack C, Weigert M. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 1991;349:331–334. doi: 10.1038/349331a0. [DOI] [PubMed] [Google Scholar]
- 5.Iliev A, Spatz L, Ray S, Diamond B. Lack of allelic exclusion permits autoreactive B cells to escape deletion. J Immunol. 1994;153:3551–3556. [PubMed] [Google Scholar]
- 6.Hertz M, Nemazee D. BCR ligation induces receptor editing in IgM+IgD− bone marrow B cells in vitro. Immunity. 1997;6:429–436. doi: 10.1016/s1074-7613(00)80286-1. [DOI] [PubMed] [Google Scholar]
- 7.Tsao BP, Chow A, Cheroutre H, Song YW, McGrath ME, Kronenberg M. B cells are anergic in transgenic mice that express IgM anti-DNA antibodies. Eur J Immunol. 1993;23:2332–2339. doi: 10.1002/eji.1830230942. [DOI] [PubMed] [Google Scholar]
- 8.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C, Nagy Z, Radic MZ, Hardy RR, Huszar D, Camper SA, Weigert M. The site and stage of anti-DNA B cell deletion. Nature. 1995;373:252–255. doi: 10.1038/373252a0. [DOI] [PubMed] [Google Scholar]
- 10.Mandik-Nayak L, Bui A, Noorchashm H, Eaton A, Erikson J. Regulation of anti–double-stranded DNA B cells in nonautoimmune mice: localization to the T–B interface of the splenic follicle. J Exp Med. 1997;186:1257–1267. doi: 10.1084/jem.186.8.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roark JH, Bui A, Nguyen K-A, Mandik L, Erikson J. Persistence of functionally compromised anti- dsDNA B cells in the periphery of non-autoimmune mice. Int Immunol. 1997;9:1615–1626. doi: 10.1093/intimm/9.11.1615. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen K-AT, Mandik L, Bui A, Kavaler J, Norvell A, Monroe JG, Roark JH, Erikson J. Characterization of anti-single-stranded DNA B cells in a non-autoimmune background. J Immunol. 1997;159:2633–2644. [PubMed] [Google Scholar]
- 13.Santulli-Marotto S, Retter MW, Gee R, Mamula MJ, Clarke SH. Autoreactive B cell regulation: peripheral induction of developmental arrest by lupus-associated B cells. Immunity. 1998;8:209–219. doi: 10.1016/s1074-7613(00)80473-2. [DOI] [PubMed] [Google Scholar]
- 14.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 16.Chu J-L, Drappa J, Parnassa A, Elkon KB. The defect in Fas mRNA expression in MRL/lpr mice is associated with insertion of the retrotransposon, ETn. J Exp Med. 1993;178:723–730. doi: 10.1084/jem.178.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pisetsky DS, Caster SA, Roths JB, Murphy ED. lprgene control of the anti-DNA antibody response. J Immunol. 1982;128:2322–2325. [PubMed] [Google Scholar]
- 18.Kishimoto H, Surh CD, Sprent J. A role for Fas in negative selection of thymocytes in vivo. J Exp Med. 1998;187:1427–1438. doi: 10.1084/jem.187.9.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kotzin BL, Babcock SK, Herron LR. Deletion of potentially self-reactive T cell receptor specificities in L3T4−, Lyt-2− T cells of lprmice. J Exp Med. 1988;168:2221–2229. doi: 10.1084/jem.168.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou T, Bluethmann H, Eldridge J, Brockhaus M, Berry K, Mountz JD. Abnormal thymocyte development and production of autoreactive T cells in T cell receptor transgenic autoimmune mice. J Immunol. 1991;147:466–474. [PubMed] [Google Scholar]
- 21.Sidman CL, Marshall JD, von Boehmer H. Transgenic T cell receptor interactions in the lymphoproliferative and autoimmune syndromes of lpr and gldmutant mice. Eur J Immunol. 1992;22:499–504. doi: 10.1002/eji.1830220231. [DOI] [PubMed] [Google Scholar]
- 22.Rathmell JC, Goodnow CC. Effects of the Lprmutation on elimination and inactivation of self-reactive B cells. J Immunol. 1994;153:2831–2842. [PubMed] [Google Scholar]
- 23.Rubio CF, Kench J, Russell DM, Yawger R, Nemazee D. Analysis of central B cell tolerance in autoimmune-prone MRL/lprmice bearing autoantibody transgenes. J Immunology. 1996;157:65–71. [PubMed] [Google Scholar]
- 24.Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med. 1990;171:265–297. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radic MZ, Mascelli MA, Erikson J, Shan H, Weigert M. Ig H and L chain contributions to autoimmune specificities. J Immunol. 1991;146:176–182. [PubMed] [Google Scholar]
- 26.Fulcher DA, Basten A. Reduced life span of anergic self-reactive B cells in a double-transgenic model. J Exp Med. 1994;179:125–134. doi: 10.1084/jem.179.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cyster JG, Hartley SB, Goodnow CC. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. 1994;371:389–395. doi: 10.1038/371389a0. [DOI] [PubMed] [Google Scholar]
- 28.Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lprmice. J Immunol. 1998;160:51–59. [PubMed] [Google Scholar]
- 29.Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-B cell stages in normal mouse bone marrow. J Exp Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glazer AN, Stryer L. Fluorescent tandem phycobiliprotein conjugates: emission wavelength shifting by energy transfer. Biophys J. 1983;43:383–386. doi: 10.1016/S0006-3495(83)84361-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl) acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roark JH, Kuntz CL, Nguyen K-A, Caton AJ, Erikson J. Breakdown of B cell tolerance in a mouse model of SLE. J Exp Med. 1995;181:1157–1167. doi: 10.1084/jem.181.3.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan EM. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv Immunol. 1989;44:93–151. doi: 10.1016/s0065-2776(08)60641-0. [DOI] [PubMed] [Google Scholar]
- 34.Losman MJ, Fasy TM, Novick KE, Monestier M. Relationships among antinuclear antibodies from autoimmune MRL mice reacting with histone H2A-H2B dimers and DNA. Int Immunol. 1993;5:513–523. doi: 10.1093/intimm/5.5.513. [DOI] [PubMed] [Google Scholar]
- 35.Raff MC, Owen JJ, Cooper MD, Lawton AR, Megson M, Gathings WE. Differences in susceptibility of mature and immature mouse B lymphocytes to anti-immunoglobulin–induced immunoglobulin suppression in vitro. Possible implications for B-cell tolerance to self. J Exp Med. 1975;142:1052–1064. doi: 10.1084/jem.142.5.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sidman CL, Unanue ER. Receptor-mediated inactivation of early B lymphocytes. Nature. 1975;257:149–151. doi: 10.1038/257149a0. [DOI] [PubMed] [Google Scholar]
- 37.Cooke MP, Heath AW, Shokat KM, Zeng Y, Finkelman FD, Linsley PS, Howard M, Goodnow CC. Immunoglobulin signal transduction guides the specificity of B cell–T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med. 1994;179:425–438. doi: 10.1084/jem.179.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartley SB, Cooke MP, Fulcher DA, Harris AW, Cory S, Basten A, Goodnow CC. Elimination of self-reactive B lymphocytes proceeds in two stages: arrested development and cell death. Cell. 1993;72:325–335. doi: 10.1016/0092-8674(93)90111-3. [DOI] [PubMed] [Google Scholar]
- 39.Fulcher DA, Lyons AB, Korn SL, Cooke MC, Koleda C, Parish C, Fazekas de St. Groth B, Basten A. The fate of self-reactive B cells depends primarily on the degree of antigen receptor engagement and availability of T cell help. J Exp Med. 1996;183:2313–2328. doi: 10.1084/jem.183.5.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cook MC, Basten A, Fazekas de St. Groth B. Outer periarteriolar lymphoid sheath arrest and subsequent differentiation of both naive and tolerant immunoglobulin transgenic B cells is determined by B cell receptor occupancy. J Exp Med. 1997;186:631–643. doi: 10.1084/jem.186.5.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mobley JL, Dailey MO. Regulation of adhesion molecule expression by CD8 T cells in vivo. I. Differential regulation of gp90MEL-14 (LECAM-1), Pgp-1, LRA-1, and VLA-4α during the differentiation of cytotoxic T lymphocytes induced by allografts. J Immunol. 1992;148:2348–2356. [PubMed] [Google Scholar]
- 42.Gallatin WM, Weissman IL, Butcher EC. A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature. 1983;304:30–34. doi: 10.1038/304030a0. [DOI] [PubMed] [Google Scholar]
- 43.Reap EA, Piecyk ML, Oliver A, Sobel ES, Waldschmidt T, Cohen PL, Eisenberg RA. Phenotypic abnormalities of splenic and bone marrow B cells in lpr and gldmice. Clin Immunol Immunopathol. 1996;78:21–29. doi: 10.1006/clin.1996.0004. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi K, Kozono Y, Waldschmidt TJ, Berthiaume D, Quigg RJ, Baron A, Holers VM. Mouse complement receptors type 1 (CR1;CD35) and type 2 (CR2; CD21): expression on normal B cell subpopulations and decreased levels during the development of autoimmunity in MRL/lpr mice. J Immunol. 1997;159:1557–1569. [PubMed] [Google Scholar]
- 45.Wilson JG, Ratnoff WD, Schur PH, Fearon DT. Decreased expression of the C3b/C4b receptor (CR1) and the C3d receptor (CR2) on B lymphocytes and of CR1 on neutrophils of patients with systemic lupus erythematosus. Arthritis Rheum. 1986;29:739–747. doi: 10.1002/art.1780290606. [DOI] [PubMed] [Google Scholar]
- 46.Levy E, Ambrus J, Kahl L, Molina H, Tung K, Holers VM. T lymphocyte expression of complement receptor 2 (CR2/CD21): a role in adhesive cell-cell interactions and dysregulation in a patient with systemic lupus erythematosus (SLE) Clin Exp Immunol. 1992;90:235–244. doi: 10.1111/j.1365-2249.1992.tb07935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marquart HV, Svendsen A, Rasmussen JM, Nielsen CH, Junker P, Svehag S-E, Leslie RGQ. Complement receptor expression and activation of the complement cascade on B lymphocytes from patients with systemic lupus erythematosus (SLE) Clin Exp Immunol. 1995;101:60–65. doi: 10.1111/j.1365-2249.1995.tb02277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen X, Martin F, Forbush KA, Perlmutter RM, Kearney JF. Evidence for selection of a population of multi-reactive B cells into the splenic marginal zone. Int Immunol. 1996;9:27–41. doi: 10.1093/intimm/9.1.27. [DOI] [PubMed] [Google Scholar]
- 49.Allman DM, Ferguson SE, Lentz VM, Cancro MP. Peripheral B cell maturation. II. Heat-stable antigenhisplenic B cells are an immature developmental intermediate in the production of long-lived marrow-derived B cells. J Immunol. 1993;151:4431–4444. [PubMed] [Google Scholar]
- 50.Nitschke L, Carsetti R, Ocker B, Kohler G, Lamers M. CD22 is a negative regulator of B-cell receptor signalling. Curr Biol. 1997;7:133–143. doi: 10.1016/s0960-9822(06)00057-1. [DOI] [PubMed] [Google Scholar]
- 51.Molina H, Kinoshita T, Inque K, Carel J-C, Holers VM. A molecular and immunochemical characterization of mouse CR2: evidence for a single gene model of mouse complement receptors 1 and 2. J Immunol. 1990;145:2974–2983. [PubMed] [Google Scholar]
- 52.Dorken B, Moldenhauer G, Pezzutto A, Schwartz R, Feller A, Kiesel S, Nadler LM. HD39 (B3), a B lineage-restricted antigen whose cell surface expression is limited to resting and activated human B lymphocytes. J Immunol. 1986;136:4470–4479. [PubMed] [Google Scholar]
- 53.Kikutani H, Suemura M, Owaki H, Nakamura H, Sato R, Yamasaki K, Barsumian E, Hardy R, Kishimoto T. Fcε receptor, a specific differentiation marker transiently expressed on mature B cells before isotype switching. J Exp Med. 1986;164:1455–1469. doi: 10.1084/jem.164.5.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kinoshita T, Thyphronitis G, Tsokos GC, Finkelman FD, Hong K, Sakai H, Inoue K. Characterization of murine complement receptor type 2 and its immunological cross-reactivity with type 1 receptor. Int Immunol. 1990;2:651–659. doi: 10.1093/intimm/2.7.651. [DOI] [PubMed] [Google Scholar]
- 55.Molina H, Kinoshita T, Webster CB, Holers VM. Analysis of C3b/C3d binding sites and factor I cofactor regions within mouse complement receptors 1 and 2. J Immunol. 1994;153:789–795. [PubMed] [Google Scholar]
- 56.Tedder TF, Inaoki M, Sato S. The CD19-CD21 complex regulates signal transduction thresholds governing humoral immunity and autoimmunity. Immunity. 1997;6:107–118. doi: 10.1016/s1074-7613(00)80418-5. [DOI] [PubMed] [Google Scholar]
- 57.Kisielow P, Bluethmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell receptor transgenic mice involves deletion of immature CD4+8+ thymocytes. Nature. 1988;333:742–746. doi: 10.1038/333742a0. [DOI] [PubMed] [Google Scholar]
- 58.Cyster JG, Goodnow CC. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity. 1995;3:691–701. doi: 10.1016/1074-7613(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 59.Lortan JE, Roobottom CA, Oldfield S, MacLennan ICM. Newly produced virgin B cells migrate to secondary lymphoid organs but their capacity to enter follicles is restricted. Eur J Immunol. 1987;17:1311–1316. doi: 10.1002/eji.1830170914. [DOI] [PubMed] [Google Scholar]
- 60.Lieberum B, Hartmann K-U. Successive changes of the cellular composition in lymphoid organs of MRL-Mp/lpr-lprmice during the development of lymphoproliferative disease as investigated in cryosections. Immunol Immunopathol. 1988;46:421–431. doi: 10.1016/0090-1229(88)90061-x. [DOI] [PubMed] [Google Scholar]
- 61.Jacobson BA, Panka DJ, Nguyen K-AT, Erikson J, Abbas AK, Marshak-Rothstein A. Anatomy of autoantibody production: dominant localization of antibody-producing cells to T cell zones in Fas-deficient mice. Immunity. 1995;3:509–519. doi: 10.1016/1074-7613(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 62.Zheng B, Han S, Kelsoe G. T helper cells in murine germinal centers are antigen-specific emigrants that downregulate Thy-1. J Exp Med. 1996;184:1083–1091. doi: 10.1084/jem.184.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith KGC, Hewitson TD, Nossal GJV, Tarlinton DM. The phenotype and fate of the antibody-forming cells of the splenic foci. Eur J Immunol. 1996;26:444–448. doi: 10.1002/eji.1830260226. [DOI] [PubMed] [Google Scholar]
- 64.van Ewijk W, Rozing J, Brons NHC, Klepper D. Cellular events during the primary immune response in the spleen. Cell Tissue Res. 1977;183:471–489. doi: 10.1007/BF00225661. [DOI] [PubMed] [Google Scholar]
- 65.Liu Y-J, Zhang J, Lane PJL, Chan EY-T, MacLennan ICM. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur J Immunol. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- 66.Jacobson BA, Rothstein TL, Marshak-Rothstein A. Unique site of IgG2a and rheumatoid factor production in MRL/lpr mice. Immunol Rev. 1997;156:103–110. doi: 10.1111/j.1600-065x.1997.tb00962.x. [DOI] [PubMed] [Google Scholar]
- 67.Giese T, Davidson WF. Evidence for early onset, polyclonal activation of T cell subsets in mice homozygous for lpr. . J Immunol. 1992;149:3097–3106. [PubMed] [Google Scholar]
- 68.Gu L, Weinreb A, Wang X, Zack DJ, Qiao J, Weisbart R, Lusis AJ. Genetic determinants of autoimmune disease and coronary vasculitis in the MRL-lpr/lprmouse model of systemic lupus erythematosus. J Immunol. 1998;161:6999–7006. [PubMed] [Google Scholar]
- 69.Vyse KJ, Kotzin BL. Genetic susceptibility to systemic lupus erythematosus. Annu Rev Immunol. 1998;16:261–292. doi: 10.1146/annurev.immunol.16.1.261. [DOI] [PubMed] [Google Scholar]
- 70.Vidal S, Kono DH, Theofilopoulos AN. Loci predisposing to autoimmunity in MRL-Fas lpr and C57BL/ 6-Fas lprmice. J Clin Invest. 1998;101:696–702. doi: 10.1172/JCI1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Watson M, Rao J, Gilkeson G, Ruiz P, Eicher E, Pisetsky D, Matsuzawa A, Rochelle J, Seldin M. Genetic analysis of MRL-lpr mice: relationship of the Fas apoptosis gene to disease manifestations and renal disease-modifying loci. J Exp Med. 1992;176:1645–1656. doi: 10.1084/jem.176.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wechsler RJ, Monroe JG. Immature B lymphocytes are deficient in expression of the src-family kinases p59fyn and p55fgr. J Immunol. 1995;154:1919–1929. [PubMed] [Google Scholar]
- 73.Wofsy D, Ledbetter JA, Hendler PL, Seaman WE. Treatment of murine lupus with monoclonal anti-T cell antibody. J Immunol. 1985;134:852–857. [PubMed] [Google Scholar]
- 74.Jevnikar AM, Grusby MJ, Glimcher LH. Prevention of nephritis in major histocompatibility complex class II–deficient MRL-lpr mice. J Exp Med. 1994;179:1137–1143. doi: 10.1084/jem.179.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chesnutt MS, Finck BK, Killeen N, Connolly MK, Goodman H, Wofsy D. Enhanced lymphoproliferation and diminished autoimmunity in CD4-deficient MRL/ lpr mice. Clin Immunol Immunopathol. 1998;87:23–32. doi: 10.1006/clin.1997.4492. [DOI] [PubMed] [Google Scholar]
- 76.Rathmell JC, Townsend SE, Xu JC, Flavell RA, Goodnow CC. Expansion or elimination of B cells in vivo: dual roles for CD40- and Fas (CD95)-ligands modulated by the B cell antigen receptor. Cell. 1996;87:319–329. doi: 10.1016/s0092-8674(00)81349-5. [DOI] [PubMed] [Google Scholar]
- 77.Rathmell JC, Cooke MP, Ho WY, Grein J, Townsend SE, Davis MM, Goodnow CC. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 1995;376:181–184. doi: 10.1038/376181a0. [DOI] [PubMed] [Google Scholar]
- 78.Schmidt KN, Cyster JG. Follicular exclusion and rapid elimination of hen egg lysozyme autoantigen-binding B cells are dependent on competitor B cells, but not on T cells. J Immunol. 1999;162:284–291. [PubMed] [Google Scholar]
- 79.Perkins DL, Glaser RM, Mahon CA, Michaelson J, Marshak-Rothstein A. Evidence for an intrinsic B cell defect in lpr/lprmice apparent in neonatal chimeras. J Immunol. 1990;145:549–555. [PubMed] [Google Scholar]
- 80.Sobel EJ, Katagiri T, Katagiri K, Morris SC, Cohen PL, Eisenberg RA. An intrinsic B cell defect is required for the production of autoantibodies in the lprmodel of murine systemic autoimmunity. J Exp Med. 1991;173:1441–1449. doi: 10.1084/jem.173.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Katagiri T, Azuma S, Toyoda Y, Mori S, Kano K, Cohen PL, Eisenberg RA. Tetraparental mice reveal complex cellular interactions of the mutant, autoimmunity-inducing lprgene. J Immunol. 1992;148:430–438. [PubMed] [Google Scholar]
- 82.Sobel ES, Cohen PL, Eisenberg RA. lpr T cells are necessary for autoantibody production in lprmice. J Immunol. 1993;150:4160–4167. [PubMed] [Google Scholar]
- 83.Chu JL, Ramos P, Rosendorff A, Nikolic-Zugic J, Lacy E, Matsuzawa A, Elkon KB. Massive upregulation of the Fas ligand in lpr and gldmice: implications for Fas regulation and the graft-versus-host disease–like wasting syndrome. J Exp Med. 1995;181:393–398. doi: 10.1084/jem.181.1.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Watanabe D, Suda T, Hashimoto H, Nagata S. Constitutive activation of the Fas ligand gene in mouse lymphoproliferative disorders. EMBO (Eur Mol Biol Organ) J. 1995;14:12–18. doi: 10.1002/j.1460-2075.1995.tb06970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity. 1998;8:451–460. doi: 10.1016/s1074-7613(00)80550-6. [DOI] [PubMed] [Google Scholar]
- 87.Dautigny N, Chabre H, Garcia C, Ezine S. Marked depletion at the late pro-B stage in the bone marrow of lprmice correlates with the development of lymphadenopathy but not autoimmunity. Eur J Immunol. 1996;26:2087–2092. doi: 10.1002/eji.1830260919. [DOI] [PubMed] [Google Scholar]