
Figure 6.
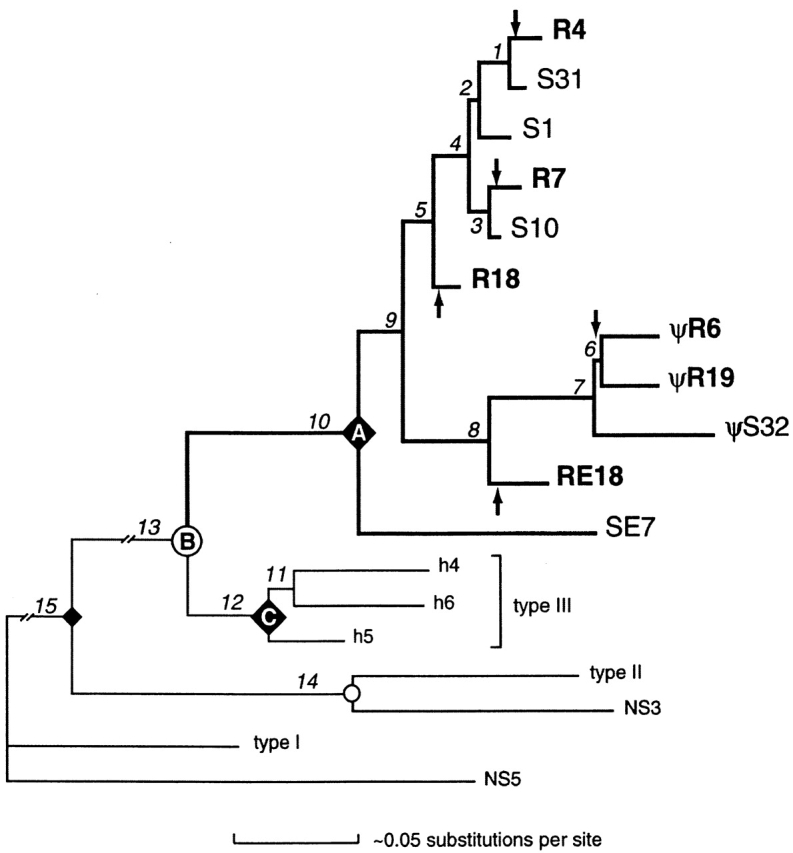
Phylogenetic tree of shark L chain V sequences. The relationships among the nurse shark NS4 family of genes are emphasized by bold branches. Other L chain types from nurse shark (NS3 and NS5) and horned shark (types I–III) were included in the analysis and used to root the tree. Branch lengths were estimated by likelihood using the HKY85+I+Γ model (reference 23), correcting for invariance and rate variation across sites as detailed in Materials and Methods. Nodes representing presumed speciation events are indicated as circles; e.g., node B corresponds to the divergence between all nurse shark NS4 genes and all horned shark type III genes. Nodes representing the most recent common ancestor of all members within a particular gene family are designated by ♦; e.g., all the NS4 genes derived from a common ancestral gene at node A and all the type III genes derived from a common ancestral gene at node C. Arrows designate branches in which germline rearrangements in NS4 genes most likely occurred. Lengths of internal branches (numbered segments) are approximately proportional to the estimated accumulation of nucleotide substitutions per site (scale at bottom), except for branches 13 and 15, which are ∼10 times as long and are truncated in the figure. The most strongly supported branches are 1, 4, 7, 8, 10, 12, 13, 14, and 15. For example, in one of many parsimony jackknife analyses of only the coding regions (assuming transversions were weighted twice that of transitions, ignoring alignment gaps, with 2,000 replications of a random selection of 50% of the nucleotide characters), each branch appeared in the following percentages of replicates (heuristic searches with 1,000 random taxon additions each): 1, 88%; 2, 61%; 3, 71%; 4, 82%; 5, 66%; 6, 51%; 7, 97%; 8, 86%; 9, 61%; 10, 95%; 11, 31%; 12, 98%; 13, 100%; 14, 100%; and 15, 100%. In a similar analysis that included noncoding regions (and excluded taxa for which these regions were not sequenced), jackknife values were (2,000 replications of exhaustive searches): 1+2, 37%; 3, 47%; 4, 75%; 5, 50%; 6, 98%; and 7+8, 94%. Horned shark cDNAs h4, h5, and h6 (available from EMBL/GenBank/DDBJ under accession nos. L25561, L25562, and L25563, respectively) were isolated by cross-hybridization with NS4 sequence. Based on separate phylogenetic analyses of V and C regions, type III was identified as a homologue of NS4 (reference 13); this conclusion is also supported by the current phylogram. All available type III sequences grouped strongly together, as did all the representative NS4 sequences (germline-rearranged R7, R4, R18, R6, R19, and RE18, and split S1, S10, S31, S32, and SE7), suggesting gene diversification after the species diverged. Pseudogenes are indicated. Other sequences are from different L chain types in horned shark (type I, X15316; type II, L25560) and nurse shark (NS3 [reference 16], NS5 [reference 39]). SE7 is the most basally divergent member of the NS4 gene family identified to date, and its sequence is not shown in Fig. 3.