

Abstract
Left ventricular (LV) remodeling after myocardial infarction (MI) results in LV dilation, a major cause of congestive heart failure and sudden cardiac death. Ischemic injury and the ensuing inflammatory response participate in LV remodeling, leading to myocardial rupture and LV dilation. Myeloperoxidase (MPO), which accumulates in the infarct zone, is released from neutrophils and monocytes leading to the formation of reactive chlorinating species capable of oxidizing proteins and altering biological function. We studied acute myocardial infarction (AMI) in a chronic coronary artery ligation model in MPO null mice (MPO−/−). MPO−/− demonstrated decreased leukocyte infiltration, significant reduction in LV dilation, and marked preservation of LV function. The mechanism appears to be due to decreased oxidative inactivation of plasminogen activator inhibitor 1 (PAI-1) in the MPO−/−, leading to decreased tissue plasmin activity. MPO and PAI-1 are shown to have a critical role in the LV response immediately after MI, as demonstrated by markedly delayed myocardial rupture in the MPO−/− and accelerated rupture in the PAI-1−/−. These data offer a mechanistic link between inflammation and LV remodeling by demonstrating a heretofore unrecognized role for MPO and PAI-1 in orchestrating the myocardial response to AMI.
Keywords: myocardial rupture, free radical, chlorination, inflammation, protease activation
Introduction
10% of all individuals over the age of 65 in the United States have been diagnosed with congestive heart failure, the vast majority secondary to left ventricular dysfunction, and LV dilation after acute myocardial infarction (AMI). The remodeling of the LV after AMI determines myocardial performance and residual left ventricle (LV)* function. In the near term (<1 wk) after AMI, the remodeling process can lead to myocardial rupture, ventricular septal defect formation, or rupture of a papillary muscle, each of which, although uncommon, has extremely high mortality rates. In the long term, remodeling leads to LV dilation, resulting in increased myocardial wall stress and work, ultimately causing decreased functional reserve and congestive heart failure (1, 2).
The molecular pathways critical to the LV remodeling process are not yet fully defined. Studies have demonstrated that mice that are null for the plasminogen activator urokinase (uPA) and matrix metalloprotease-9 have decreased myocardial rupture (3, 4) and LV dilation (4). However, the mechanisms responsible for activation of these enzymes in response to AMI are still under study.
Myeloperoxidase (MPO) is contained in the granules of neutrophils and monocytes and is released upon leukocyte activation, resulting in altered biological function of surrounding proteins and lipids by the generation of chlorinating, nitrating, and other oxidizing species (5–8). Leukocytes release MPO both within the infarct zone after AMI and at sites of ventricular rupture (3). To determine whether MPO-generated oxidants participate in LV rupture and the remodeling process, we studied AMI in the MPO null (MPO−/−) mouse (9).
Materials and Methods
Left Anterior Descending Coronary Artery (LAD) Ligation.
All animal protocols were approved by the Animal Research Committee and all animals were housed in the American Association for Accreditation of Laboratory Animal Care–approved animal facility of the Cleveland Clinic Foundation. Anterior wall myocardial infarction (MI) was induced in ∼20–25-g male littermate WT (C57BL/6J), MPO−/− (9), or plasminogen activator inhibitor 1 (PAI-1)−/− (C57BL/6J background; Jackson ImmunoResearch Laboratories; reference 10) mice by ligation of the LAD (3, 4). Animals were intubated and ventilated with room air at 100 breaths per minute using a rodent ventilator (Harvard Apparatus). Sternotomy was performed and the proximal LAD was identified using a surgical microscope (Leica M500) after retraction of the left atrium and ligated with 7-0 prolene. Blanching and dysfunction of the anterior wall verified LAD ligation. Mice were included in survival studies if they were alive the morning after surgery. During our study, we had an ∼30% mortality in the WT and MPO−/− mice, a 40% mortality in the PAI-1−/− mice on the day of surgery, and a 10% mortality during the first night after surgery in all groups.
Immunohistochemistry.
Mice were anesthetized and perfusion fixed with HistoChoice at physiological pressures. Fixed hearts were embedded in paraffin and sectioned at 4 μm. Immunohistochemistry for CD45+ cells was performed using rat anti–mouse CD45 antibody and an FITC-labeled rabbit anti–rat IgG secondary antibody (Santa Cruz Biotechnology, Inc.).
Tissue Processing for Biochemical Analysis.
Protein extracts for biochemical analyses were obtained from tissue from the infarct zone of hearts that were rapidly perfused with PBS in situ and frozen in liquid nitrogen. Tissues were minced frozen and extracts were made in 300 μl buffer containing 10 mM sodium phosphate, pH 7.2, 150 mM sodium chloride, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, and 0.2% sodium azide for 1 h at 4°C (11, 12) with constant rocking and frequent vortexing. Samples were centrifuged at 10,000 rpm and supernatants were immediately divided into 10-μl aliquots and frozen at −80°C. Protein content was determined using the modified Lowery method (13). An aliquot was used only once and before its use, it was diluted with additional extraction buffer to obtain a final total protein concentration of 5 μg/μl.
Collagen Content.
Collagen content was determined by picrosirius red polarization microscopy for detection of interstitial collagen according to Junqueira's method (14). We used the protocol of Ducharme et al. (4) that has been successfully implemented in the mouse AMI model. The birefringence under illumination of myocardium with polarized light identifies collagen, including types I and III (14). Fresh 5-μM frozen sections of hearts that were flash frozen after perfusion with PBS from control mice and mice 12 and 21 d after AMI were fixed in 10% formalin and after rinsing in distilled water were incubated with 0.1% picrosirius red F3BA in saturated picric acid for 90 min. Tissues were treated with 0.01 N HCl for 1 min before being immersed in distilled water. Tissue sections were then dehydrated, coverslipped, and imaged under polarized light. Images were captured with the same exposure time for all tissues and the percent area containing collagen (yellow birefringence) was determined using NIH Image software.
Echocardiography.
2D echocardiography was performed 3–5 d before AMI as well as on days 3 and 21 after AMI using a 15-MHz linear array transducer interfaced with a Sequoia C256 (Acuson). Mice were routinely handled to allow for echocardiography in unsedated mice. We digitally recorded 2D clips and m-Mode images in a short axis view from the mid-LV just below the level of the papillary muscles so that we could consistently obtain measurements from the same anatomical location. Measurements were made off-line using MedArchive Viewer (version 2.1). Each measurement in each animal was made six times from three randomly chosen m-Mode clips out of five recorded by two observers blinded to the strain of the mice. As a measure of LV function, the shortening fraction was calculated from m-Mode recordings. Shortening fraction (%) = (LV end diastolic dimension [LVEDD]−LV end systolic dimension)/LVEDD*100. Dimensions were measured between the anterior wall and posterior wall from the short axis view just below the level of the papillary muscle.
Zymography.
SDS-PAGE was performed under nondenaturing conditions using 10% acrylamide gels impregnated with 1 mg/ml gelatin (as a substrate for matrix metalloproteinase [MMP]-2 and MMP-9; reference 15) or casein (as a plasmin substrate; reference 16). 10–20 μg protein extract from the infarct zone was separated over ∼2 h at 4°C. Gels were incubated in 2.5% Triton X-100 for 1 h and then in CaCl2 containing solution overnight at 37°C. Gels were then fixed (methanol/acetic acid 40:10%) and developed with 10% Coomassie blue. Gels were digitally stored and bands were quantified in arbitrary units using NIH Image software.
Tissue PAI-1 Activity.
PAI-1 activity was determined by a two-stage, indirect enzyme assay system using polylysine-stimulated glu-plasminogen activation (kit 101201; American Diagnostica, Inc.; references 17) or by reverse zymography (18, 19).
Two-Stage Indirect Assay for PAI-1.
In this assay, 1 U PAI-1 activity is defined as the amount of PAI-1 that inhibits one international unit of human single chain tissue plasminogen activator (tPA). In brief, 150 μg total protein extract from the infarct zone is treated with 40 IU tPA. The reaction mixture is then acidified to destroy plasmin inhibitors that might be present. PAI-1 activity was determined by quantifying the residual tPA activity through the addition of plasminogen and a chromogenic substrate (Spectrozyme PL; American Diagnostica, Inc.) for plasmin. No significant signal was obtained if the tPA was not added, suggesting that the level of plasmin activity within the total protein extract did not contribute to the measured signal. As a control, we verified that recombinant mouse PAI-1 (Molecular Innovations) was successfully detected using this system. However, the standard curve used to determine PAI-1 activity was based on serial dilutions of tPA.
Reverse Zymography for PAI-1.
PAI-1 activity was determined by reverse zymography (18, 19) in SDS-PAGE gel. We separated 20 μg total protein extracts at 4°C on a 12% SDS-PAGE gel containing 1 mg/ml casein, 10 μg/ml human plasminogen (provided by E. Plow, Cleveland Clinic Foundation, Cleveland, OH) and 0.5 mU/ml human uPA (American Diagnostica, Inc.). Gels were incubated in 2.5% Triton X-100 for 1 h and then in CaCl2 containing solution overnight at 37°C. Gels were then fixed (water/methanol/acetic 50:40:10%) and developed with 10% Coomassie blue. Gels were digitally stored and bands were quantified in arbitrary units using NIH Image software. This protocol was able to detect the activity of <1 ng murine PAI-1 (Molecular Innovations).
MPO-mediated Oxidation of PAI-1.
0.25 μg murine PAI-1 was treated with 25 nM MPO and H2O2 in the presence of 140 mM NaCl, pH 7.4, for 15 min. Treatment with MPO or H2O2 alone had no effect on PAI-1 activity. Oxidation was quenched with 1 mM dithiothreitol for 5 min and then uPA (1 unit; American Diagnostica, Inc.) and CaCl2 (final concentration of 50 μM) were added to the PAI-1 sample. We assayed the reaction for residual uPA activity by adding a uPA-specific chromogenic substrate (Spectrozyme UK; American Diagnostica, Inc.) and CaCl2 (final concentration of 400 μM). Absorbance was measured at 405 nm as a function of time at 25°C.
Quantification of PAI-1 Chlorotyrosine Content.
PAI-1 chlorotyrosine content was quantified by stable isotope dilution gas chromatography-mass spectrometry (7). For in vivo samples, 50 μg myocardial homogenate at 0, 1, and 3 d after LAD ligation of WT (n = 4, 7, and 3, respectively) and MPO−/− (n = 4, 4, and 3, respectively) was separated by SDS-PAGE. After transfer to PVDF, the band corresponding to intact PAI-1 as determined by Western blot analysis (see below) of adjacent lanes, was cut out and subjected to acid hydrolysis and then mass spectrometry analysis.
Western Blot Analysis.
A lane from each end of the PVDF was blocked with 5% milk powder with 0.1% Tween 20. The blots were incubated in primary antibody to PAI-1 (SC-6644; Santa Cruz Biotechnology, Inc.) in 5% milk powder with 0.1% Tween 20 for 2 h, washed, and then incubated in secondary antibody in 5% milk powder with 0.1% Tween 20 for 1 h. After washing three times, the peroxidase was visualized on X-ray film using enhanced chemiluminescence.
Statistical Analysis.
Data are presented as mean ± SD. Comparisons between groups were made by Student's t test.
Results
Effect of MPO on Leukocyte Infiltration after AMI.
AMI of the anterior wall of WT and MPO−/− mice was induced by chronic ligation of the proximal LAD. WT mice begin to die from myocardial rupture on day 4 after LAD ligation in this model (3). Therefore, we initially studied if MPO had significant effects on the myocardial response to AMI 72 h after LAD ligation. 3 d after AMI, MPO−/− exhibited preservation of myocardial architecture and significantly decreased leukocyte infiltration when compared with WT mice (Figs. 1 and 2) . Rather, the infarct zone in the MPO−/− consists of enucleated (dead) myocytes. Note there was no significant leukocyte infiltration in the posterior wall, a site remote from the ischemic anterior wall in either WT or MPO−/− (Fig. 1, C and D).
Figure 1.
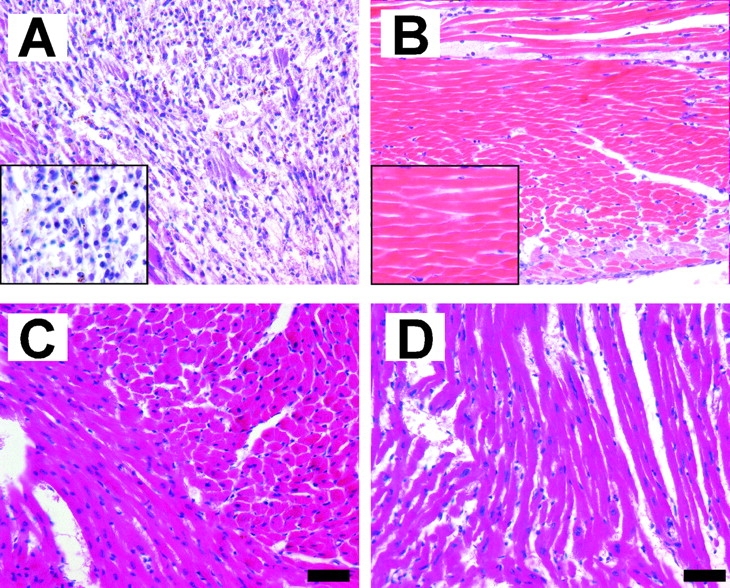
Hematoxylin and eosin staining of representative cross sections from the mid-ventricle of (A and C) WT and (B and D) MPO−/− 3 d after LAD ligation. The figures represent (A and B) the infarct zone and (C and D) the non-infarcted posterior wall. The insets of A and B show higher power images from the infarct zone revealing dead enucleated myocytes in the MPO−/− and inflammatory infiltrate in the WT infarct zones. Bar, 50 μM.
Figure 2.

Quantification of CD45 immunoreactive cells within the infarct zone of WT and MPO−/− 3 d after LAD ligation (n = 6) in each group. Data represent mean ± SD of five fields per animal.
Effect of MPO on LV Remodeling and Function after MI.
Cardiac size and function in WT and MPO−/− were monitored by 2D echocardiography after AMI. No significant differences in cardiac dimensions nor performance were observed in uninjured WT and MPO−/− mice (Fig. 3 , 0 time points). 3 d after AMI, significant thinning of the anterior (infarcted) wall was evident in both WT and MPO−/− (Fig. 3 A), without any significant change noted in either the thickness of the non-infarcted posterior wall (Fig. 3 B) nor the extent of LVEDD (Fig. 3 C). In contrast, echocardiography at 21 d after AMI demonstrated that myocardium from WT possessed significantly thinned anterior wall with accompanying marked LV chamber dilation, compared with that observed in the MPO−/− (Fig. 3, A, C, and D). The differences in dimensions observed at 21 d were due to an apparent arrest in myocardial thinning and dilation in the MPO−/− compared with the ongoing LV remodeling observed in the WT. LV remodeling in the MPO−/− was accompanied by changes in non-infarct tissue as evidenced by hypertrophy of the posterior wall in the MPO−/− at 21 d after AMI (Fig. 3 B).
Figure 3.

Echocardiographic analysis of LV size in WT and MPO−/− as a function of time after AMI. (A) Anterior wall thickness, (B) posterior wall thickness, and (C) LVEDD before and 3 and 21 d after AMI in WT (○, n = 8 at each time point) and MPO−/− (•, n = 8 at 0 and 3 d and n = 7 at 21 d after AMI) mice. There were no significant differences in measured parameters 3 d after AMI. At 21 d the anterior and posterior walls were thicker (P < 0.05 and P < 0.01, respectively) and the ventricle was less dilated (P < 0.01) in the MPO−/− mice. Data represent mean ± SD. (D) Representative m-mode recording from a WT and MPO−/− animal 21 d after AMI. *, P < 0.05, WT versus MPO−/−.
No significant difference in LV function as measured by shortening fraction was observed between the non-infarcted WT and MPO−/− mice nor at 3 d after AMI. However, the combination of decreased LV dilation and posterior wall hypertrophy resulted in a significant increase (∼85%; Table I) in LV function at 21 d after AMI in the MPO−/− compared with WT.
Table I.
LV Shortening Fraction as a Function of Time after AMI in WT and MPO−/− Mice
| Days after AMI | WT | MPO−/− |
|---|---|---|
| 0 | 58.9 ± 2.0 | 59.6 ± 3.1 |
| 3 | 25.4 ± 4.8 | 21.5 ± 2.9 |
| 21 | 12.9 ± 3.1 | 23.9 ± 4.8a |
P < 0.005, MPO versus WT at specified time.
WT, n = 8 at each time point.
MPO−/−, n = 8 at 0 and 3 d and n = 7 at 21 d.
Role for MPO on Activation of Plasminogen after MI.
Decreased leukocyte infiltration after AMI has been reported in the uPA−/− mouse model (3). Therefore, we postulated that MPO release after AMI might result in urokinase activation. The activity of urokinase in the infarct zone after AMI was determined by quantifying tissue plasmin activity by zymography. No significant differences in plasmin activity were observed in uninjured hearts from WT and MPO−/− (not depicted), and baseline plasmin activity was approximately equal to that observed in MPO−/− 3 d after LAD ligation (Fig. 4, A and C) . There is significantly more plasmin activity after MI in the WT myocardium. (Fig. 4, A and C) Of interest, the increase in plasmin activity noted in WT myocardium was not accompanied by increased MMP-2 nor MMP-9 activity (Fig. 4, B and D). No significant MMP-2 nor MMP-9 activities were observed in uninjured myocardium from WT nor MPO−/− (not depicted). Quite unexpectedly, our data demonstrate that both MMP-2 (∼60 kD) and proMMP-9 (94 kD) were significantly more up-regulated in the MPO−/− 3 d after AMI compared with WT. To verify that the MMP-9 band observed was in fact the pro-form, myocardial extracts were treated with 4-aminophenylmercuric acetate (20), a chemical agent that cleaves the proMMP-9 into its active form. The appropriate shift in electrophoretic mobility was observed, confirming conversion from proMMP-9 to MMP-9 (82 kD; not depicted). We also verified that the bands observed were not secondary to plasmin degradation of gelatin by demonstrating that the band identified as MMP-2 and pro/partially active MMP-9 were not sensitive to the addition of the plasmin inhibitor aprotinin (5 μg/ml) to the CaCl2 reaction solution.
Figure 4.

Protease activation within the infarct zone of WT and MPO−/−. (A) Representative casein and (B) gelatin zymograms of protein extracts from the infarct zones of two representative WT and two representative MPO−/− mice 3 d after AMI. Quantification of the density of (C) plasmin activity band and the (D) MMP-2 (open bars) and proMMP-9 (black bars) bands in the infarct zone of WT (n = 6) and MPO−/− (n = 6) mice 3 d after AMI. No significant differences in plasmin activity in non-infarct myocardium between strains were seen, and no MMP activity was seen in non-infarcted tissue from either strain.
PAI-1 Activity Is Preserved in MPO−/− Mice.
We postulated that the increase in urokinase activity in the WT myocardium could be due to the decreased activity of its inhibitor, PAI-1, because its activity is known to be oxidant sensitive (21–23). Therefore, we quantified PAI-1 mass and activity in protein extracts from myocardium of WT and MPO−/− mice using reverse zymography for PAI-1 (Fig. 5, A and B) and a two-stage plasmin-based PAI-1 activity assay (Fig. 5 C). Western blot and activity analyses of PAI-1 in non-infarcted myocardium revealed similar PAI-1 mass (not depicted) and activity (Fig. 5 B) between the two stains. Remarkably, 1 d after AMI, PAI-1 activity was significantly inhibited in the infarct zone of WT tissue compared with non-infarcted myocardium by ∼30% as measured by reverse zymography (Fig. 5 B) and a two-stage plasmin-based activity assay (Fig. 5 C). However, no significant inhibition of PAI-1 activity was seen in the myocardial infarct zone of MPO−/− mice (Fig. 5, B and C).
Figure 5.
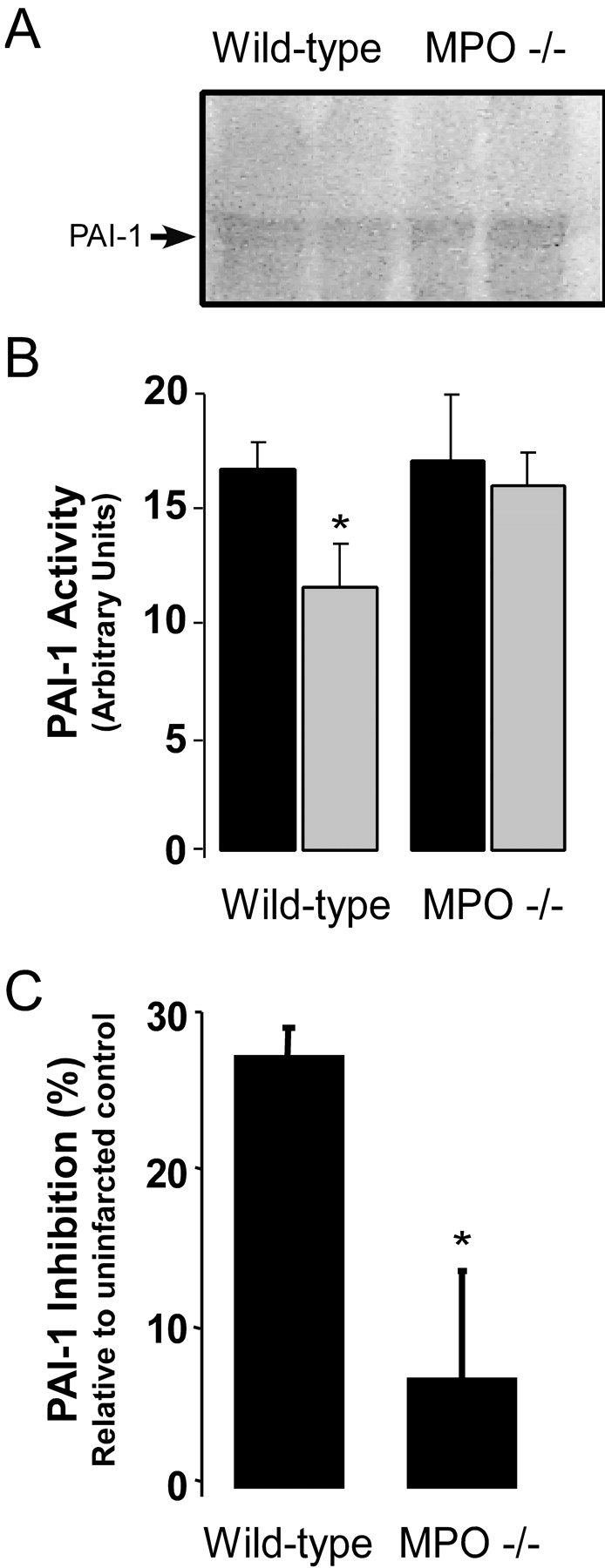
PAI-1 activity 1 d after AMI in WT and MPO−/−. (A) Representative reverse casein zymograms for PAI-1 activity in protein extracts from the infarct zones of two representative WT and two MPO−/− mice with no MI (filled bars) or one representative MPO−/−. (B) PAI-1 activity in WT and MPO−/− 1 d after AMI (open bars) as quantified in arbitrary units from reverse zymograms (n = 4 animals per group per time point). *, P < 0.02, WT versus MPO−/− 1 d after AMI. (C) Inhibition of PAI-1 activity (%) in the infarct zone relative to PAI-1 activity in strain-matched non-infarcted tissue as measured by two-stage indirect plasmin-based assay for PAI-1 in WT (n = 6) and MPO−/− (n = 6) myocardium 1 d after LAD ligation. Data represent mean ± SD. *, P < 0.01.
Effect of MPO on Myocardial Healing.
To determine if abnormal healing occurred in the MPO−/− mice, we quantified collagen content in the infarct zone as percent area containing collagen as a function of time after AMI using picrosirius red staining (4). Our data demonstrate that there is no difference in collagen content in control hearts from WT and MPO−/− (Fig. 6) . However, at 12 d after AMI, there is significantly less collagen in the infarct zone of the MPO−/− compared with WT. Interestingly, by 21 d after AMI, the percent area of collagen deposition in the MPO−/− is near that of the WT (Fig. 6). This observation is consistent with the increased cellularity of the infarct zone at 21 d after AMI seen in the MPO−/− (not depicted), but not the plasminogen−/−, MMP-9−/−, nor uPA−/− mouse models (3, 4, 24).
Figure 6.

Collagen content in the infarct zone was quantified as the percent area that was birefringent under illumination with polarized light after staining with picrosirius red. Collagen content was quantified from five areas within the infarct zone from three frozen sections in three animals per time point in both the WT and MPO−/−. Data represents mean ± SD. *, P < 0.02, WT versus MPO at given time point.
MPO-mediated Oxidation Inhibits PAI-1 Activity.
The above data suggest that in response to AMI, MPO inhibits PAI-1 activity, leading to increased urokinase activity and plasmin generation. To test if MPO-generated oxidants can inhibit PAI-1 activity we used an in vitro PAI-1 assay system. PAI-1 activity was quantified by assessing its ability to inhibit uPA cleavage of chromogenic substrate. Incubation of PAI-1 with the MPO-H2O2-Cl− system resulted in dose-dependent inactivation of PAI-1 (Fig. 7 A) and was accompanied by increased PAI-1 content of chlorotyrosine (Fig. 7 B), a specific posttranslational modification of proteins formed by action of MPO-generated oxidation of target proteins (6, 7). In contrast, incubation of PAI-1 with either MPO or H2O2 alone had no effect on PAI-1 activity (not depicted). Thus, MPO-generated oxidants are capable of inhibiting PAI-1. To determine if MPO promotes oxidative modification of PAI-1 in vivo after AMI, we quantified the level of chlorotyrosine in PAI-1 recovered from WT and MPO−/− myocardium at baseline, 24 and 72 h after AMI. PAI-1 was significantly chlorinated in WT mice 24 and 72 h after LAD ligation (Fig. 7 B). In marked contrast, no significant chlorotyrosine was detected in PAI-1 recovered from WT nor MPO−/− non-infarcted myocardium, nor in myocardium recovered from MPO−/− mice at any time after AMI.
Figure 7.
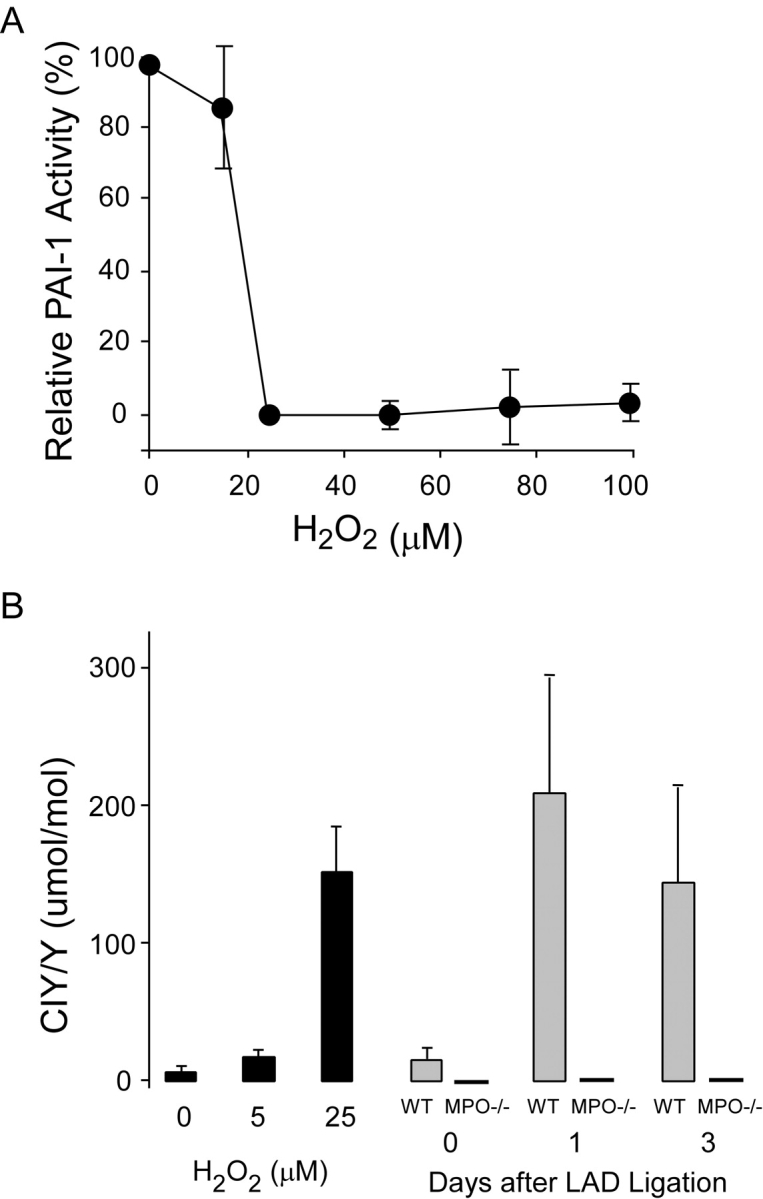
Effect of MPO-mediated oxidation on PAI-1 activity in vitro and in vivo. (A) Inhibition of PAI-1 activity as a function of increasing hydrogen peroxide concentration in the presence of 25 nM human MPO. PAI-1 activity was quantified as residual urokinase activity after incubation with MPO-H2O2-Cl–treated PAI-1. Zero percent PAI-1 inhibition was defined as urokinase activity after incubation with PAI-1 treated with H2O2 or MPO alone and 100% PAI-1 inhibition was defined as urokinase activity in the absence of PAI-1. Data represent mean ± SD of three samples per H2O2 concentration. (B) Level of protein-bound 3-chlorotyrosine in PAI-1 as measured by gas chromatography-mass spectrometry after (gray bars) treatment with MPO-H2O2-Cl− at the indicated H2O2 concentrations, or in non-infarcted myocardium or in the infarct zone of WT (n = 7) and MPO−/− (n = 4) 1 and 3 d after LAD ligation. Data represent mean ± SD.
Integral Role for MPO and PAI-1 in Regulating Tissue Degradation after MI.
To further demonstrate a critical role for PAI-1 regulation of the uPA-mediated activation of plasmin after MI, we performed LAD ligation in PAI-1−/− mice. Non-infarcted PAI-1−/− demonstrated similar cardiac function and wall dimensions as WT and MPO−/− (not depicted). 3 d after LAD ligation, the heart of the PAI-1−/− had significant leukocyte infiltration and areas of intramyocardial hemorrhage, a feature that was not observed in infarcted myocardium from WT nor MPO−/−.
To further investigate the importance of MPO effects on PAI-1 activity after AMI, we studied the survival of WT, MPO−/−, and PAI-/1−/− mice as a function of time after LAD ligation by a surgeon blinded to the identity of the mice. Death by myocardial rupture was defined by criteria similar to that used by other investigators (4). The animals had to (a) be healthy the day before death, (b) have blood in the cavity at necropsy, and (c) clot on the surface of the heart, typically at the infarct border zone.
The structural changes in myocardium from MPO−/− mice seen 3 d after LAD ligation were accompanied by a significant decrease in early myocardial rupture (Fig. 8) . No MPO−/− mice died before day 10 after LAD ligation compared with an overall 40% mortality observed in WT mice over the same time interval. Long-term (21 d) MPO−/− mice demonstrated similar mortality rates to WT with a late phase of myocardial rupture beginning on day 10 in the MPO−/− mice. In stark contrast to the MPO−/−, myocardial rupture in the PAI-1−/− was significantly accelerated compared with WT, and all PAI-1−/− mice died of myocardial rupture by day 6 after AMI. Because patients deficient in PAI-1 have increased bleeding after surgery (25), we performed sham LAD ligation that consisted of placement of suture around the LAD without impeding blood flow. None of these animals died within the 8 d after surgery. Together, these results support our hypothesis that MPO-mediated inactivation of PAI-1 plays a significant role in determining the myocardial response to AMI.
Figure 8.

Survival of WT (solid line, n = 17), MPO−/− (dotted line, n = 10), and PAI-1−/− (dashed line, n = 6) as a function of time after LAD ligation. No animals died in any group between days 15 and 21.
Discussion
This study provides evidence suggesting a critical role for MPO and PAI-1 in LV remodeling after MI and offers a mechanistic link between inflammation and LV dilation after AMI. In the AMI model of chronic LAD ligation used, we observed that in MPO−/− mice there is decreased leukocyte infiltration, decreased LV dilation, enhanced ventricular function (shortening fraction), and delayed early death due to myocardial rupture. Decreased leukocyte infiltration with preservation of the myocardial structure has previously been observed in uPA−/− and plasminogen−/− mouse models of MI (3, 24). Distinct from the uPA−/− model in which cardiac rupture was completely inhibited (3), cardiac rupture was markedly delayed in the MPO−/−.
Despite the decrease in myocardial rupture in the uPA−/− (3) and MMP-9−/− (4) mice, studies using these mice have described abnormal myocardial healing due to mummification of the dead cardiac myocytes. Failure to clear the necrotic debris of the infarct zone appears to lead to (a) decreased collagen content, (b) increased erythemogenic potential, and (c) decreased responsiveness to inotropic agents (3, 4). In the MPO−/− we observed that myocardial healing, as quantified by collagen content, is only delayed, not inhibited, because collagen content in the infarct zone is similar between the WT and MPO−/− at 21 d after AMI. Given the overall improved LV function at 21 d, this suggests that transient early inhibition of leukocyte recruitment after AMI does not necessitate abnormal healing and can lead to improved LV remodeling and function.
Leukocyte recruitment in the MPO−/− has been shown to be no different than that in WT mice in a thioglycollate peritonitis model (7), suggesting that leukocyte response to chemotactic factors is preserved in the MPO−/− and that the decreased leukocyte infiltration we observe in the MPO−/− is not due to decreased leukocyte recruitment, but rather decreased leukocyte entry into the infarct zone. The mechanism for MPO modulation of leukocyte infiltration and LV remodeling of the infarct zone appears to be via activation of the uPA pathway, ultimately leading to increased plasmin generation. Our findings indicate that the increased plasmin activity in the infarcted myocardium from the WT mice is due to a decrease in the activity of the uPA inhibitor, PAI-1. The activity of many members of the serpin family including PAI-1 (23), but not PAI-2 (26), have been shown to be oxidant sensitive. In this study, we demonstrated in an in vitro assay system that PAI-1 activity is sensitive to oxidative inactivation by MPO-generated oxidants. Moreover, the chlorotyrosine content of PAI-1 recovered from infarct zone of WT myocardium was in excess of that formed in PAI-1 oxidatively inactivated by exposure to MPO-generated HOCl, in vitro (Fig. 7). Thus, MPO-generated oxidants appear to function as a physiological signal for the inactivation of PAI-1 activity (Fig. 5, B and C) in the post-AMI setting.
Our observation that the posterior wall hypertrophies in MPO−/− mice in response to acute anterior wall MI might be a result of the decreased inflammatory response. As seen in Fig. 1, C and D, there was no significant leukocyte infiltration into the posterior wall of WT nor MPO−/− 3 d after LAD ligation. Potential mediators of remote LV dysfunction that are released during the inflammatory response, such as TNF-α (27), have been shown to have direct negative inotropic effects on cardiac myocytes as well as lead to myocardial thinning (28). Thus, in the MPO−/−, the posterior wall may hypertrophy due to the decreased inflammation in the myocardium in response to the anterior wall ischemia.
The finding that the MPO−/− mice had increased expression of proMMP-9 and MMP-2 was unexpected. The effect of MPO deficiency on MMP expression in vivo has not been previously reported. The mechanism for this finding is unknown but may reflect a critical role for MPO in modulating H2O2 exposure to myocardium in WT mice. Indeed, H2O2 has been shown to increase MMP expression in cell culture (29). That MPO−/− had increased MMP expression but exhibited decreased LV dilation after AMI, suggests that plasmin, and not MMP's, may have a more important role in LV remodeling after MI. This concept is corroborated by the data in the literature that demonstrate incomplete protection from myocardial rupture after LAD ligation in MMP-9 null mice but complete protection in uPA null mice (3, 4). The possibility exists that the increased MMP expression in the MPO−/− mice may participate in the late phase of myocardial rupture observed in these animals after AMI.
The ability of PAI-1 to inhibit uPA activity has been shown to be redox sensitive (21–23). Nonphysiological models of oxidation such as exposure to caustic oxidants/chemical agents have documented reduction in PAI-1 activity (22). Although the oxidation pathways operational in vivo have never been examined, a role for oxidative modification of methionine(s) to methionine sulfoxide during inactivation of PAI-1 has been suggested (23). HOCl, major reactive oxidant formed by the MPO-H2O2-Cl− system of leukocytes, rapidly scavenges methionine as a primary target in biological matrices (30).
This study demonstrates that leukocyte-mediated oxidation reactions play a critical role in LV remodeling after MI. They also identify MPO as an enzymatic participant in the oxidant-sensitive pathways responsible. Paradoxically, angiotensin converting enzyme inhibitors (ACE-I) lower endothelial cell production of PAI-1 (31, 32) as well as circulating PAI-1 levels (33), and this is one of the postulated mechanisms by which ACE-I may lead to smaller infarct size after MI (34). Whether the improved outcomes in patients on ACE-I with lower circulating PAI-1 levels are due to a decrease in the thrombotic potential of the vascular wall secondary to increased fibrinolysis or some PAI-1–independent effect of ACE-I remains to be determined (35).
It is interesting to note that the current findings may suggest potential genetic links for the myocardial response to AMI. MPO levels vary significantly in individuals, are elevated in subjects with a family history of coronary artery disease (36), and polymorphisms in the MPO promoter and circulating MPO levels have recently been shown to be associated with a higher prevalence of coronary artery disease (37). Similarly, there are multiple known polymorphisms of PAI-1 such as the 4G/4G genotype in the PAI-1 promoter that is associated with increased PAI-1 expression (38). Although the role of PAI-1 and MPO have been intensely studied with respect to the development and presence of atherosclerosis and plaque rupture (39–41), the effects of these molecules on LV size or the development of congestive heart failure after AMI have not. However, it is intriguing to postulate based on our data that an interaction of MPO and PAI-1 contributes to the early myocardial response to AMI. Additional elucidation of the mechanism of MPO inhibition of PAI-1 may lead to a deeper understanding of the role of the inflammatory process on LV remodeling after MI, as well as identify the molecular targets to augment myocardial performance after cardiac myocyte death.
Acknowledgments
Supported by National Institutes of Health grants HL62526 and HL70621 to S.L. Hazen.
A.T. Askari and M.-L. Brennan contributed equally to this work.
Footnotes
Abbreviations used in this paper: ACE-I, angiotensin converting enzyme inhibitors; AMI, acute myocardial infarction; LAD, left anterior descending coronary artery; LV, left ventricle; LVEDD, LV end diastolic dimension; MI, myocardial infarction; MMP, matrix metalloproteinase; MPO, myeloperoxidase; tPA, tissue plasminogen activator; uPA, plasminogen activator urokinase.
References
- 1.St. John, S.M., M.A. Pfeffer, L. Moye, T. Plappert, J.L. Rouleau, G. Lamas, J. Rouleau, J.O. Parker, M.O. Arnold, B. Sussex, et al. 1997. Cardiovascular death and left ventricular remodeling two years after myocardial infarction: baseline predictors and impact of long-term use of captopril: information from the Survival and Ventricular Enlargement (SAVE) trial. Circulation. 96:3294–3299. [DOI] [PubMed] [Google Scholar]
- 2.Groenning, B.A., J.C. Nilsson, L. Sondergaard, T. Fritz-Hansen, H.B. Larsson, and P.R. Hildebrandt. 2000. Antiremodeling effects on the left ventricle during beta-blockade with metoprolol in the treatment of chronic heart failure. J. Am. Coll. Cardiol. 36:2072–2080. [DOI] [PubMed] [Google Scholar]
- 3.Heymans, S., A. Luttun, D. Nuyens, G. Theilmeier, E. Creemers, L. Moons, G.D. Dyspersin, J.P. Cleutjens, M. Shipley, A. Angellilo, et al. 1999. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 5:1135–1142. [DOI] [PubMed] [Google Scholar]
- 4.Ducharme, A., S. Frantz, M. Aikawa, E. Rabkin, M. Lindsey, L.E. Rohde, F.J. Schoen, R.A. Kelly, Z. Werb, P. Libby, et al. 2000. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 106:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hazen, S.L., J.R. Crowley, D.M. Mueller, and J.W. Heinecke. 1997. Mass spectrometric quantification of 3-chlorotyrosine in human tissues with attomole sensitivity: a sensitive and specific marker for myeloperoxidase-catalyzed chlorination at sites of inflammation. Free Radic. Biol. Med. 23:909–916. [DOI] [PubMed] [Google Scholar]
- 6.Eiserich, J.P., M. Hristova, C.E. Cross, A.D. Jones, B.A. Freeman, B. Halliwell, and A. van der Vliet. 1998. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 391:393–397. [DOI] [PubMed] [Google Scholar]
- 7.Brennan, M.L., W. Wu, X. Fu, Z. Shen, W. Song, H. Frost, C. Vadseth, L. Narine, E. Lenkiewicz, M.T. Borchers, et al. 2002. A tale of two controversies: i) defining the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase deficient mice; and ii) defining the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 277:17415–17427. [DOI] [PubMed] [Google Scholar]
- 8.Baldus, S., J.P. Eiserich, A. Mani, L. Castro, M. Figueroa, P. Chumley, W. Ma, A. Tousson, C.R. White, D.C. Bullard, et al. 2001. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J. Clin. Invest. 108:1759–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brennan, M.L., M.M. Anderson, D.M. Shih, X.D. Qu, X. Wang, A.C. Mehta, L.L. Lim, W. Shi, S.L. Hazen, J.S. Jacob, et al. 2001. Increased atherosclerosis in myeloperoxidase-deficient mice. J. Clin. Invest. 107:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmeliet, P., L. Kieckens, L. Schoonjans, B. Ream, A. van Nuffelen, G. Prendergast, M. Cole, R. Bronson, D. Collen, and R.C. Mulligan. 1993. Plasminogen activator inhibitor-1 gene-deficient mice. I. Generation by homologous recombination and characterization. J. Clin. Invest. 92:2746–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carmeliet, P., L. Moons, V. Ploplis, E. Plow, and D. Collen. 1997. Impaired arterial neointima formation in mice with disruption of the plasminogen gene. J. Clin. Invest. 99:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lijnen, H.R., B. Van Hoef, F. Lupu, L. Moons, P. Carmeliet, and D. Collen. 1998. Function of the plasminogen/plasmin and matrix metalloproteinase systems after vascular injury in mice with targeted inactivation of fibrinolytic system genes. Arterioscler. Thromb. Vasc. Biol. 18:1035–1045. [DOI] [PubMed] [Google Scholar]
- 13.Lowery, O.H., and N.J. Rosenbrough. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275. [PubMed] [Google Scholar]
- 14.Junqueira, L.C., G. Bignolas, and R.R. Brentani. 1979. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 11:447–455. [DOI] [PubMed] [Google Scholar]
- 15.Romanic, A.M., R.F. White, A.J. Arleth, E.H. Ohlstein, and F.C. Barone. 1998. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 29:1020–1030. [DOI] [PubMed] [Google Scholar]
- 16.Mulligan-Kehoe, M.J., R. Wagner, C. Wieland, and R. Powell. 2001. A truncated plasminogen activator inhibitor-1 protein induces and inhibits angiostatin (kringles 1-3), a plasminogen cleavage product. J. Biol. Chem. 276:8588–8596. [DOI] [PubMed] [Google Scholar]
- 17.Pinsky, D.J., H. Liao, C.A. Lawson, S.F. Yan, J. Chen, P. Carmeliet, D.J. Loskutoff, and D.M. Stern. 1998. Coordinated induction of plasminogen activator inhibitor-1 (PAI-1) and inhibition of plasminogen activator gene expression by hypoxia promotes pulmonary vascular fibrin deposition. J. Clin. Invest. 102:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoop, A.A., F. Lupu, and H. Pannekoek. 2000. Colocalization of thrombin, PAI-1, and vitronectin in the atherosclerotic vessel wall: a potential regulatory mechanism of thrombin activity by PAI-1/vitronectin complexes. Arterioscler. Thromb. Vasc. Biol. 20:1143–1149. [DOI] [PubMed] [Google Scholar]
- 19.Reidy, M.A., C. Irvin, and V. Lindner. 1996. Migration of arterial wall cells. Expression of plasminogen activators and inhibitors in injured rat arteries. Circ. Res. 78:405–414. [DOI] [PubMed] [Google Scholar]
- 20.Todor, D.R., I. Lewis, G. Bruno, and D. Chyatte. 1998. Identification of a serum gelatinase associated with the occurrence of cerebral aneurysms as pro-matrix metalloproteinase-2. Stroke. 29:1580–1583. [DOI] [PubMed] [Google Scholar]
- 21.Zhao, W., D.R. Spitz, L.W. Oberley, and M.E. Robbins. 2001. Redox modulation of the pro-fibrogenic mediator plasminogen activator inhibitor-1 following ionizing radiation. Cancer Res. 61:5537–5543. [PubMed] [Google Scholar]
- 22.Strandberg, L., D.A. Lawrence, L.B. Johansson, and T. Ny. 1991. The oxidative inactivation of plasminogen activator inhibitor type 1 results from a conformational change in the molecule and does not require the involvement of the P1′ methionine. J. Biol. Chem. 266:13852–13858. [PubMed] [Google Scholar]
- 23.Lawrence, D.A., and D.J. Loskutoff. 1986. Inactivation of plasminogen activator inhibitor by oxidants. Biochemistry. 25:6351–6355. [DOI] [PubMed] [Google Scholar]
- 24.Creemers, E., J. Cleutjens, J. Smits, S. Heymans, L. Moons, D. Collen, M. Daemen, and P. Carmeliet. 2000. Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am. J. Pathol. 156:1865–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fay, W.P., A.C. Parker, L.R. Condrey, and A.D. Shapiro. 1997. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. 90:204–208. [PubMed] [Google Scholar]
- 26.Baker, M.S., S.P. Green, N. Goss, M. Katrantzis, and W.F. Doe. 1990. Plasminogen activator inhibitor 2 (PAI-2) is not inactivated by exposure to oxidants which can be released from activated neutrophils. Biochem. Biophys. Res. Commun. 166:993–1000. [DOI] [PubMed] [Google Scholar]
- 27.Kalra, D.K., X. Zhu, M.K. Ramchandani, G. Lawrie, M.J. Reardon, D. Lee-Jackson, W.L. Winters, N. Sivasubramanian, D.L. Mann, and W.A. Zoghbi. 2002. Increased myocardial gene expression of tumor necrosis factor-alpha and nitric oxide synthase-2: a potential mechanism for depressed myocardial function in hibernating myocardium in humans. Circulation. 105:1537–1540. [DOI] [PubMed] [Google Scholar]
- 28.Sivasubramanian, N., M.L. Coker, K.M. Kurrelmeyer, W.R. MacLellan, F.J. DeMayo, F.G. Spinale, and D.L. Mann. 2001. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 104:826–831. [DOI] [PubMed] [Google Scholar]
- 29.Siwik, D.A., P.J. Pagano, and W.S. Colucci. 2001. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 280:C53–C60. [DOI] [PubMed] [Google Scholar]
- 30.Harris, J.E., and J. Schultz. 1976. Studies on the chlorinating activity of myeloperoxidase. J. Biol. Chem. 251:1371–1374. [PubMed] [Google Scholar]
- 31.Nishimura, H., H. Tsuji, H. Masuda, K. Nakagawa, Y. Nakahara, H. Kitamura, T. Kasahara, T. Sugano, M. Yoshizumi, S. Sawada, et al. 1997. Angiotensin II increases plasminogen activator inhibitor-1 and tissue factor mRNA expression without changing that of tissue type plasminogen activator or tissue factor pathway inhibitor in cultured rat aortic endothelial cells. Thromb. Haemost. 77:1189–1195. [PubMed] [Google Scholar]
- 32.Mehta, J.L., D.Y. Li, H. Yang, and M.K. Raizada. 2002. Angiotensin II and IV stimulate expression and release of plasminogen activator inhibitor-1 in cultured human coronary artery endothelial cells. J. Cardiovasc. Pharmacol. 39:789–794. [DOI] [PubMed] [Google Scholar]
- 33.Pahor, M., L.V. Franse, S.R. Deitcher, W.C. Cushman, K.C. Johnson, R.I. Shorr, K. Kottke-Marchant, R.P. Tracy, G.W. Somes, and W.B. Applegate. 2002. Fosinopril versus amlodipine comparative treatments study: a randomized trial to assess effects on plasminogen activator inhibitor-1. Circulation. 105:457–461. [DOI] [PubMed] [Google Scholar]
- 34.Raggi, P., N.R. Dickson, M. Boyne, R. Pereira, B. Cooil, N. Wattanasuwan, and D.C. Russell. 1998. Influence of prior ACE inhibitor therapy on morbidity and mortality following acute myocardial infarction. Ann. Pharmacother. 32:1141–1146. [DOI] [PubMed] [Google Scholar]
- 35.Lapointe, N., C. Blais, Jr., A. Adam, T. Parker, M.G. Sirois, H. Gosselin, R. Clement, and J.L. Rouleau. 2002. Comparison of the effects of an angiotensin-converting enzyme inhibitor and a vasopeptidase inhibitor after myocardial infarction in the rat. J. Am. Coll. Cardiol. 39:1692–1698. [DOI] [PubMed] [Google Scholar]
- 36.Gasic, S., O.F. Wagner, P. Fasching, C. Ludwig, M. Veitl, S. Kapiotis, and B. Jilma. 1999. Fosinopril decreases levels of soluble vascular cell adhesion molecule-1 in borderline hypertensive type II diabetic patients with microalbuminuria. Am. J. Hypertens. 12:217–222. [DOI] [PubMed] [Google Scholar]
- 37.Nikpoor, B., G. Turecki, C. Fournier, P. Theroux, and G.A. Rouleau. 2001. A functional myeloperoxidase polymorphic variant is associated with coronary artery disease in French-Canadians. Am. Heart J. 142:336–339. [DOI] [PubMed] [Google Scholar]
- 38.Gardemann, A., J. Lohre, N. Katz, H. Tillmanns, F.W. Hehrlein, and W. Haberbosch. 1999. The 4G4G genotype of the plasminogen activator inhibitor 4G/5G gene polymorphism is associated with coronary atherosclerosis in patients at high risk for this disease. Thromb. Haemost. 82:1121–1126. [PubMed] [Google Scholar]
- 39.Eitzman, D.T., R.J. Westrick, Z. Xu, J. Tyson, and D. Ginsburg. 2000. Plasminogen activator inhibitor-1 deficiency protects against atherosclerosis progression in the mouse carotid artery. Blood. 96:4212–4215. [PubMed] [Google Scholar]
- 40.Sjoland, H., D.T. Eitzman, D. Gordon, R. Westrick, E.G. Nabel, and D. Ginsburg. 2000. Atherosclerosis progression in LDL receptor-deficient and apolipoprotein E-deficient mice is independent of genetic alterations in plasminogen activator inhibitor-1. Arterioscler. Thromb. Vasc. Biol. 20:846–852. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, R., M.L. Brennan, X. Fu, R.J. Aviles, G.L. Pearce, M.S. Penn, E.J. Topol, D.L. Sprecher, and S.L. Hazen. 2001. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 286:2136–2142. [DOI] [PubMed] [Google Scholar]