

Abstract
Osteoclasts are multinucleated cells that resorb bones, and are derived from hematopoietic cells of the monocyte/macrophage lineage. The receptor activator of NF-κB ligand (RANKL, also called ODF/TRANCE/OPGL) stimulates both osteoclast differentiation from osteoclast progenitors and activation of mature osteoclasts. To identify genes responsible for osteoclast differentiation, we used a molecular indexing technique. Here, we report a clone of one of these genes whose transcription is induced by soluble RANKL (sRANKL) in both the RAW264.7 cells of the mouse macrophage cell line and the mouse primary bone marrow cells. The predicted protein was found to be a mouse homologue of Jun dimerization protein 2 (JDP2), a member of the AP-1 family of transcription factors, containing a basic region-leucine zipper motif. Transient transfection experiments revealed that overexpression of JDP2 leads to activation of both tartrate-resistant acid phosphatase (TRAP) and cathepsin K gene promoters in RAW264.7 cells. Infection of mouse primary bone marrow cells with retroviruses expressing JDP2-facilitated sRANKL-mediated formation of TRAP-positive multinuclear osteoclasts. Importantly, antisense oligonucleotide to JDP2 strongly suppressed sRANKL-induced osteoclast formation of RAW264.7 cells. Our findings suggest that JDP2 may play an important role in the RANK-mediated signal transduction system, especially in osteoclast differentiation.
Keywords: bone resorption, signal transduction, osteoclastogenesis, molecular indexing, mitogen-activated protein kinase
Introduction
Osteoclasts are multinucleated cells derived from hematopoietic progenitors of the monocyte/macrophage lineage. The receptor activator of NF-κB (RANK)* ligand (RANKL; also known as ODF/TRANCE/OPGL) is a newly discovered member of the TNF superfamily of cytokines that stimulate osteoclast differentiation from osteoclast progenitor cells in the presence of macrophage colony-stimulating factor (M-CSF; 1, 2). Bone marrow stroma/osteoblasts produce RANKL on their cell surface in response to several osteotropic factors, whereas osteoclast progenitors express a receptor for RANKL, RANK, which is a member of the TNF receptor superfamily (1, 3). Recently, it was reported that mice lacking RANKL or RANK also lack osteoclasts. These mice have profound defects in bone resorption and exhibit an osteopetrotic phenotype (4, 5). Transgenic mice that overexpress a soluble RANK–Fc fusion protein have severe osteopetrosis because of a reduction in osteoclasts, which is similar to OPG/osteoclastogenesis-inhibitory factor transgenic mice (6). Therefore, the RANKL/RANK system is found to be physiologically crucial for osteoclast formation. It is likely that dysfunction or modulation of the RANKL/RANK system is associated with diseases such as postmenopausal osteoporosis (7–9), destruction of bone in rheumatoid arthritis (10), and bone metastasis of cancer cells (11).
RANK, like other TNF receptor superfamily proteins, is known to activate a cascade of intracellular signaling events, including interactions with TNF receptor-associated factors and activation of both c-Jun NH2-terminal kinase (JNK) and NF-κB pathway (12, 13). So far, only a limited number of intracellular signaling molecules, such as NF-κB (12), c-Fos (14), and Fra-1 (15) have been shown to be involved in osteoclast differentiation. Identification of other factors that participate in osteoclast differentiation and their functions are needed to elucidate the detailed molecular signaling of RANK, which induces osteoclast differentiation.
Here, we describe the isolation and characterization of a member of the AP-1 family of transcription factors, Jun dimerization protein 2 (JDP2). We present evidence, based on expression profiles and signaling capability to activate osteoclast differentiation of mouse RAW264.7 and primary bone marrow cells, that JDP2 may be a key regulator of osteoclast differentiation.
Materials and Methods
Preparation of a Soluble RANKL (sRANKL).
Recombinant sRANKL was expressed in Escherichia coli strain TOP10 (Invitrogen) as a fusion protein of an extracellular domain of mouse RANKL (Gln137 to Asp316) and the COOH-terminal end of glutathione S-transferase, and purified by affinity chromatography through a glutathione-Sepharose 4B gel, anion exchange chromatography, and gel filtration chromatography.
Cell Culture.
RAW264.7 and J774A.1 cells were purchased from Dainippon Pharmaceutical. The cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in DMEM supplemented with 10% heat-inactivated (56°C for 30 min) FCS, 100 µg/ml streptomycin, and 100 U/ml penicillin (all from Life Technologies). Mouse primary bone marrow cells were obtained from femurs and tibias of 8-wk-old ddY mice.
Molecular Indexing (16).
Two cDNA pools were synthesized from total RNA (30 µg) isolated from untreated and sRANKL-treated (100 ng/ml, 48 h) RAW264.7 cells, with SuperScript II RNaseH− reverse transcriptase (Life Technologies). The primer for the first strand cDNA synthesis was a mixture of the following three primers: 5′-GGATCCTTTTTTTTTTTTTTTTA-3′ (DAPA1), 5′-CAGCTGTTTTTTTTTTTTTTTTC-3′ (DAPC1), and 5′-CTCGAGTTTTTTTTTTTTTTTTG-3′ (DAPG1). The two cDNA pools were digested with FokI (Takara Bio Inc.) and ligated with either one of the following adaptors: C1T adaptor, 5′-biotin-GTACATATTGTCGTTAGAACGCT-3′ (sense), 5′-NXYZAGCGTTCTAACGACAATATGTAC-3′ (antisense); and C1G adaptor, 5′-biotin-GTACATATTGTCGTTAGAACGCG-3′ (sense), 5′-NXYZCGCGTTCTAACGACAATATGTAC-3′ (antisense). The ligated products were recovered by Dynabeads M-280 streptavidin (Dynal) and reconstituted to 50 µl with 5 U of Stoffel fragment (Applied Biosystems). Using TAMRA-labeled C1S (5′-GTACATATTGTGTTAGAACGC-3′; Applied Biosystems) and one of the anchored oligo (dT) primers (DAPA1, DAPC1, or DAPG1), PCR was performed under the following conditions: predenaturation at 94°C for 2 min, 35 cycles of DNA amplification (at 94°C for 30 s, 55°C for 1 min, and 72°C for 1 min), and final elongation at 72°C for 10 min. Supernatant was loaded on a denaturing polyacrylamide gel, and the bands were analyzed by Molecular Imager Fx (Bio-Rad Laboratories). Bands, which appeared to represent differentially expressed genes on the basis of their relative intensities on the gels, were purified for cloning into a pCR2.1 (Invitrogen) vector and sequencing.
cDNA Cloning and Expression Vectors.
RAW264.7 cDNA library was constructed in pCR3.1 (Invitrogen) with 5 µg poly(A)+ RNA from RAW264.7 cells that were treated with 100 ng/ml sRANKL for 48 h using the ZAP Express cDNA synthesis kit (Stratagene). Based on the sequences of the two mouse EST clones (AI847606 and AI450325), an oligonucleotide (5′-TCCACCCACCAGTTAAGGCCATT-3′ [sense]) was synthesized, biotinylated, and used for the enrichment of corresponding cDNA clones from the library by using the GeneTrapper™ cDNA positive selection system (Life Technologies) according to the manufacturer's instructions. Similarly, a human homologue of mouse JDP2 cDNA was isolated from the human spleen cDNA library (Life Technologies), using an oligonucleotide (5′-CAGCTCGGCCCTGACTGTGGAG-3′ [sense]) based on a reported human genomic sequence (GenBank/EMBL/DDBJ accession no. AF111167). cDNA fragments of JDP2 and RANK were obtained by PCR amplification and cloned into plasmid pCR3.1. Each construct was confirmed by DNA sequencing and Western blot analysis.
Northern Blot Analysis.
Total RNA of 15 µg was loaded and separated on 1% agarose gel containing 6% formaldehyde and transferred onto Hybond-N+ (Amersham Biosciences) nylon membranes. The 75-bp fragment of the 3′-terminal in the mouse JDP2 cDNA or PCR fragments of mouse tartrate-resistant acid phosphatase (TRAP), cathepsin K, and β-actin cDNAs were 32P-labeled using a Random Primer DNA Labeling kit (Takara Bio Inc.), and the membrane was hybridized with these probes in ExpressHyb buffer (CLONTECH Laboratories, Inc.) according to the manufacturer's instructions. The expression levels of each mRNA were determined using a bioimage analyzer (Fujix BAS2000; Fuji Co., Ltd.).
Quantitative Real-time RT-PCR Analysis.
The mRNA expression levels of TRAP, cathepsin K, JDP2, and ribosomal 18S RNA as an endogenous reference, were quantified by a real-time PCR method (TaqMan Chemistry) on a sequence detection system (ABI Prism 7900HT; Applied Biosystems). The quantitative real-time RT-PCR was performed in a total reaction volume of 50 µl containing 1× Master Mix without UNG, 1× MultiScribe™, RNase inhibitor mix (Applied Biosystems), 200 nM forward and reverse primers, 140–200 nM TaqMan Probe, and 100 ng total RNA. Specific primers and probes used for amplification were as follows: TRAP forward, 5′-CGACCATTGTTAGCCACATACG-3′; TRAP reverse, 5′-CACATAGCCCACACCGTTCTC-3′; TRAP probe, 5′-CATGACCACAACCTGCAGTATCTTCAGGAC-3′; cathepsin K forward, 5′-GGGAACGAGAAAGCCCTGA-3′; cathepsin K reverse, 5′-GTAAAACTGGAAAGATGCCAAGC-3′; cathepsin K probe, 5′-CCATCTCTGTGTCCATCGACGCAAG-3′; JDP2 forward, 5′-CGCTGACATCCGCAACATT-3′; JDP2 reverse, 5′-GGCCTCTTGCCCAGTTTCA-3′; and JDP2 probe, 5′-TGCGCCCTTGCACTTCCTGGAG-3′. For detection of ribosomal 18S RNA, the primers and probe were purchased from Applied Biosystems. Samples were heated at 48°C for 30 min, 95°C for 10 min, and amplified for 40 cycles of 15 s at 95°C and 1 min at 60°C.
Antibodies and Western Blot Analysis.
A polyclonal antibody against mouse JDP2 was generated in rabbits using a synthetic peptide with a sequence, CVKLGKRPQPVKSELD (Cys plus amino acids 55–69 of mouse JDP2), identical to that of the corresponding human peptide. Anti-PU.1 and horseradish peroxidase–linked anti–rabbit IgG were obtained from Santa Cruz Biotechnology, Inc. and Amersham Biosciences, respectively. To prepare whole cell extracts, the cells were washed with PBS, lysed in RIPA buffer (1% NP-40, 0.5% deoxycholic acid, and 0.1% SDS in PBS) supplemented with complete protease inhibitor cocktail (Roche Diagnostics K. K.), and centrifuged. Nuclear extracts were prepared by essentially the same method as described by Schreiber et al. (17), except for the absence of dithiothreitol in buffers used for cell lysis and extraction of nuclear proteins. Equal amounts of protein (10–15 µg) were separated in 10–20% gradient SDS-PAGE and electrotransferred onto a polyvinylidene difluoride membrane (Bio-Rad Laboratories). Detection was performed using an ECL Plus Western blotting detection system (Amersham Biosciences) according to the manufacturer's instructions.
Preparation of Retrovirus Stocks and Infection of Mouse Primary Bone Marrow Cells.
A cDNA of mouse JDP2 was inserted into pMX-puro (18) and pMX–internal ribosome entry site (IRES)–enhanced green fluorescent protein (EGFP; 19) vectors to construct retrovirus vectors carrying a cDNA for JDP2. The resulting plasmid (2 µg) was transfected into packaging cell line Bosc 23 (2 × 106 cells) using FuGENE6 (Roche). After 48 h, the culture supernatants were collected and used as viral stocks. Bone marrow cells (106) obtained from both femora and tibiae of 6-wk-old ddY mice were cultured in α-MEM containing 10% FCS and 10 ng/ml M-CSF overnight in 24-well plates. The medium was replaced with the viral supernatant containing 4 µg/ml polybrene, and the cells were incubated for 4 h at 37°C. Infected cells were cultured for 2 d in the presence of M-CSF (10 ng/ml), and for additional 4 d in the presence of both M-CSF (10 ng/ml) and sRANKL (0, 10, or 30 ng/ml). Adherent cells were fixed with 10% formaldehyde, treated with ethanol–acetone (50:50), and stained for TRAP activity.
Luciferase Constructs, DNA Transfection, and Luciferase Assay.
A 1.9-kbp fragment containing the 5′-flanking sequence of mouse TRAP gene that spans nucleotides –1846 to +2 from the ATG codon (20), which was obtained by PCR amplification using RAW264.7 cell genomic DNA as a template. The 5′-flanking sequence of the mouse cathepsin K gene that spans nucleotides –1670 to +5 from the start of the first exon (21) was also amplified by PCR. These PCR products were subcloned into the pGL3-Basic vector (Promega) to place the TRAP or cathepsin K promoter region upstream of the firefly luciferase gene and generate pGL3-TRAP or pGL3-CTSK, respectively. The fidelity of the constructs was verified by nucleotide sequencing. When each construct was permanently transfected into RAW264.7 cells, the luciferase activities in both cell lines were significantly increased by exposure to sRANKL for 24–72 h, although weak or little enhancement in the activity was observed in cells stimulated by LPS or PMA, respectively (unpublished data).
RAW264.7 cells (3–3.5 × 106) were cotransfected by the DEAE-dextran (0.5 mg/ml) method with indicated expression plasmids (3 µg) and a firefly luciferase reporter plasmid (1 µg), together with a Renilla luciferase reporter plasmid pRL-SV40 (0.02 µg; Promega), for the normalization of transfection efficiency. The next day, cells were incubated with indicated stimuli for indicated periods, and cell extracts were prepared using Passive Lysis Buffer (Promega). Firefly and Renilla luciferase activities in the cell lysates were determined using a Dual-Luciferase® Assay System (Promega) according to the supplier's instruction.
Results and Discussion
Molecular Indexing and Cloning of JDP2.
Exposure to sRANKL causes the mouse macrophage RAW264.7 cells to differentiate into osteoclast-like TRAP-positive cells (6). In an attempt to isolate genes that are induced in RAW264.7 cells by sRANKL stimulation, and are responsible for osteoclast differentiation, we used a molecular indexing technique (16). With this technique, we obtained 10 cDNA clones whose expressions were induced in RAW264.7 cells by sRANKL stimulation for 48 h (Fig. 1 A and not depicted). One of them revealed 100% identity to two mouse EST clones (AI847606 and AI450325) by BLASTN analysis of the sequence, and is further characterized in this paper. The full-length cDNA of this gene, which was obtained from a cDNA library of RAW264.7 cells, has an open reading frame encoding 163 amino acids whose sequence is identical to that of the rat cDNA clone, JDP2 (GenBank/EMBL/DDBJ accession no. U53449, version GI:2833639), indicating that this gene is a mouse homologue of JDP2. Mouse JDP2 cDNA has also been cloned by yeast two-hybrid screening, using activating transcription factor-2 (ATF-2) as the bait (22). Structurally, JDP2 proteins have three parts: (a) an NH2-terminal part; (b) a central part that corresponds to a basic region-leucine zipper domain required for DNA binding and heterodimerization; and (c) a COOH-terminal part. Previous papers reported JDP2 as a member of the AP-1 group that can heterodimerize with c-Jun, other Jun proteins (23), and activating transcription factor-2 (ATF-2; reference 22).
Figure 1.

Expression of JDP2 and osteoclast-specific genes at mRNA and protein levels in RAW264.7 and mouse bone marrow cells. (A) Northern blot analysis of total RNA (15 µg/lane) prepared from RAW264.7 cells stimulated with 100 ng/ml sRANKL for indicated periods. The blots were probed with mouse jdp2 (top), TRAP (center), or cathepsin K (bottom) cDNA. The same membranes were rehybridized with β-actin probe. (B) Western blot analysis of RAW264.7 cells stimulated with 100 ng/ml sRANKL for indicated periods. Whole cell extracts were prepared and subjected (15 µg/lane) using specific antibodies for JDP2 (top) or PU.1 (bottom). (C) Total RNA was extracted from mouse bone marrow primary cultures treated with either 25 ng/ml M-CSF alone or in combination with 100 ng/ml sRANKL for indicated periods. Expression of mRNA for JDP2, TRAP, or cathepsin K was analyzed by real-time RT-PCR using ribosomal 18S RNA as an endogenous control.
Expression of JDP2 in RAW264.7 and Mouse Bone Marrow Cells.
To elucidate the role of JDP2 in sRANKL-induced osteoclast differentiation, we analyzed the time course of accumulation of mRNA for JDP2 and some osteoclast-specific genes such as TRAP, cathepsin K, and calcitonin receptor. Expression of mRNA for JDP2 was induced within 5 h after sRANKL stimulation, reached the peak level at 24 h, and declined thereafter (Fig. 1 A). In contrast, the mRNA levels of TRAP, cathepsin K, and calcitonin receptor (unpublished data) were increased at 24–48 h after the stimulation, and remained elevated until 96 h (Fig. 1 A). The results indicate that the up-regulation of JDP2 mRNA by sRANK proceeds induction of expression of osteoclast-specific marker genes during osteoclast differentiation. On the other hand, LPS only weakly stimulated JDP2 gene expression and failed to induce expression of TRAP and cathepsin K genes (unpublished data). Western blot analysis using polyclonal antibody raised against the peptide sequence of JDP2 also showed that the level of JDP2 protein was transiently increased by sRANKL (Fig. 1 B). Importantly, up-regulation of JDP2 mRNA was not detected in mouse macrophage cell line J774A.1 cells, which lack osteoclastogenic potential in spite of the fact that they have the same ability to activate the NF-κB pathway as did RAW264.7 (unpublished data). These results suggest an association between sRANKL-induced up-regulation of JDP2 and the osteoclast differentiation.
To test this further, we assessed JDP2 gene expression in mouse bone marrow cells during the differentiation into TRAP-positive mono- and multinucleated cells. Real-time quantitative RT-PCR analysis revealed that treatment with sRANKL in the presence of M-CSF induced an increase in the levels of mRNA for TRAP and cathepsin K (Fig. 1 C). In parallel with the induction of these osteoclast-specific genes, the expression of JDP2 mRNA was found to be up-regulated by the addition of sRANKL (Fig. 1 C).
Infection with JDP2-expressing Retrovirus Promotes Osteoclast Differentiation In Vitro.
To examine the functional role of JDP2 in regulating osteoclast differentiation, mouse bone marrow primary cells were infected with recombinant retrovirus carrying JDP2 cDNA in vitro. Under our experimental conditions, we could not detect any TRAP-positive both mononuclear and multinuclear osteoclasts in the absence of sRANKL, even after the virus infection (Fig. 2 A). However, in the presence of a low dose of sRANKL (10 ng/ml) that failed to induce differentiation to the TRAP-positive cells, infection with JDP2 retrovirus clearly enhanced the formation of TRAP-positive mono- and multinuclear cells (Fig. 2 A). This enhancement was also observed in the presence of 30 ng/ml sRANKL (Fig. 2 A). Quantitative analysis showed that the number of the TRAP-positive multinuclear osteoclasts was synergistically increased by infection with JDP2 retrovirus in combination with 10–30 ng/ml sRANKL (Fig. 2 B), suggesting enhancement of osteoclast differentiation by JDP2. To detect the cells infected with the retrovirus encoding JDP2, we used a bicistronic expression system using the IRES and the EGFP as a selection marker gene, both of which were placed downstream of JDP2 cDNA. We found that cells expressing EGFP were preferentially differentiated into TRAP-positive multinuclear cells (Fig. 2 C). These results suggest that JDP2 may act as an intermediary to transduce sRANKL-induced osteoclast formation of mouse bone marrow primary cells.
Figure 2.
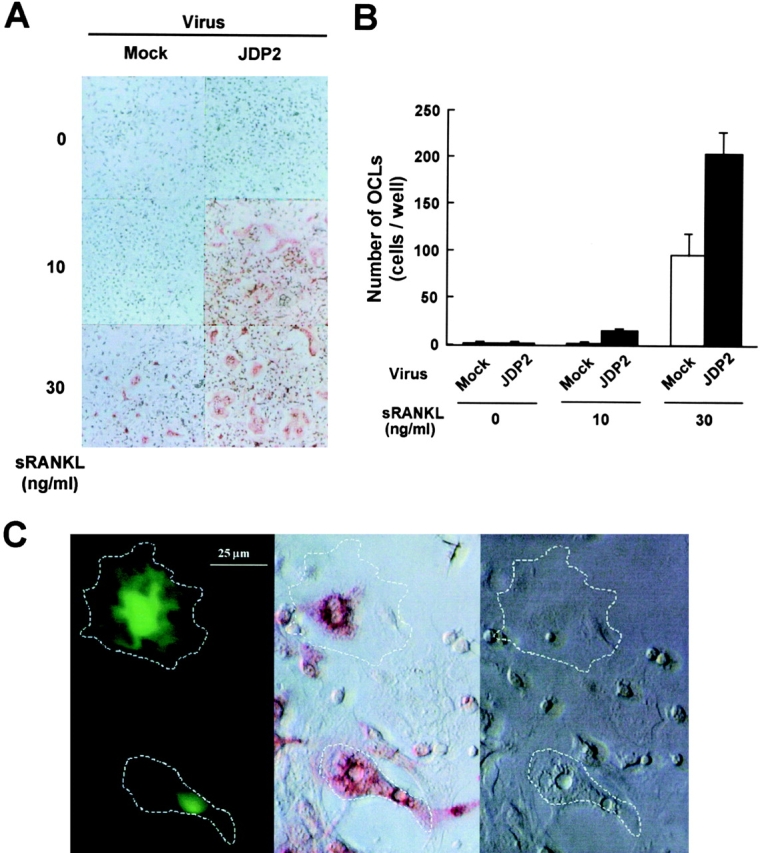
Infection with JDP2-expressing retrovirus promotes osteoclast generation of mouse bone marrow cells in vitro. Bone marrow cells prepared from 6-wk-old ddY mice were infected with retrovirus vector carrying no insert or JDP2 cDNA. The infected cells were treated with the indicated concentration of sRANKL and stained for TRAP activity (A). TRAP-positive multinucleated cells containing more than three nuclei were counted as osteoclasts (B). (C) Bone marrow cells were infected with IRES–EGFP retrovirus vector carrying JDP2 cDNA and stimulated with 10 ng/ml sRANKL. The infected cells were detected by EGFP fluorescence (left) and stained for TRAP activity (center). The corresponding phase-contrast image is also shown (right). The boundaries of EGFP-positive multinucleated cells are marked by dotted lines.
Suppression of Osteoclast Differentiation by Antisense Oligonucleotide to JDP2.
To investigate the essential role of JDP2 in sRANKL-induced osteoclastogenesis, we attempted to interrupt this process by using antisense oligonucleotide targeted at JDP2. As shown in Fig. 3 A, the induction of JDP2 protein by sRANKL was largely suppressed in RAW264.7 cells exposed to JDP2 antisense oligonucleotide. Treatment with the JDP2 antisense oligonucleotide resulted in a strong inhibition of sRANKL-induced formation of TRAP-positive mono- and multinuclear cells (Fig. 3 B). Quantitative analysis showed that sRANKL-induced increase in the number of the TRAP-positive multinuclear osteoclasts was significantly suppressed by the JDP2 antisense oligonucleotide in a dose-dependent manner, but not suppressed by the sense oligonucleotide (Fig. 3 C). These results indicate that JDP2 acts as an essential factor to induce osteoclast differentiation by sRANKL.
Figure 3.

Suppression of osteoclast formation by antisense oligonucleotide to JDP2 in RAW264.7 cells. (A) RAW264.7 cells (106 cells/well) cultured in DMEM containing 10% FCS in 60-mm-diameter dishes were treated with 10 µg/ml S-oligonucleotides (antisense, 5′-ATCATAGCAGGAGGAG-3′; sense, 5′-CTCCTCCTGCTATGAT-3′) in the presence of 100 ng/ml sRANKL for 24 h. Nuclear extracts were prepared and subjected to Western blot analysis using antibody for JDP2. (B) RAW264.7 cells (104 cells/well) cultured in DMEM containing 5% FCS in 96-well plates were treated with the sense or antisense S-oligonucleotide at 100 µg/ml in the presence of 30 ng/ml sRANKL for 4 d. The cells were fixed and stained for TRAP activity. (C) RAW264.7 cells were treated with indicated concentrations of the sense or antisense S-oligonucleotide in the presence of sRANKL (30 ng/ml) for 4 d, fixed, and stained for TRAP activity. TRAP-positive multinucleated cells containing more than three nuclei were counted as osteoclasts.
JDP2 Activates the Osteoclastogenic Pathway in RAW264.7 Cells.
To examine the functional role of JDP2 in facilitating osteoclast differentiation at the molecular level, we developed reporter assay systems to detect osteoclastogenic activity of the protein. RAW264.7 cells were transiently cotransfected with the expression vector of mouse JDP2 along with the TRAP or cathepsin K promoter–luciferase construct and the luciferase activity was measured. As shown in Fig. 4 A, mouse JDP2 enhanced TRAP or cathepsin K promoter activity in these cells to a level comparable in magnitude to that observed with mouse RANK, which is a receptor of RANKL. These elevated activities were further enhanced by treatment with sRANKL, indicating that JDP2 facilitated sRANKL-induced activation of these gene expressions. These increases in luciferase activity by JDP2 were dependent on the dose of JDP2 plasmid used (Fig. 4 B). Similarly, human JDP2 had the ability to activate both TRAP and cathepsin K promoters in RAW264.7 cells (unpublished data). Removal of the NH2-terminal region including the basic region and two leucine zippers of JDP2 protein completely abolished the activity to stimulate TRAP and cathepsin K promoters (unpublished data), suggesting that DNA binding and/or homo- or heterodimerization are necessary for the activity of JDP2. These data, along with our findings of suppression of osteoclast formation by the disruption of expression of JDP2 by antisense oligonucleotide (Fig. 3), suggest that JDP2 is a crucial factor to activate the osteoclastogenic pathway leading to enhancement of TRAP and cathepsin K gene expressions, especially in combination with sRANKL, in RAW264.7 cells.
Figure 4.
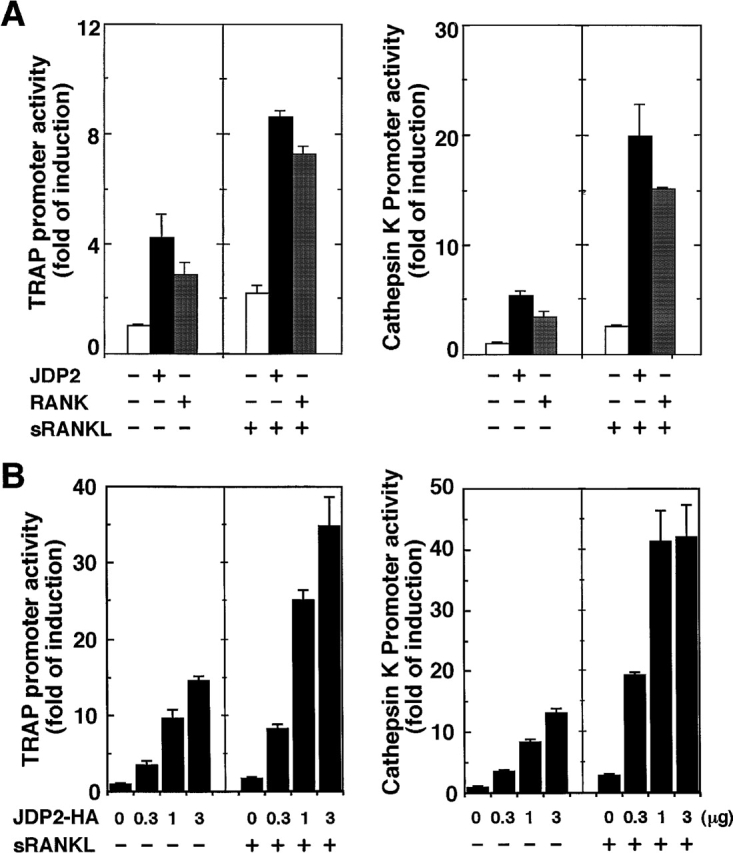
Overexpression of JDP2 stimulates TRAP and cathepsin K gene promoters in RAW264.7 cells. (A) RAW264.7 cells were cotransfected with indicated expression vector for JDP2 or RANK, together with the firefly luciferase reporter construct (pGL3-TRAP or pGL3-CTSK) and pRL-SV40. Cells were either untreated or treated for 24 h with 100 ng/ml sRANKL before being collected. Relative luciferase activity was determined, and the level of induction is indicated as a ratio with respect to cells transfected with the empty vector in the absence of sRANKL. Values shown are means ± SD of a representative of at least two independent experiments performed in triplicate. (B) RAW264.7 cells were cotransfected with the indicated dose of expression vectors for JDP2 with a COOH-terminal HA epitope (JDP2-HA), together with the firefly luciferase reporter construct (pGL3-TRAP or pGL3-CTSK) and pRL-SV40. The total amount of DNA in each transfection was kept constant by adding empty vector (pCR3.1) DNA. Cells were either untreated or treated for 24 h with 100 ng/ml sRANKL before being collected. Relative luciferase activity was determined as described in A. Values shown are means ± SD of a representative experiment performed in triplicate.
Requirement of JNK- and p38 Mitogen-activated Protein Kinase (MAPK)–mediated Signals for Inducing JDP2 Expression and Osteoclast Differentiation in RAW264.7 Cells.
RANKL has been demonstrated to activate JNK, which phosphorylates the AP-1 components to potentiate its transcriptional activity, through RANK in osteoclastic cells (6, 13, 24, 25), whereas p38 MAPK-mediated signals have been shown to regulate osteoclast differentiation (26). To determine whether the JNK and p38 MAPK pathways could couple RANK to JDP2 expression, we analyzed JDP2 mRNA levels of RAW264.7 cells using specific inhibitors for JNK and p38 MAPK, SP600125 (27), and SB203580 (28), respectively. As shown in Fig. 5 , both inhibitors suppressed sRANKL-induced expression of JDP2 mRNA, as well as formation of TRAP-positive mono- and multinuclear cells. These results indicate that the JNK and p38 MAPK pathways play an important role for regulating JDP2 expression induced by sRANKL. These results further support the notion that JDP2 is a crucial factor for RANKL-mediated osteoclast formation. Although recent studies showed the ability of JNK and p38 MAPK to phosphorylate JDP2 at Thr-148 (29, 30), JDP2 was found in low abundance in resting cells but its levels were highly induced upon sRANKL stimulation (Fig. 1). Thus, JDP2 activity appears to be mainly regulated at the transcriptional level through JNK and p38 MAPK pathways.
Figure 5.
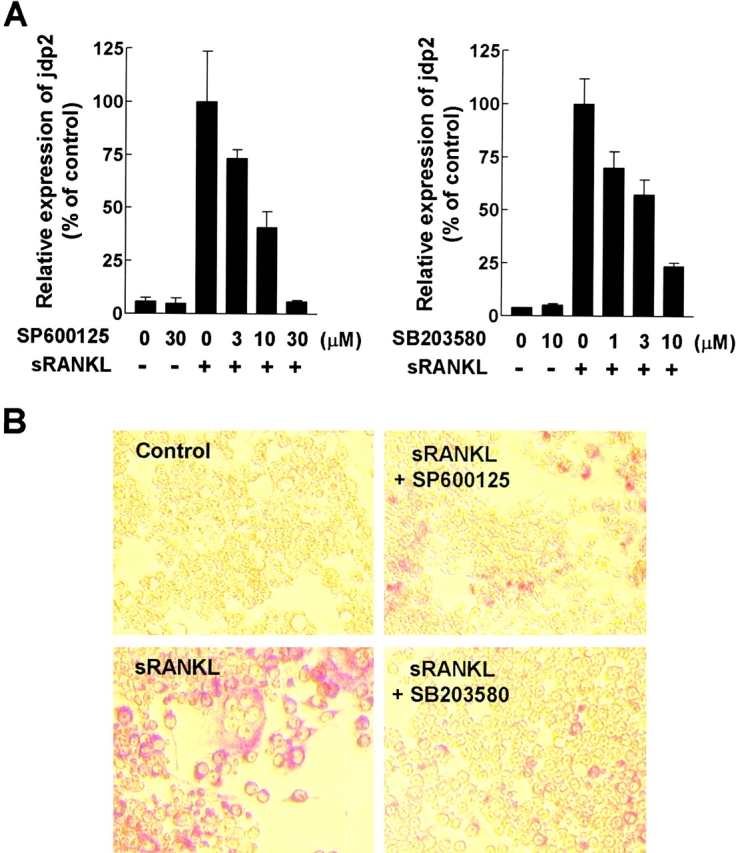
Suppression of sRANKL-induced JDP2 expression and osteoclast formation by SP600125 and SB203580 in RAW264.7 cells. (A) RAW264.7 cells (5 × 105 cells/well) cultured in DMEM containing 10% FCS in 6-well plates were pretreated with indicated concentrations of either SP600125 or SB203580 for 30 min and stimulated with 100 ng/ml sRANKL for 24 h. Expression of mRNA for jdp2 was analyzed by real-time RT-PCR using ribosomal 18S RNA as an endogenous control. Similar results were obtained from two independent experiments. (B) RAW264.7 cells (5 × 103 cells/well) cultured in DMEM containing 10% FCS in 96-well plates were pretreated with 10 µM of either SP600125 or SB203580 for 30 min and stimulated with 100 ng/ml sRANKL for 3 d. The cells were fixed and stained for TRAP activity.
In conclusion, we identified JDP2 as an important signaling molecule for RANK-induced osteoclast differentiation. Further studies on JDP2, including identification of its partner proteins and generation of JDP2-deficient mice, will elucidate the precise role of JDP2 in vivo. Such information on JDP2 will give us not only new insights into the molecular mechanism regulating bone resorption, especially osteoclastogenesis, but also give new opportunities for developing therapeutics to treat diseases of the bone.
Acknowledgments
We thank Dr. Toshio Kitamura (The University of Tokyo) for the pMX-IRES–EGFP vector, Kenji Wakabayashi for the preparation of recombinant sRANKL, and Yohei Ozeki for excellent assistance in homology search of various DNA sequence databases. We also thank Chiaki Tanimoto and Kyoko Kobayashi-Miura for their technical assistance in DNA sequencing.
This work was supported by Grants-in-Aid from the Ministry of Education, Science, Sports, and Culture of Japan, and by the Health Science Research grants from the Ministry of Health and Welfare of Japan given to S. Tanaka.
R. Kawaida and T. Ohtsuka contributed equally to this work.
Footnotes
Abbreviations used in this paper: EGFP, enhanced green fluorescent protein; JDP2, Jun dimerization protein 2; JNK, c-Jun NH2-terminal kinase; IRES, internal ribosome entry site; MAPK, mitogen-activated protein kinase; M-CSF, macrophage colony-stimulating factor; RANK, receptor activator of NF-κB; RANKL, RANK ligand; sRANKL, soluble RANKL; TRAP, tartrate-resistant acid phosphatase.
References
- 1.Yasuda, H., N. Shima, N. Nakagawa, K. Yamaguchi, M. Kinosaki, S. Mochizuki, A. Tomoyasu, K. Yano, M. Goto, A. Murakami, et al. 1998. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA. 95:3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lacey, D.L., E. Timms, H.L. Tan, M.J. Kelley, C.R. Dunstan, T. Burgess, R. Elliott, A. Colombero, G. Elliott, S. Scully, et al. 1998. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 93:165–176. [DOI] [PubMed] [Google Scholar]
- 3.Nakagawa, N., M. Kinosaki, K. Yamaguchi, N. Shima, H. Yasuda, K. Yano, T. Morinaga, and K. Higashio. 1998. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 253:395–400. [DOI] [PubMed] [Google Scholar]
- 4.Kong, Y.Y., H. Yoshida, I. Sarosi, H.L. Tan, E. Timms, C. Capparelli, S. Morony, A.J. Oliveira-dos-Santos, G. Van, A. Itie, et al. 1999. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 397:315–323. [DOI] [PubMed] [Google Scholar]
- 5.Li, J., I. Sarosi, X.Q. Yan, S. Morony, C. Capparelli, H.L. Tan, S. McCabe, R. Elliott, S. Scully, G. Van, et al. 2000. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA. 97:1566–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu, H., D.L. Lacey, C.R. Dunstan, I. Solovyev, A. Colombero, E. Timms, H.L. Tan, G. Elliott, M.J. Kelley, I. Sarosi, et al. 1999. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. USA. 96:3540–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yasuda, H., N. Shima, N. Nakagawa, S.I. Mochizuki, K. Yano, N. Fujise, Y. Sato, M. Goto, K. Yamaguchi, M. Kuriyama, et al. 1998. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 139:1329–1337. [DOI] [PubMed] [Google Scholar]
- 8.Khosla, S. 2001. Minireview: The OPG/RANKL/RANK system. Endocrinology. 142:5050–5055. [DOI] [PubMed] [Google Scholar]
- 9.Hofbauer, L.C., and A.E. Heufelder. 2001. Role of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in bone cell biology. J. Mol. Med. 79:243–253. [DOI] [PubMed] [Google Scholar]
- 10.Kotake, S., N. Udagawa, M. Hakoda, M. Mogi, K. Yano, E. Tsuda, K. Takahashi, T. Furuya, S. Ishiyama, K.J. Kim, et al. 2001. Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum. 44:1003–1012. [DOI] [PubMed] [Google Scholar]
- 11.Thomas, R.J., T.A. Guise, J.J. Yin, J. Elliott, N.J. Horwood, T.J. Martin, and M.T. Gillespie. 1999. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 140:4451–4458. [DOI] [PubMed] [Google Scholar]
- 12.Iotsova, V., J. Caamano, J. Loy, Y. Yang, A. Lewin, and R. Bravo. 1997. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat. Med. 3:1285–1289. [DOI] [PubMed] [Google Scholar]
- 13.Darnay, B.G., V. Haridas, J. Ni, P.A. Moore, and B.B. Aggarwal. 1998. Characterization of the intracellular domain of receptor activator of NF-kappaB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-kappaB and c-Jun N-terminal kinase. J. Biol. Chem. 273:20551–20555. [DOI] [PubMed] [Google Scholar]
- 14.Wang, Z.Q., C. Ovitt, A.E. Grigoriadis, U. Mohle-Steinlein, U. Ruther, and E.F. Wagner. 1992. Bone and haematopoietic defects in mice lacking c-fos. Nature. 360:741–745. [DOI] [PubMed] [Google Scholar]
- 15.Matsuo, K., J.M. Owens, M. Tonko, C. Elliott, T.J. Chambers, and E.F. Wagner. 2000. Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat. Genet. 24:184–187. [DOI] [PubMed] [Google Scholar]
- 16.Kato, K. 1995. Description of the entire mRNA population by a 3′ end cDNA fragment generated by class IIS restriction enzymes. Nucleic Acids Res. 23:3685–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schreiber, E., P. Matthias, M.M. Muller, and W. Schaffner. 1989. Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucleic Acids Res. 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitamura, T. 1998. New experimental approaches in retrovirus-mediated expression screening. Int. J. Hematol. 67:351–359. [DOI] [PubMed] [Google Scholar]
- 19.Chida, D., O. Miura, A. Yoshimura, and A. Miyajima. 1999. Role of cytokine signaling molecules in erythroid differentiation of mouse fetal liver hematopoietic cells: functional analysis of signaling molecules by retrovirus-mediated expression. Blood. 93:1567–1578. [PubMed] [Google Scholar]
- 20.Reddy, S.V., T. Scarcez, J.J. Windle, R.J. Leach, J.E. Hundley, J.M. Chirgwin, J.Y. Chou, and G.D. Roodman. 1993. Cloning and characterization of the 5′-flanking region of the mouse tartrate-resistant acid phosphatase gene. J. Bone Miner. Res. 8:1263–1270. [DOI] [PubMed] [Google Scholar]
- 21.Li, Y.P., and W. Chen. 1999. Characterization of mouse cathepsin K gene, the gene promoter, and the gene expression. J. Bone Miner. Res. 14:487–499. [DOI] [PubMed] [Google Scholar]
- 22.Jin, C., H. Ugai, J. Song, T. Murata, F. Nili, K. Sun, M. Horikoshi, and K.K. Yokoyama. 2001. Identification of mouse Jun dimerization protein 2 as a novel repressor of ATF-2. FEBS Lett. 489:34–41. [DOI] [PubMed] [Google Scholar]
- 23.Aronheim, A., E. Zandi, H. Hennemann, S.J. Elledge, and M. Karin. 1997. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell. Biol. 17:3094–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee, Z.H., K. Kwack, K.K. Kim, S.H. Lee, and H.H. Kim. 2000. Activation of c-Jun N-terminal kinase and activator protein 1 by receptor activator of nuclear factor kappaB. Mol. Pharmacol. 58:1536–1545. [PubMed] [Google Scholar]
- 25.David, J.P., K. Sabapathy, O. Hoffmann, M.H. Idarraga, and E.F. Wagner. 2002. JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J. Cell Sci. 115:4317–4325. [DOI] [PubMed] [Google Scholar]
- 26.Li, X., N. Udagawa, K. Itoh, K. Suda, Y. Murase, T. Nishihara, T. Suda, and N. Takahashi. 2002. p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology. 143:3105–3113. [DOI] [PubMed] [Google Scholar]
- 27.Bennett, B.L., D.T. Sasaki, B.W. Murray, E.C. O'Leary, S.T. Sakata, W. Xu, J.C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, et al. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA. 98:13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, J.C., S. Kumar, D.E. Griswold, D.C. Underwood, B.J. Votta, and J.L. Adams. 2000. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology. 47:185–201. [DOI] [PubMed] [Google Scholar]
- 29.Katz, S., R. Heinrich, and A. Aronheim. 2001. The AP-1 repressor, JDP2, is a bona fide substrate for the c-Jun N-terminal kinase. FEBS Lett. 506:196–200. [DOI] [PubMed] [Google Scholar]
- 30.Katz, S., and A. Aronheim. 2002. Differential targeting of the stress mitogen-activated protein kinases to the c-Jun dimerization protein 2. Biochem. J. 368:939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]