

Abstract
The molecular basis of CD28-dependent costimulation of T cells is poorly understood. Bcl-xγ is a member of the Bcl-x family whose expression is restricted to activated T cells and requires CD28-dependent ligation for full expression. We report that Bcl-xγ–deficient (Bcl-xγ−/−) T cells display defective proliferative and cytokine responses to CD28-dependent costimulatory signals, impaired memory responses to proteolipid protein peptide (PLP), and do not develop PLP-induced experimental autoimmune encephalomyelitis (EAE). In contrast, enforced expression of Bcl-xγ largely replaces the requirement for B7-dependent ligation of CD28. These findings identify the Bcl-xγ cytosolic protein as an essential downstream link in the CD28-dependent signaling pathway that underlies T cell costimulation.
Keywords: Bcl-xγ, CD28, T cell, costimulation, EAE
Introduction
The T cell response to antigen requires costimulatory signals that promote efficient T cell expansion and cytokine secretion. Transduction of costimulatory signals by the CD28 receptor is a pivotal element in the full stimulation of T cells and prevention of anergy in vitro (1). CD28-dependent signals are also required for effective in vivo T cell responses to allografts (2) and for development of experimental autoimmune encephalomyelitis (EAE;* reference 3). Recent studies have suggested that CD28-dependent effects on T cell survival and proliferation may represent biochemically distinct pathways that originate from different sites on the CD28 molecule's cytoplasmic domain. One pathway, marked by Bcl-xL expression, may contribute to T cell survival upon growth factor withdrawal in vitro (4) and may enhance survival of mature thymocytes (5, 6) and peripheral T cells (7, 8). A second pathway, proposed to account for the biologic signature of CD28-dependent costimulation-enhanced proliferation and cytokine production, has not been well characterized (9, 10).
Bcl-xγ, a recently identified isoform of the Bcl-x gene, is expressed after TCR and CD28 ligation and may contribute to successful proliferation and IL-2 secretion after stimulation by antigen (11). Importantly, the expression of Bcl-xγ is mainly confined to the T cell compartment (11), while Bcl-xL has a much wider tissue distribution (4). These findings opened the possibility that Bcl-xγ function may be more specifically tailored to T cell activation. In contrast, Bcl-xL, which exerts antiapoptotic activity in a wide variety of different cell types (4), can enhance the survival, but not proliferation of activated T cells (8). Therefore, it was of interest to determine whether Bcl-xγ function might overlap with Bcl-xL and potentiate cell survival or exert a distinctive and complementary role in T cell activation. To this end, we generated mice that either lack or constitutively express Bcl-xγ in T cells. We find that Bcl-xγ–deficient T cells fail to respond to CD28-dependent costimulatory signals according to cell proliferation and cytokine expression, while enforced expression of Bcl-xγ largely replaces the requirement for CD28-dependent costimulatory signals; manipulation of Bcl-xγ expression had no significant effect on T cell survival.
Materials and Methods
Generation of Bcl-xγ Tg Mice.
Bcl-xγ Tg mice containing full-length Bcl-xγ cDNA under control of the LckDis (p56lck) promoter (12) were generated and backcrossed to C57BL/6 for eight generations. Enforced expression of Bcl-xγ (in a T cell–specific manner) in these transgenic mice was confirmed by Western blot analysis as well as by RT-PCR, using oligos that distinguish transgenic and endogenous RNA transcripts. C57BL/6 Bcl-xγ Tg mice were also crossed with Balb/c DO11.10 TCR Tg mice to generate C57BL/6 × Balb/c Bcl-xγ Tg DO11.10 TCR Tg mice and Bcl-xγ wild-type (wt) DO11.10 TCR Tg control littermates.
Transfection and Screening of Embryonic Stem Cells and Generation of Bcl-xγ −/− Mice.
TC1 embryonic stem (ES) cells were electroporated with 15 μg of PvuI-linearized Bcl-xγ−/− vector DNA and selected in media containing G418 and Gancyclovir as described previously (14). Bcl-xγ+/− ES cells were identified by Southern blot analysis using the 5′ knockout probe on HindIII-digested DNA and confirmed by using the 3′ knockout probe on BglII-digested DNA. Bcl-xγ+/− ES cells were transiently transfected by electroporation with 30 μg/ml of pMC-CreN, and clones were isolated and screened by Southern blot analysis for Cre-loxP–mediated deletion of the neor gene. Targeted TC1 ES cells were used to generated chimeric mice. Bcl-xγ+/− ES cells were injected into C57BL/6 blastocysts, and chimeric mice capable of transmitting the Bcl-xγ+/− allele to offspring were obtained from two independent clones. Offspring from these mice were bred to 129sv to generate Bcl-xγ+/+, Bcl-xγ+/−, and Bcl-xγ−/− mice.
Flow Cytometry.
Single-cell suspensions were prepared from thymus, spleen, and LNs, where indicated. Erythrocytes were lysed with ammonium chloride lysis buffer, and cells were washed in 2% FBS/PBS. Cell surface molecules were detected by staining 106 viable cells with indicated conjugates for 20 min at 4°C in 2% FBS/PBS. Cells were stained with FITC or R-phycoerythrin or Cy-Chrome–conjugated antibodies. Data from ∼50,000 size-gated cells were acquired on a Coulter Epics XL flow cytometer (Beckman Coulter).
Southern Blot Hybridization.
Genomic DNA was isolated and Southern blotting was performed as described previously (13), using Zetaprobe membranes (Bio-Rad Laboratories) and probes generated by random hexamer priming (Boehringer Mannheim) using (γ-32P)dCTP (NEN Life Science Products). The 5′ knockout probe is a 1-kb NotI-EcoRI genomic fragment, and 3′ knockout probe is a 600-bp MscI-XbaI genomic DNA fragment.
RT-PCR and Touchdown PCR Analysis.
Total RNA was prepared from cultured T cells and mouse thymus using RNAzole (Tel-Test, Inc.). RT-PCR and PCR cloning were performed as described previously (4). In brief, total RNA from mouse thymus was reverse transcribed in a GeneAmp PCR 9600 (Perkin-Elmer) using a GeneAmp RNA-PCR kit (Perkin-Elmer) at 45°C for 60 min, followed by 95°C for 3 min, and then placed on ice. For Bcl-xγ detection, primers (e4f: 5′-GGA ATT CAG TGG AGG TAC ACC CCT CAG ATCT-3′ and e4b: 5′-CCA CTC CTG GCA CTT GAA CAA GCCT-3′) specific for the 5′ and 3′ ends of exon 4 were used (see Fig. 1 A). To detect Bcl-xL, primers to exon 2 (3e: 5′-TCG CTC GCC CAC ATC CCA GCT TCA CAT AAC CCC-3′) and exon 3 (15e: 5′-CCA CCA ACA AGA CAG GCT-3′) were used (see Fig. 1 A). Each three-step thermal cycle for routine PCR analysis consisted of 30 s at 95°C, 30 s at 60°C, and 60 s at 72°C. Additional PCR reactions were performed for 35–40 cycles using Advantage cDNA Polymerase Mix (CLONTECH). PCR products were visualized by agarose gel electrophoresis and ethidium bromide staining, and were positively identified by size markers. A negative control containing all reagents except cDNA was included in each PCR analysis.
Figure 1.

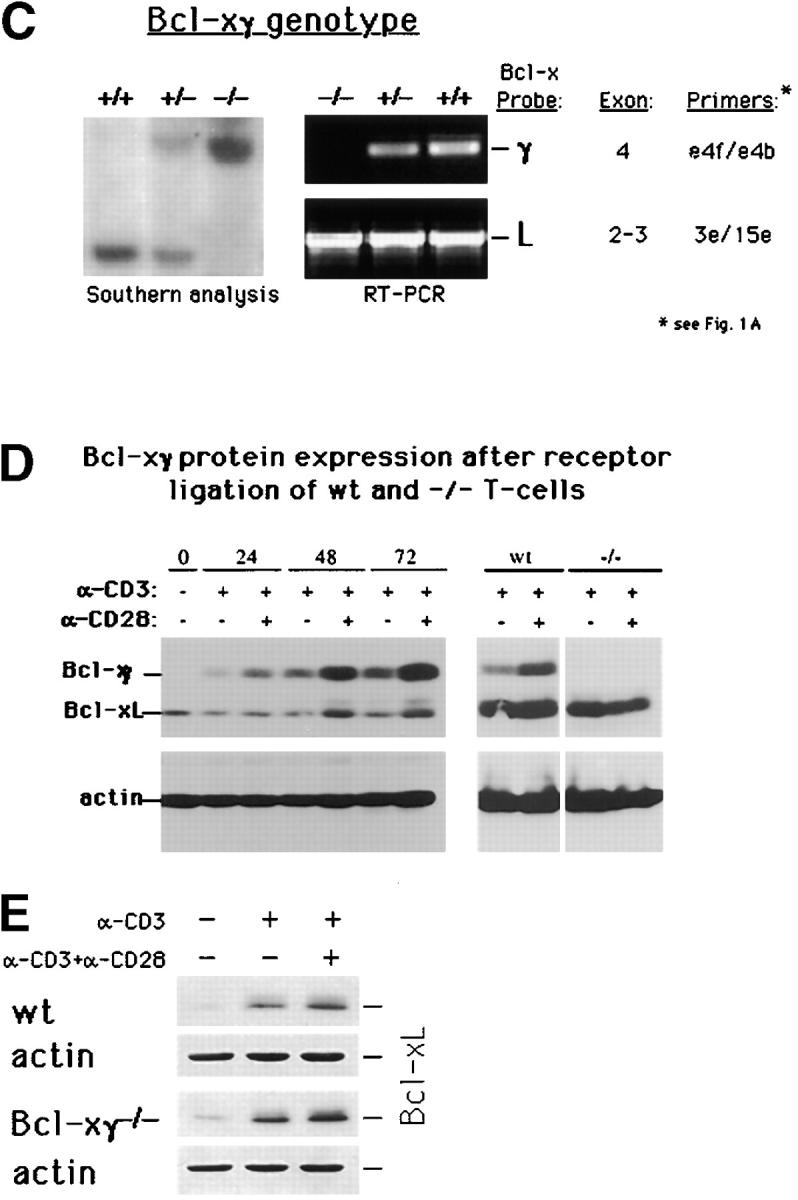
Generation of Bcl-xγ2/− mice. (A) Schematic representation of the coding and noncoding exons of Bcl-xγ and Bcl-xL. Exon 1 encodes the distal portion of the 5′ untranslated region (UTR), and exon 2 encodes the remainder of the 5′ UTR and the common NH2 terminus of Bcl-xL and Bcl-xγ proteins. Exon 3 encodes the COOH terminus of Bcl-xL, as well as the 3′ UTR of Bcl-xL. Exon 4 encodes the unique COOH terminus of Bcl-xγ protein and alternative splicing of exons 5–8 regulates expression of the Bcl-xγ 3′ UTR. (B) Strategy used to generate Bcl-xγ–deficient mice is shown based on targeted disruption of exon 4, the coding region for the unique COOH terminus of Bcl-xγ, as described in Materials and Methods. The genomic structure surrounding exon 4 and the structure of the targeting vector and mutant gene is shown. (C, left) Southern blot of murine Bcl-xγ (+/+, +/−, and −/−) (see Materials and Methods); (right) RT-PCR analysis of RNA (1 μg per sample) derived from Con A-activated LN T cells of Bcl-xγ wt, +/−, and −/− mice. PCR primers used to detect Bcl-xγ and Bcl-xL are indicated in Fig. 1 A and detailed in Materials and Methods. (D) Expression of Bcl-xγ and Bcl-xL protein after CD3/CD28 ligation. (left) Time-course analysis of Bcl-xγ and Bcl-xL protein in T cells from wt mice after CD3 and CD28 ligation. T cells isolated from wt and −/− mice were incubated for the indicated intervals in the presence of plate-bound anti-CD3 antibody (0.2 μg/ml) or anti-CD3 (0.2 μg/ml) and anti-CD28 antibody (1.0 μg/ml); 100 μg of after nuclear supernatant (PNS) loaded per lane. (right) Bcl-xγ and Bcl-xL protein levels in T cells derived from wt and −/− mice activated as described above for 48 h. The upper band (corresponding to Bcl-xγ) was confirmed by reprobing the same blots with an antibody specific to the unique COOH terminus of Bcl-xγ (see Materials and Methods). The immuno-blots shown are representative of one of three independent experiments and were overexposed to provide sensitive detection of trace amounts of Bcl-xγ, while increased levels of Bcl-xL in CD3 and CD3/CD28 cells are noted. Increasing the concentration of plate-bound anti-CD3 alone (up to 5 μg/ml) was not sufficient to induce significant levels of Bcl-xγ in the absence of CD28 ligation. The blots for actin served as a control for protein loading. (E) Expression of Bcl-xL in wt and Bcl-xγ2/− LN after anti-CD3 and anti-CD28 ligation. LN from wt and Bcl-xγ−/− mice were stimulated with plate-bound anti-CD3 or anti-CD3 plus anti-CD28 for 48 h before Bcl-xL expression was confirmed by Western blot analysis with anti–Bcl-x antibody. Expression of Bcl-xL in both strains is similar, indicating that loss of the Bcl-xγ gene does not affect expression of Bcl-xL. Actin was used as a loading control, after membranes were stripped and reprobed with anti-actin antibody.
Western Blot Analysis.
For immunoblot detection of Bcl-xγ and Bcl-xL protein, 100 μg of primary T cell PNS (per lane) were resolved on SDS-polyacrylamide gels containing 10% acrylamide and 0.27% N,N′-methylene bis-acrylamide under nonreducing conditions. To obtain better separation of Bcl-xγ and Bcl-xL proteins, a full-length gel apparatus of 20 × 20 cm was used. After electrophoresis, proteins were transferred to PVDF membranes (Millipore). Membranes were blocked, washed, and probed with an affinity-purified rabbit anti–Bcl-x antibody (R&D Systems) at a final concentration of 0.3 μg/ml. To confirm the identity of the (top) Bcl-xγ band, membranes were stripped and reprobed with an affinity-purified rabbit antibody (1.0 μg/ml) specific to the unique COOH terminus of Bcl-xγ. HRP-conjugated secondary antibodies and chemiluminescence detection reagents were from Amersham Pharmacia Biotech.
IL-2 Production and T Cell Proliferation.
CD4+ T cells were isolated from LNs of Bcl-xγ+/+, Bcl-xγ−/−, or Bcl-xγ Tg mice by negative section. In brief, LN cells were incubated with rat anti–mouse B220, Mac-1, Gr-1, CD8, and NK1.1 antibodies (BD PharMingen) for 30 min, washed, and incubated with magnetic beads coated with sheep anti–rat antibody (Dynal) for 45 min. Finally, antibody-coated cells were removed by magnetic separation and the isolated T lymphocytes were washed again. LN T cells (5 × 105 cells per well) were cultured in triplicates in round-bottomed 96-well plates (Costar) in 200 μl of freshly prepared Iscove's DMEM (10% FCS, 50 μM 2-mercaptoethanol). T cells were activated by culturing in the presence of plate-bound anti-CD3 (145.2C11 clone) and anti-CD28 antibodies (BD PharMingen) at final concentrations of 0.2 and 1.0 μg/ml, respectively. Alternatively, soluble anti-CD3 (1 μg/ml) and IL-2 (BD PharMingen) were used at the indicated concentrations. T cells were cultured for 48 h, and 1 μCi 3[H]thymidine was added to the wells for the last 18 h of culture. T cell culture supernatants were assayed for IL-2 in triplicates at indicated time points by ELISA. Where indicated, supernatants were harvested after 24, 48, and 72 h of culture. IL-2 levels were determined by sandwich ELISA, according to the manufacturer's instructions (BD PharMingen).
EAE Induction Using Proteolipid Protein Peptide.
EAE was induced in mice by immunization with a 12mer peptide representing amino acids 172–183 (PVYIYFNTWTTC) of proteolipid protein peptide (PLP)172–183. Per mouse, 150 μg of PLP peptide and 300 μg of killed Mycobacterium tuberculosis in CFA were injected (subcutaneously), by means of three injections over the flanks. In addition, 400 ng of pertussis toxin was injected, intraperitoneally on days 0 and 2. Disease was scored as follows: score 0, normal mouse with no sign of disease; score 1, limp tail; score 2, limp tail and partial hind limb weakness; score 3, partial hind limb paralysis; score 4, complete hind limb paralysis; and score 5, moribund state per death by EAE.
Apoptosis Assays.
24- or 96-well plates containing plate-bound anti-CD3 antibody (0.1–0.2 μg/ml, clone 145.2C11; BD PharMingen) were washed three times with PBS before addition of cells. Cells (either 5 × 106 or 5 × 105 cells per well) were washed twice with PBS at the indicated times, and resuspended in 1× Annexin V binding buffer. Cells in the early stages of apoptosis measured positive for fluorescein-Annexin V (Annexin V-FITC; BD PharMingen) as determined in an Epics XL FACScan™ using standard settings in semilogarithmic mode, and these cells contained higher forward scattering levels (for gating) than dead cells in the samples. Fully activated T cells from Bcl-xγ−/− mice exhibited a 10–20% increase in apoptosis versus control (wt) cells while the levels of apoptosis in nonactivated Bcl-xγ Tg+ T cells were only 10–30% lower compared with nonactivated control T cells. In certain experiments, cells were stained with FITC-conjugated anti-CD3 antibody or propidium iodide (PI) (BD PharMingen) before measurement of cell survival.
PI Fluorescence.
The percentage of cells in G0/G1, S, and G2/M phases was determined by cytofluorometric analysis after PI staining.
For PI staining, ∼106 cells were suspended in 100 μl PBS and 1 ml of ice-cold 80% ethanol was added drop-wise, while vortexing. Samples were then incubated for 1 h on ice. Cells were then washed gently twice with PBS, and resuspended in 250 μl of PBS containing 12.5 μg of RNase. Cells were incubated at 37°C for 30 min before staining cellular DNA with 250 μl of a PI solution (50 μg/ml) in PBS for 1 h at 37°C. Cells were then immediately place on ice and analyzed using FACScan™. The 1n chromosome-containing lymphocyte peak (main peak) was set to identical values on the semilogarithmic scale at the beginning of each analysis. Cell yields from cultures of Bcl-xγ wt, −/−, or Bcl-xγ Tg T cells were not significantly different at the time of analysis (36 h after stimulation).
Adoptive Transfer and CFSE Labeling of Donor Cells.
Peripheral and mesenteric LN lymphocytes from Bcl-xγ wt and −/− mice were isolated and washed in 2% FBS/PBS. To monitor the proliferation of these donor cells in recipient (syngenic Bcl-xγ wt) mice, CFSE (Molecular Probes) labeling was performed by incubating 107 cells per milliliter in PBS/0.1% BSA with CFSE (final concentration 10 μM) for 10 min at 37°C. Labeled cells were washed twice with PBS/0.1% BSA before they were resuspended in sterile PBS for tail vein injection. Approximately 5 × 106 of CFSE-labeled cells were injected, intravenously, per mouse.
Results
Generation of Bcl-xγ–deficient Mice.
We selectively disrupted this isoform within the Bcl-x locus to generate Bcl-xγ–deficient mice. Analysis of Bcl-xγ and Bcl-x genomic structure (14) has indicated that mature Bcl-xγ RNA transcripts are comprised of coding exons 1, 2, and 4 (Fig. 1 A) joined with alternatively-spliced combinations of exons 5–8 that comprise several distinct noncoding 3′-UTR (14). We generated Bcl-xγ–deficient mice (Materials and Methods) using a Cre/LoxP-mediated gene targeting vector to delete exon 4 in ES cells as outlined in Fig. 1 B. Southern blot analysis of tail cut DNA from ES cell–injected mice showed that exon 4 had been successfully disrupted (Fig. 1 C, left panel). Additional RT-PCR analysis with primers specific for exon 4 (for Bcl-xγ) or exons 2 and 3 (for Bcl-xL) confirmed that Bcl-xγ mRNA was no longer expressed while Bcl-xL mRNA expression was unchanged in the same activated T cells in three independent experiments (Fig. 1 C). To document the effect of this mutation on Bcl-xγ and Bcl-xL protein expression, Western blotting of Bcl-xγ and Bcl-xL after coligation of CD3 and CD28 was performed. These experiments confirmed other studies (14) that CD28 coligation is essential for full and early expression of Bcl-xγ in activated T cells (Fig. 1 D, left panel and legend). They also showed that CD3/CD28 coligation failed to induce detectable Bcl-xγ protein in Bcl-xγ−/− T cells after overexposure of Western blots (Fig. 1 D, right panel). At the same time, expression of Bcl-xL protein after CD3 or CD3/CD28 ligation was indistinguishable in Bcl-xγ−/− and Bcl-xγ wt T cells according to normally exposed Western blots (Fig. 1 E).
Proliferative Responses of Bcl-xγ–deficient and Bcl-xγ Tg T Cells In Vitro.
We measured the effects of CD3 and CD28 coligation on the proliferative and IL-2 responses of Bcl-xγ−/− T cells (15, 16). As expected, CD3/CD28 coligation of wt T cells resulted in a 4–6-fold increase in 3[H]thymidine incorporation and IL-2 production compared with anti-CD3 alone; however, CD28 coligation failed to augment the response of T cells from Bcl-xγ−/− mice (Fig. 2, A and B), suggesting that the Bcl-xγ mutation nullifies CD28-dependent costimulation.
Figure 2.


Proliferation profile and cytokine production of T cells from Bcl-xγ wt and −/− mice after CD3 and CD28 ligation. (A) Proliferation of activated T cells from wt and −/− mice. T cells were isolated from wt and −/− mice and cultured for 48 h in the presence of plate-bound anti-CD3 and/or anti-CD28 antibodies. Cells were then pulsed with 3[H]thymidine and cultured for an additional 18–20 h before harvesting. Data (±SEM) represents the mean thymidine uptake of triplicate cultures as described in Materials and Methods. (B) Time-course of IL-2 production from activated T cells. Supernatants from T cells activated as described in (A) were harvested at the indicated times and analyzed for IL-2 by ELISA. Values are the average of triplicate determinations (±SE) from one of three representative and independent experiments as described in Materials and Methods.
Then, we asked whether enforced expression of Bcl-xγ in T cells might replace the need for CD28-dependent costimulation. Expression of a Bcl-xγ transgene under the control of the Lck distal promoter resulted in constitutive overexpression of Bcl-xγ in peripheral T cells independent of overt activation (Fig. 3 A). Enforced Bcl-xγ expression did not enhance survival of unstimulated T cells, in contrast to the reported effects of Bcl-xL (Fig. 3 B, legend). However, proliferative and IL-2 responses of T cells from Bcl-xγ Tg+ mice after CD3 ligation were 3–4-fold greater than T cells from nontransgenic littermates and were not significantly increased by CD28 coligation (Fig. 3, B and C). Moreover, enforced expression of Bcl-xγ restored the response of CD4 DO11.10+ T cells to OVA peptide presented by B7.1/B7.2−/− APC to levels provoked by B7.1/B7.2+ APC (Fig. 3 D).
Figure 3.
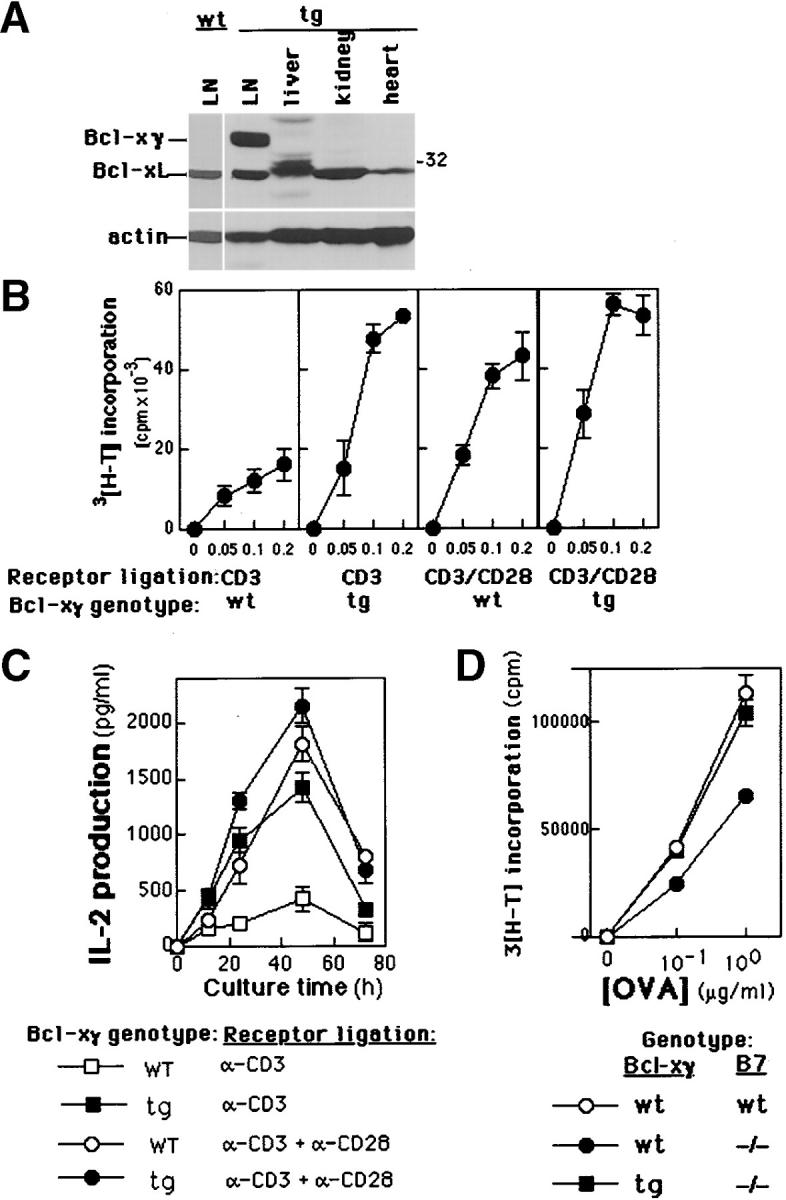
Expression and proliferation profiles of Bcl-xγ in cells derived from Tg mice. (A) Expression of Bcl-xγ and Bcl-xL in different murine tissues from Tg mice (see Materials and Methods). Immunoblot analysis was performed as described in Materials and Methods and is representative of data in one of two independent experiments; the level of actin serves as a control for protein loading. (B) Proliferation of activated T cells from wt and Tg mice. T cells were isolated from Tg or wt mice and cultured for 48 h in the presence of plate-bound anti-CD3 and/or anti-CD28 antibodies. Cells were then pulsed with 3[H]thymidine and cultured for an additional 18 h before harvesting. In the absence of antibody-dependent activation, survival of T cells from Bcl-xγ Tg and wt mice was not significantly different after 24–72 h in culture. (C) Time-course of IL-2 production from activated T cells. Supernatants from T cells activated as described above were harvested at the indicated times and analyzed for IL-2 by ELISA. All values are the average of triplicate determinations (±SEM) from one of four representative and independent experiments. (D) Proliferative response of T cells from Bcl-xγ and DO11.10 TCR Tg to OVA peptide (323–339). Purified Bcl-xγ transgenic and wt T cells (4 × 104) from DO11.10+ littermates (B6.129 × Balb/c genotype) were incubated with irradiated (5 × 105) splenocytes from either Balb/c mice or from B7.1−/−B7.2−/−Balb/c mice for 48 h before addition of 1 μCi 3[H]thymidine to each well. Proliferation (cpm) of triplicate cultures was measured after 16 h.
Effects of Bcl-xγ Deficiency or Forced Expression on Cell Cycling.
CD28 costimulation induces cell cycle progression through IL-2–dependent and IL-2–independent mechanisms (17–19). Although earlier studies of T cell lines that contained Bcl-xγ antisense constructs indicated a critical role in successful T cell activation, it was not possible to distinguish between a primary effect on cellular proliferation versus antigen-induced apoptosis (11) since the effects of Bcl-xγ expression could not be evaluated in nontransformed T cells. We find that the proliferative response of Bcl-xγ−/− T cells to CD3/CD28 coligation was reduced by >80% according to 3[H]thymidine incorporation and IL-2 expression (e.g., Fig. 2), while levels of apoptosis changed <15%. Levels of apoptosis and cell death of nonactivated wt. Bcl-xγ Tg or Bcl-xγ−/− T cells were indistinguishable over a 96-h incubation period (not shown).
The proliferative response of Bcl-xγ Tg, −/−, and wt T cells after TCR ligation was analyzed for alterations in progress through the cell cycle. Coligation of CD3/CD28 that resulted in a doubling of progression of wt T cells into G2/M did not enhance progression of Bcl-xγ−/− T cells (Fig. 4 A). This observation was complemented by the finding that enforced Bcl-xγ expression largely replaced the need for CD28 ligation for efficient entry into G2/M (Fig. 4 A). A role for Bcl-xγ in the IL-2–independent component of CD28-dependent regulation of T cell proliferation was underlined by the inability of IL-2 to fully restore the Bcl-xγ−/− T cell response to CD3 or CD3/CD28 coligation (Fig. 4, C and D). Finally, we asked whether the reduced ability of Bcl-xγ−/− T cells to proliferate after antibody-dependent receptor ligation in vitro was also apparent in vivo. Analysis of CFSE-labeled Bcl-xγ−/− T cells in mice stimulated with the superantigen staphylococcal enterotoxin A (SEA; references 20 and 21) revealed that SEA-dependent replication was reduced by ∼60% (Fig. 4 B).
Figure 4.

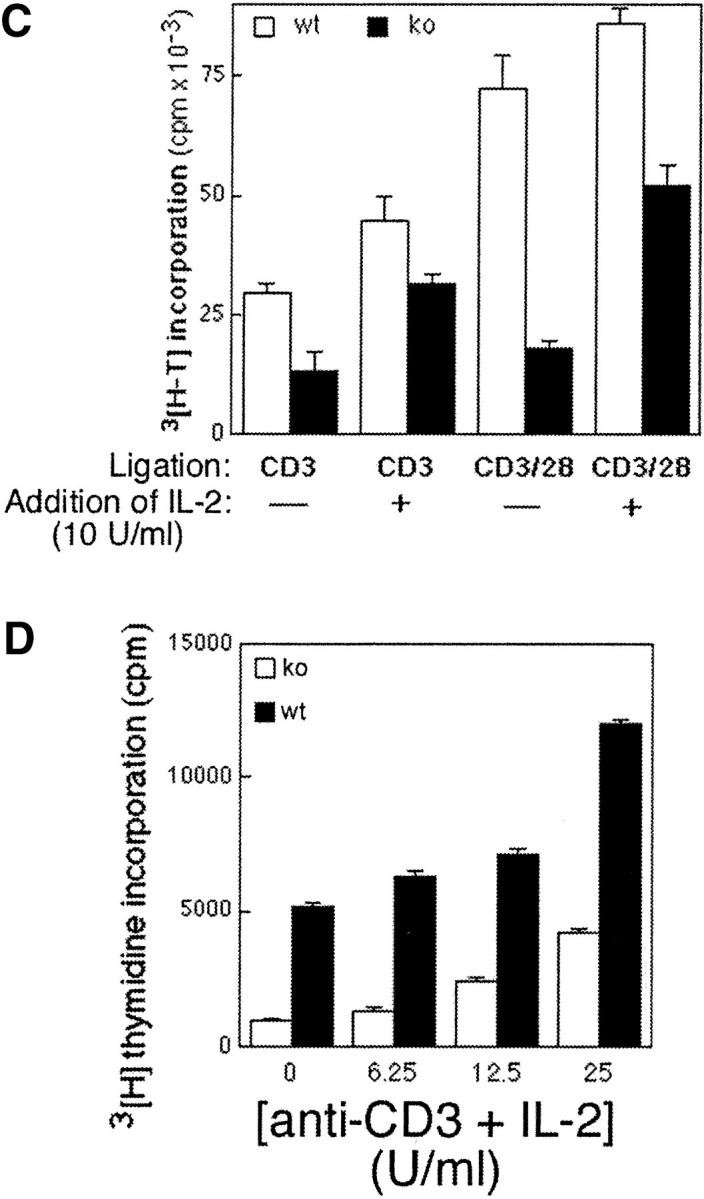
Cell cycle analysis of T cells after CD3 and CD28 ligation. (A) In vitro T cells from Bcl-xγ wt and −/− (left) and Bcl-xγ wt and transgenic mice (right) were cultured for 36 h in the presence of plate-bound anti-CD3 and/or anti-CD28 antibodies in 24-well dishes before harvesting. The percentage of T cells from these mice in G0/G1, and G2/M phases was determined by cytofluorometric analysis after PI staining as described in Materials and Methods. Results from one representative animal in each group are shown (from one of three independent experiments). Levels of annexin-positive cells were not significantly different in these groups and were <15%. (B) In vivo proliferation of T cells from Bcl-xγ wt and −/− mice after superantigen administration. LN and splenic T cells from donor Bcl-xγ wt and −/− mice were labeled with a fluorescent chloromethyl derivative, CFSE. Labeled cells were then transferred intravenously into syngenic, recipient Bcl-xγ wt mice as described in Materials and Methods. The following day, SEA toxin was injected intraperitoneally, and mice in each group were killed at 3 and 5 d after superantigen exposure. Labeled T cells were analyzed by flow cytometry as described in Materials and Methods. Results representative of one of two independent experiments are shown. (C and D) Definition of the contribution of altered IL-2 production to the altered activation pattern of T cells from Bcl-xγ2/− mice. The response of T cells from Bcl-xγ−/− or wt mice to the indicated antibody-dependent ligation in the presence or absence of exogenous IL-2 is shown (see the legend to Fig. 2 and Materials and Methods).
Overexpression of Bcl-xγ Does Not Protect from Apoptosis or Activation-induced Cell Death.
Bcl-xL has been characterized as an essential molecule in the regulation of cell survival and death (4). It was shown that overexpression of Bcl-xL could restore cell survival in CD28-deficient T cells, which normally fail to upregulate this molecule (8). Similarly, when Bcl-xγ was first described, this new isoform of Bcl-x appeared to modulate the sensitivity of CTLL-2 cells to apoptosis after TCR ligation (11). However, levels of apoptosis were found to differ only slightly between Bcl-xγ–deficient and wt T cells after TCR stimulation (unpublished data). To further investigate the possible role of Bcl-xγ in the regulation of cell survival and death, Bcl-xγ–deficient T lymphocytes were compared with Bcl-xγ Tg cells in their response to cytotoxic agents. We found no difference in survival between the two cell types after either irradiation (1,000 rad), treatment with dexamethasone (5 μM) or culture in the absence of stimulation for 96 h (Fig. 5 A). A comparison of the response of T cells from Bcl-xγ Tg/ DO11.10 TCR Tg mice with cells from Bcl-xγ wt/ DO11.10 TCR Tg littermates after successive stimulation with OVA 323–339 peptide followed by TCR religation with anti-CD3 antibody indicated that overexpression of Bcl-xγ did not protect cells from activation-induced cell death (AICD); both groups of T cells displayed increased cell death and dramatic reduction of the proliferative response after TCR religation (Fig. 5 B). Together, these results suggest that the proliferative advantage conferred to T cells by the Bcl-xγ transgene is not mediated through enhanced survival, and that Bcl-xγ does not seem to play an obvious inhibitory role in the process of apoptosis.
Figure 5.


Effect of Bcl-xγ on apoptosis and AICD. (A) Effect of Bcl-xγ expression on survival to cytotoxic treatments. Splenocytes from Bcl-xγ−/− and Bcl-xγ Tg mice (106 cells per well on a 24-well plate) were cultured in complete medium either after irradiation (1,000 rad), addition of DEX (5 μM), or no addition. Cells were collected at the time points shown and stained for CD4 expression and PI incorporation. Percentages of dead CD4+ T cells were determined by flow cytometry (percentage of PI-positive cells in the CD4+ population). (B) Effect of Bcl-xγ overexpression on AICD. Splenocytes from Bcl-xγ Tg and wt mice (3 × 105) were stimulated with OVA 323–339 (0.01, 0.1, and 1 μM) or cultured without peptide. After 48 h, the cells were transferred onto fresh microtiter plates which were either uncoated or coated with anti-CD3 (1 μg/ml at 4°C for 16 h). 3[H]thymidine was added at the same time and proliferation was measured 18 h later (left). Alternatively, no 3[H]thymidine was added, and the cells were collected for flow cytometry (stained for CD4 expression and PI incorporation) 20 h after transfer. The percentages of PI-positive cells within the CD4+ population are shown (right). Results are representative of two similar experiments.
Effects of Bcl-xγ Deficiency on the Development of Autoimmunity.
CD28-dependent costimulatory signaling is essential for development of several T cell–mediated autoimmune diseases (3, 22, 23) including experimental autoimmune encephalomyelitis (EAE), which depends on efficient expansion of T cells that recognize peptides derived from myelin proteins (for a review, see reference 24). In view of the defective CD28-dependent costimulatory response of Bcl-xγ−/− T cells noted above, we defined EAE susceptibility of Bcl-xγ−/− mice after immunization with PLP172–183. Bcl-xγ−/− mice failed to develop significant EAE compared with substantial disease displayed by Bcl-xγ+/+ or +/− littermates (Fig. 6 A) and displayed defective development of T cell memory to PLP172–183. T cells from PLP-immunized Bcl-xγ−/− mice exhibited a substantial (50–100-fold) reduction in secondary response as judged by PLP-specific proliferation and IL-2 production after in vitro restimulation by PLP172–183 peptide (Fig. 6 B).
Figure 6.
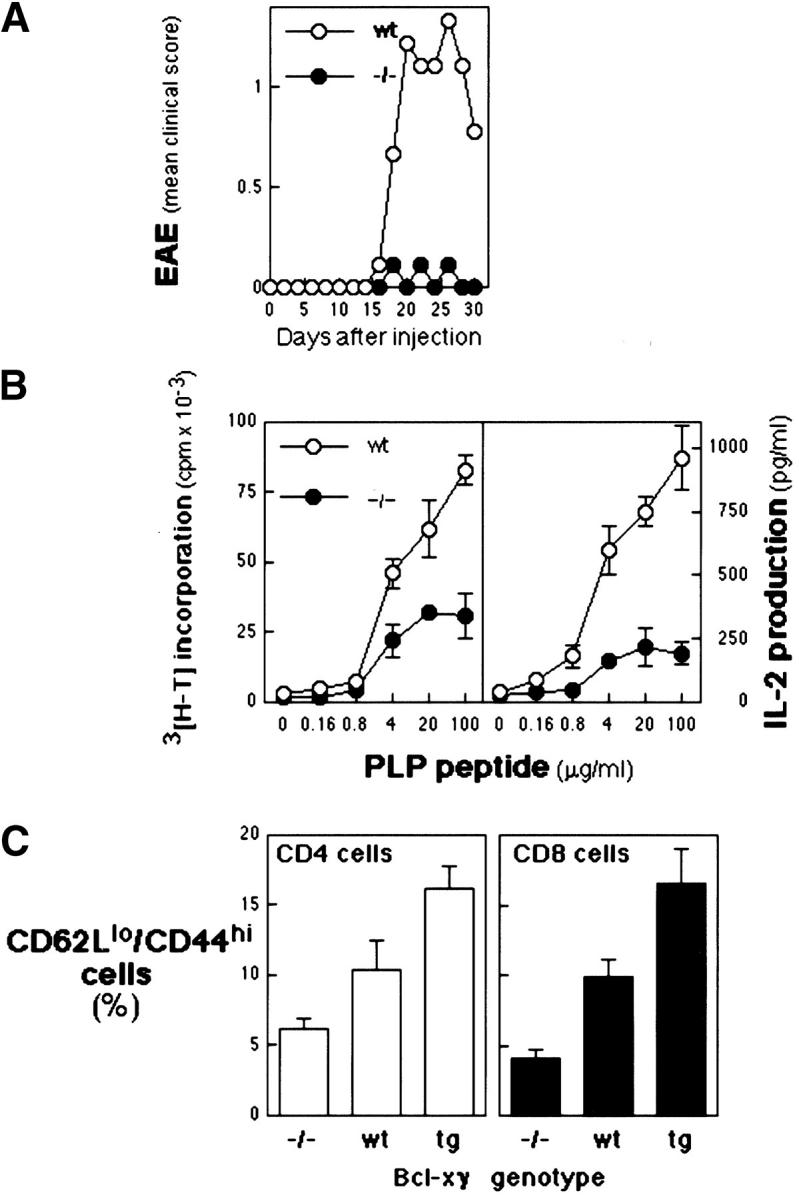
Induction of EAE in Bcl-xγ wt and −/− mice. (A) Mean clinical scores. Animals were immunized with the a PLP, PLP172–183; wt, n = 9, and −/−, n = 9. Scoring of disease development in (C57BL/6 × 129)F2 littermates that carried the Bcl-xγ−/− (−/−) or wt (+/+ and +/−) genotype is shown based on the mean scores of two independent experiments (±SEM) as described in Materials and Methods. (B, left) Proliferative responses of PLP peptide-specific T cells from wt and −/− mice. T cells were stimulated in vitro with PLP peptide in the presence of irradiated APCs. Data (±SE) represents the mean thymidine uptake of triplicate cultures. (Right) PLP peptide-specific IL-2 production (measured by ELISA) from draining LN cells of Bcl-xγ wt and −/− mice. T cells were activated in vitro with PLP peptide in the presence of irradiated APCs. Values are the average of triplicate determinations (±SEM). (C) FACS® analysis of CD4 and CD8 T cells from Bcl-xγ−/−, Bcl-xγ Tg, and Bcl-xγ wt mice according to expression of CD44hi, CD62L, and CD45RB. LN cells from Bcl-xγ Tg, −/−, and control (littermate) mice were isolated and analyzed by flow cytometry (16) for the indicated cell surface/activation markers expressed by LN cells (3.5 × 107 cells per animal). Data shown are the average values from four mice (for each genotype) that were 6 mo of age (± 1 wk) and expressed as a percentage of wt values.
Although development of thymocytes and T cells from Bcl-xγ−/− and Bcl-xγ Tg mice was not obviously dysregulated according to FACS® analysis (Table I), levels of memory T cells, as judged by expression of CD62L and CD44, were decreased by ∼50% in Bcl-xγ−/− mice and increased by this amount in Bcl-xγ Tg mice (Fig. 6 C). These findings suggest a direct correlation between Bcl-xγ expression and successful generation of memory T cells secondary to experimental (Fig. 6, A and B) or environmental (Fig. 6 C) antigenic stimuli.
Table I.
FACS® Analysis of Lymphoid Cells from Bcl-xγ−/− and Bcl-xγ Tg Mice
| Thymus | Bcl-xγ wt | Bcl-xγ2/− | Bcl-xγ Tg |
|---|---|---|---|
| % | |||
| DN | 2.7 | 1.9 | 2.4 |
| DP | 82.1 | 86.5 | 85.3 |
| CD4 SP | 11.5 | 8.2 | 9.7 |
| CD8 SP | 3.8 | 3.4 | 2.6 |
| TCR-β+ | 18 | 17.5 | 19.4 |
| CD3+ | 14.4 | 12.7 | 19.7 |
| HAS+ | 90.3 | 92.4 | 93.2 |
| CD69+ | 15.6 | 14.1 | 13.4 |
| Spleen (%) | |||
| CD4 T cell | 60.3 | 62.8 | 62.1 |
| CD8 T cell | 30.7 | 27.2 | 31.2 |
| DN T cell | 9 | 9.5 | 6.7 |
| CD19/B220 | 28.2 | 29.6 | 27.7 |
| B220/CD8 | 6.4 | 6.4 | 7.8 |
| NK1.1 | 1.2 | 1.1 | 1.1 |
| TCR-β+ | 56.2 | 58.5 | 61.2 |
| TCR-γ+ | 0.6 | 0.7 | 0.4 |
| LN (%) | |||
| CD4 T cell | 64.9 | 60.9 | 64 |
| CD8 T cell | 32.7 | 36.3 | 33.4 |
| DN T cell | 2.5 | 2.8 | 2.3 |
| CD19/B220 | 8.8 | 10.1 | 9.7 |
| B220/CD8 | 1.9 | 1.5 | 1.4 |
| NK1.1 | 1.5 | 1.2 | 0.8 |
| TCR-β | 87.9 | 87 | 91.2 |
| TCR-γ | 0.8 | 0.6 | 0.5 |
Data shown are representative of one of three independent experiments with groups of three to four mice (8–12 wk of age).
Discussion
An efficient T cell response to antigen requires costimulatory signals provided by the CD28 receptor that promote cell expansion and cytokine secretion. Although the importance of CD28 ligation to complete T cell activation is well documented (25), its molecular basis remains poorly understood. Since expression of the Bcl-xγ isoform is tightly coupled to CD28/CD3 coligation, we investigated its potential role in expression of the costimulatory phenotype using mice containing T cells that specifically lack or overexpress this member of the Bcl-x family.
LoxP-dependent excision of exon 4 of the Bcl-x gene eliminated Bcl-xγ expression without a detectable effect on expression of Bcl-xL at the RNA or protein level (Fig. 1). This genetic lesion was associated with defective T cell memory in vivo; Bcl-xγ−/− mice displayed reduced numbers of CD45RB+ CD44hi T cells, reduced secondary responses after restimulation with PLP172–183 peptide, and failed to develop PLP-induced EAE. Analysis of the response of Bcl-xγ−/− T cells in vitro mapped this phenotype to a defect in CD28-dependent costimulation, since CD3-dependent activation was unimpaired while CD28 coligation failed to enhance 3[H]thymidine incorporation, IL-2 production, and entry into G2/S. These findings were complemented by studies showing that enforced expression of Bcl-xγ largely replaced the need for CD28-dependent costimulation and the response of Bcl-xγ Tg T cells to CD3 ligation was similar to the response of nonTg T cells to CD3/CD28 coligation.
Ligation of CD28 enhances T cell proliferation through increased expression of IL-2 as well as IL-2–independent acceleration of entry into cell cycle (17, 25, 26). Bcl-xγ appears to contribute to both elements of costimulation; although Bcl-xγ−/− T cells display reduced IL-2 production, reduced proliferation is only partially remedied by exogenous IL-2 and IL-2–independent entry of Bcl-xγ–deficient T cells into G2 is impaired (Fig. 4). Since expression of other Bcl-x isoforms including Bcl-xL is unaltered in these cells (Fig. 1), these experiments underline the essential intracellular role of Bcl-xγ in expression of CD28-dependent costimulation.
Initial studies of Bcl-xγ showed that this Bcl-x isoform is expressed in T cells that successfully proliferate after engagement of the TCR by peptide antigen, whereas expression of Bcl-x antisense constructs in T cell lines reduced proliferation and increased apoptosis (11). However, analysis of the effects of dysregulated Bcl-xγ gene expression in transformed cells did not delineate the potential role of this cytosolic protein in T cell proliferation and CD28-dependent costimulation. The finding that CD28-dependent costimulation depends on expression of the cytosolic Bcl-xγ protein supports an emerging view that CD28-dependent enhancement of T cell survival and T cell proliferation to antigen may reflect separate signaling pathways originating from distinct regions of the CD28 cytoplasmic domain (9, 10). Other studies have indicated that Bcl-xL expression marks a costimulatory signaling pathway that contributes to T cell survival (4, 7) but not proliferation or cytokine expression after antigen stimulation (8–10). Our studies suggest that Bcl-xγ expression is essential for enhanced entry into cell cycle and IL-2 production after TCR ligation, but not for protection from AICD or irradiation or dexamethasone-induced apoptosis. Furthermore, overexpression of Bcl-xγ did not enhance cell survival in the absence of stimuli or after TCR ligation. These findings support a division of labor among the Bcl-x isoforms that relies on Bcl-xγ enhancement of T cell proliferation and Bcl-L to potentiate cell survival after CD28 ligation.
Unlike Bcl-xL, which is tethered to the outer surface of mitochondrial membranes, the Bcl-xγ protein locates in the cytosol, lacks a hydrophobic tail, and may be phosphorylated at serine/threonine (14). A two-hybrid screen using the unique COOH terminus of Bcl-xγ has suggested that the Jun-binding protein QM (27, 28) specifically interacts with Bcl-xγ, and not Bcl-xL (unpublished data), opening the possibility that CD28-dependent induction of Bcl-xγ may facilitate DNA binding of AP-1 or Jun/Jun dimers. Alternatively, Bcl-xγ may interact with caspases and/or caspase-activating molecules in a similar fashion to Bcl-xL (29). According to the latter view, Bcl-xγ might specifically block negatively acting caspases while sparing stimulatory caspases (30, 31). Insufficient Bcl-xγ induction after incomplete T cell activation would thus allow negatively acting caspases to dampen T cell activation. Although further experiments are required to precisely define the mechanism of Bcl-xγ−dependent enhancement of the T cell response, definition of the complementary roles of Bcl-xγ and Bcl-xL in CD28-dependent costimulation represents an important step toward understanding this activation process.
Acknowledgments
We thank Dr. L. Klein for advice and reagents and Ms. Alison Angel for kind assistance in the preparation of this manuscript.
This work was supported in part by National Institutes of Health research grants A13600, A112184, and AI48125 to H. Cantor, AI10468 to L.A. Trimble, AI07386 to X.-F. Yang, and AI20047 and AI35714 to F.W. Alt, who is also supported by the Howard Hughes Medical Institute; B.P. Sleckman is a recipient of a Burroughs Wellcome Fund Career Development Award; B. Press is a fellow of the Cancer Research Institute; C.H. Bassing is a fellow of the Irvington Institute; and M. Jansson is a fellow of the Swedish Foundation for International Cooperation in Research and Higher Education (STINT).
Q. Ye and B. Press contributed equally to this work.
Footnotes
Abbreviations used in this paper: AICD, activation-induced cell death; EAE, encephalomyelitis; ES, embryonic stem; PLP, proteolipid protein peptide; wt, wild-type.
References
- 1.Harding, F.A., J.G. MacArthur, J.A. Gross, D.H. Raulet, and J.P. Allison. 1992. CD28-mediated signalling costimulates murine T-cells and prevents induction of anergy in T-cell clones. Nature. 356:607–609. [DOI] [PubMed] [Google Scholar]
- 2.Turka, L.A., P.S. Linsley, H. Lin, W. Brady, J.M. Leiden, R.Q. Wei, M.L. Gibson, X.G. Zheng, S. Myrdal, D. Gordon, et al. 1992. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc. Natl. Acad. Sci. USA. 89:11102–11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang, T.T., C. Jabs, R.A. Sobel, V.K. Kuchroo, and A.H. Sharpe. 1999. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J. Exp. Med. 190:733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boise, L.H., M. Gonzalez-Garcia, C.E. Postema, L.Y. Ding, T. Lindsten, L.A. Turka, X.H. Mao, G. Nunez, and C.B. Thompson. 1993. Bcl-x, a Bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 74:597–608. [DOI] [PubMed] [Google Scholar]
- 5.Chao, D.T., G.P. Linette, L.H. Boise, L.S. White, C.B. Thompson, and S.J. Korsmeyer. 1995. Bcl-XL and Bcl-2 repress a common pathway of cell death. J. Exp. Med. 182:821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grillot, D.A.M., R. Merino, and G. Nunez. 1995. Bcl-xL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J. Exp. Med. 182:1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boise, L.H., A.J. Minn, P.J. Noel, C.H. June, M.A. Accavitti, T. Lindsten, and C.B. Thompson. 1995. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xL. Immunity. 3:87–98. [DOI] [PubMed] [Google Scholar]
- 8.Dahl, A.M., C. Klein, P.G. Andres, C.A. London, M.P. Lodge, R.C. Mulligan, and A.K. Abbas. 2000. Expression of bcl-X(L) restores cell survival, but not proliferation off effector differentiation, in CD28-deficient T lymphocytes. J. Exp. Med. 191:2031–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burr, J.S., N.D. Savage, G.E. Messah, S.L. Kimzey, A.S. Shaw, R.H. Arch, and J.M. Green. 2001. Cutting edge: distinct motifs within CD28 regulate T cell proliferation and induction of Bcl-xL. J. Immunol. 166:5331–5335. [DOI] [PubMed] [Google Scholar]
- 10.Okkenhaug, K., L. Wu, K.M. Garza, J. La Rose, W. Khoo, B. Odermatt, T.W. Mak, P.S. Ohashi, and R. Rottapel. 2001. A point mutation in CD28 distinguishes proliferative signals from survival signals. Nat. Immunol. 2:325–332. [DOI] [PubMed] [Google Scholar]
- 11.Yang, X.F., G.F. Weber, and H. Cantor. 1997. A novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity. 7:629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen, J.M., K.A. Forbush, and R.M. Perlmutter. 1992. Functional dissection of the lck proximal promoter. Mol. Cell Biol. 12:2758–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laird, P.W., A. Zijderveld, K. Linders, M.A. Rudnicki, R. Jaenisch, and A. Berns. 1991. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 19:4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang, X.F., Q. Ye, B. Press, R.-H. Han, C.H. Bassing, B.P. Sleckman, F.W. Alt, and H. Cantor. 2002. Analysis of the complex genomic structure of Bcl-x and its relationship to Bcl-xγ expression after CD28-dependent costimulation. Mol. Immunol. In press. [DOI] [PubMed] [Google Scholar]
- 15.Gimmi, C.D., G.J. Freeman, J.G. Gribben, K. Sugita, A.S. Freedman, C. Morimoto, and L.M. Nadler. 1991. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc. Natl. Acad. Sci. USA. 88:6575–6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mueller, D.L. 2000. T cells: a proliferation of costimulatory molecules. Curr. Biol. 10:R227–R230. [DOI] [PubMed] [Google Scholar]
- 17.Lucas, P.J., I. Negishi, K. Nakayama, L.E. Fields, and D.Y. Loh. 1995. Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J. Immunol. 154:5757–5768. [PubMed] [Google Scholar]
- 18.Appleman, L.J., A. Berezovskaya, I. Grass, and V.A. Boussiotis. 2000. CD28 costimulation mediates T cell expansion via IL-2-independent and IL-2-dependent regulation of cell cycle progression. J. Immunol. 164:144–151. [DOI] [PubMed] [Google Scholar]
- 19.Boonen, G.J., A.M. van Dijk, L.F. Verdonck, R.A. van Lier, G. Rijksen, and R.H. Medema. 1999. CD28 induces cell cycle progression by IL-2-independent down-regulation of p27kip1 expression in human peripheral T lymphocytes. Eur. J. Immunol. 29:789–798. [DOI] [PubMed] [Google Scholar]
- 20.Muraille, E.M., G. De Becker, M. Bakkus, K. Thielemans, J. Urbain, M. Moser, and O. Leo. 1995. Co-stimulation lowers the threshold for activation of naive T cells by bacterial superantigens. Int. Immunol. 7:295–304. [DOI] [PubMed] [Google Scholar]
- 21.Stoffel, B., P. Bauer, M. Nix, K. Deres, and W. Stoffel. 1998. Ceramide-independent CD28 and TCR signaling but reduced IL-2 secretion in T cells of acid sphingomyelinase-deficient mice. Eur. J. Immunol. 28:874–880. [DOI] [PubMed] [Google Scholar]
- 22.Oliveira-dos-Santos, A.J., A. Ho, Y. Tada, J.J. Lafaille, S. Tonegawa, T.W. Mak, and J.M. Penninger. 1999. CD28 costimulation is crucial for the development of spontaneous autoimmune encephalomyelitis. J. Immunol. 162:4490–4495. [PubMed] [Google Scholar]
- 23.Perrin, P.J., C.H. June, J.H. Maldonado, R.B. Ratts, and M.K. Racke. 1999. Blockade of CD28 during in vitro activation of encephalitogenic T cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 163:1704–1710. [PubMed] [Google Scholar]
- 24.Anderson, T.J., M. Klugmann, C.E. Thomson, A. Schneider, C. Readhead, K.A. Nave, and I.R. Griffiths. 1999. Distinct phenotypes associated with increasing dosage of the PLP gene: implications for CMT1A due to PMP22 gene duplication. Annu. NY Acad. Sci. 883:234–246. [DOI] [PubMed] [Google Scholar]
- 25.Chambers, C.A., and J.P. Allison. 1999. Costimulatory regulation of T-cell function. Curr. Opin. Cell Biol. 11:203–210. [DOI] [PubMed] [Google Scholar]
- 26.Shahinian, A., K. Pfeffer, K.P. Lee, T.M. Kundig, K. Kishihara, A. Wakeham, K. Kawai, P.S. Ohashi, C.B. Thompson, and T.W. Mak. 1993. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 261:609–612. [DOI] [PubMed] [Google Scholar]
- 27.Monteclaro, F.S., and P.K. Vogt. 1993. A Jun-binding protein related to a putative tumor suppressor. Proc. Natl. Acad. Sci. USA. 90:6726–6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imafuku, I., T. Masaki, M. Waragai, S. Takeuchi, M. Kawabata, S. Hirai, S. Ohno, L.E. Nee, C.F. Lippa, I. Kanazawa, et al. 1999. Presenilin 1 suppresses the function of c-Jun homodimers via interaction with QM/Jif-1. J. Cell Biol. 147:121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu, Y., M.A. Benedict, D. Wu, N. Inohara, and G. Nunez. 1998. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc. Natl. Acad. Sci. USA. 95:4386–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alam, A., L.Y. Cohen, S. Aouad, and R.P. Sekaly. 1999. Early activation of caspases during T lymphocyte stimulation results in selective substrate cleavage in nonapoptotic cells. J. Exp. Med. 190:1879–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy, N.J., T. Kataoka, J. Tschopp, and R.C. Budd. 1999. Caspase activation is required for T cell proliferation. J. Exp. Med. 190:1891–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]