

Abstract
Immune and inflammatory systems are controlled by multiple cytokines, including interleukins (ILs) and interferons. These cytokines exert their biological functions through Janus tyrosine kinases and signal transducer and activator of transcription (STAT) transcription factors. We recently identified two intrinsic Janus kinase (JAK) inhibitors, JAK binding protein (JAB; also referred to as suppressor of cytokine signaling [SOCS1]/STAT-induced STAT inhibitor [SSI1]) and cytokine-inducible SH2 protein (CIS)3 (or SOCS3/SSI3), which play an essential role in the negative regulation of cytokine signaling. We have investigated the role of STATs and these JAK inhibitors in intestinal inflammation. Among STAT family members, STAT3 was most strongly tyrosine phosphorylated in human ulcerative colitis and Crohn's disease patients as well as in dextran sulfate sodium (DSS)-induced colitis in mice. Development of colitis as well as STAT3 activation was significantly reduced in IL-6–deficient mice treated with DSS, suggesting that STAT3 plays an important role in the perpetuation of colitis. CIS3, but not JAB, was highly expressed in the colon of DSS-treated mice as well as several T cell–dependent colitis models. To define the physiological role of CIS3 induction in colitis, we developed a JAB mutant (F59D-JAB) that overcame the inhibitory effect of both JAB and CIS3 and created transgenic mice. DSS induced stronger STAT3 activation and more severe colitis in F59D-JAB transgenic mice than in their wild-type littermates. These data suggest that hyperactivation of STAT3 results in severe colitis and that CIS3 plays a negative regulatory role in intestinal inflammation by downregulating STAT3 activity.
Keywords: Janus kinase, CIS/SOCS, interleukin 6, ulcerative colitis, negative regulation
Introduction
Proinflammatory cytokines have been demonstrated to play a crucial role in the pathogenesis of inflammatory bowel diseases including Crohn's disease (CD). It has been shown that in addition to IFN-γ, IL-6 is one of the main cytokines secreted by lamina propria cells, and that CD patients produce substantially higher amounts of IL-6 than control patients 1 2 3 4. Strong expression of IL-6 has also been reported in acute bowel inflammation induced by dextran sulfate sodium (DSS) in mice 5. A recent study using anti–IL-6 receptor antibodies demonstrated that IL-6 plays a critical role in the development of Th1 cell–mediated chronic colitis 6 7. IL-6 also plays a central role in arthritis, which is confirmed by recent reports of IL-6 gene–disrupted mice that were shown to be resistant to antigen-induced arthritis 8 9 and collagen-induced arthritis 10. IL-6 and its related cytokines, including leukemia inhibitory factor (LIF), oncostatin M (OSM), and IL-11, preferentially activate Janus kinase (JAK) tyrosine kinases and the signal transducer and activator of transcription (STAT)3 transcription factor. Furthermore, antiinflammatory cytokine IL-10 mainly utilizes STAT3 11. Therefore, the regulatory mechanism of the JAK-STAT3 pathway is extremely important for the understanding of inflammation.
We have reported a new family of cytokine-inducible SH2 proteins (CIS) that are involved in the negative regulation of cytokine signals, especially JAKs and STATs (for a review, see reference 12). The first identified CIS gene, CIS1, has been shown to be a negative feedback regulator of the STAT5 pathway 13 14. We recently cloned other CIS family members, JAK binding protein (JAB) and CIS3, which directly bind to the JAK2 tyrosine kinase domain and inhibit JAK tyrosine kinase activity 15. Overexpression of JAB and CIS3 resulted in the suppression of cytokine signaling by using JAKs, including IL-6 and LIF 16. Two other groups have reported on related genes: one referred to JAB, CIS2, and CIS3 as suppressor of cytokine signaling (SOCS)1, SOCS2, and SOCS3, respectively 17, whereas the other referred to them as STAT-induced STAT inhibitor (SSI)1, SSI2, and SSI3, respectively 18. Gene disruption studies have demonstrated that JAB/SOCS1/SSI1 and CIS3/SOCS3/SSI3 negatively regulate IFN-γ signaling 19 20 21 22 and fetal liver hematopoiesis 23, respectively. However, the physiological function of CIS3 in adult tissues remains to be determined, as CIS3 gene disruption was embryonically lethal 23. Many reports have indicated that CIS3 is induced by various inflammatory and antiinflammatory cytokines such as IL-6, IFN-γ, and IL-10 and that it negatively regulates those cytokine actions as well as STAT functions 12. However, these studies used primary cultured cells or cell lines, and few of them focused on the relationship between CIS3 and STATs in vivo. In this report, we examined the expression of CIS3 in colitis and its relationship to STAT3 activation using mouse colitis models. First, we demonstrated that IL-6 plays a crucial role in the development of DSS-induced colitis, suggesting that STAT3 activation is involved in the perpetuation of colitis. We found that CIS3 was induced as colitis developed, whereas a high level of CIS3 expression rather suppressed STAT3 activation. Forced expression of a dominant negative form of JAB/CIS3 in mice resulted in a more severe colitis induced by DSS. Thus, it is highly possible that expression of CIS3 negatively regulates the pathogenesis of inflammation by modulating STAT3 activity.
Materials and Methods
Cells.
The human colon carcinoma cell line, HCT, and the murine intestinal-derived epithelial cell line, CMT-93, were cultured in DMEM containing 10% FCS 24. CMT-93 cells were obtained from American Type Culture Collection (no. CCL 223). NIH-3T3 and 293 cells were cultured in DMEM containing 10% calf serum (CS). Human and mouse hematopoietic cell lines (Raji, HEL, Ba/F3, and 32D) were cultured in RPMI containing 10% FCS, as described 16.
Mice.
7–8-wk-old female BALB/c, C57BL6 mice were purchased from SLC Co., Ltd., and the TCR-α chain (TCR-α−/− mice) were obtained from Charles River Laboratories. IL-6−/−, IL-10−/−, and IFN-γ−/− mice in a C57BL6 background were obtained from The Jackson Laboratory. Mice with a cell type–specific disruption of the STAT3 gene in macrophages and neutrophils (Mφ-STAT3−/−) have been described previously 25. To create transgenic (Tg) mice, a pCAGGS-F59D-JAB expression vector was constructed by subcloning the F59D-JAB cDNA 15 into the EcoRI cloning site of the pCAGGS as described 14. After digestion with SalI and HindIII, the fragment-containing promoter, F59D-JAB cDNA as well as the 3′ noncoding region was used for microinjection into zygotes of C57BL6XC3H (B6C3-F1) parents. Eggs surviving microinjection were transferred into the oviducts of recipient pseudo-pregnant females as described 14. Tg mice were identified by PCR analysis of tail genomic DNA, and expression was confirmed by reverse transcription (RT)-PCR using primer sets (GATCCCATCGAATTCGAGTAG and GTGATGCGCCGGTAATCGGA). Four independent lines were established and B6C3XC57BL6-F2 mice were used for experiments. All animal studies were approved by the institutional review board for animal experiments of Kurume University.
Patients and Samples.
Mucosal samples were taken from freshly obtained intestinal resection specimens of patients with ulcerative colitis (UC) whose disease had been classified as active by endoscopic and histopathological analyses. Mucosal samples were also taken from involved areas of CD patients. The disease was active in patients, as defined by a CD activity index (CDAI) of >150. In these patients, the primary site of involvement was ileocolonic. In all four patients, indication for surgery was a chronic active course poorly responsive to corticosteroid treatment or other medical treatment. Biopsy samples were obtained from the diseased colonic mucosa of a patient with infectious colitis (IC) during colonoscopy. Microscopically unaffected areas of colon cancer specimens were used as normal control colon. The collection of surgical samples was approved by the ethical committee and the institutional review board of Kurume University.
DSS-induced Colitis.
Colitis was induced by means of drinking water supplemented with 2–4% DSS (mol wt, 40,000; ICN Biomedicals) as described previously 26. Control mice were treated in a similar manner with drinking water without DSS. The DAI and histological score were assessed in accordance with established criteria 27, which combined scores of weight loss, consistency, and bleeding divided by 3, and acute clinical symptoms with diarrhea and/or extremely bloody stools. Colonic sections were stained with hematoxylin and eosin and used for histological analysis. IL-6 and IL-10 levels were measured by ELISA assay kits (Endogen) using the colon organ culture of DSS-treated mice as described 28.
Antigen-induced and T Cell–induced Colitis.
The 2,4, 6-trinitrobenzene sulfonic acid (TNBS)-induced colitis has been described elsewhere 29. In brief, 1 wk before the induction of colitis, mice were immunized with 1 mg of TNBS (Sigma-Aldrich) in saline by intrarectal administration. Colitis was induced by twice intrarectal administration of 0.5 mg TNBS in 50% ethanol at days 0 and 7. Mice were killed on day 14.
Induction of colitis by transfer of CD4+ T cells into SCID mice was done as described previously 7. CD62L+CD4+ T cells were isolated from normal Balb/c mice spleen by using anti-CD62L and anti-CD4 monoclonal antibodies and magnetic beads. Purified CD62L+CD4+ T cells expressed high levels of CD45RB antigen. CD62L+CD4+ splenic T cells (106) were injected intravenously into C.B-17 SCID mutant mice. Mice were killed on day 21.
Immunoblot Analysis.
Colon and other tissues obtained from mice treated with DSS were immediately frozen in liquid nitrogen and stored at −80°C. The whole colon of mice and intestinal resection specimens of human patients were homogenized in 2–4 ml of a lysis buffer containing 50 mM Tris-HCl (pH 8.0)-0.5% NP-40, 1 mM EDTA, 150 mM NaCl, 10% glycerol, 1 mM sodium vanadate, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, and 1 mM phenylmethylsulfonyl fluoride with a protease inhibitor cocktail (Sigma-Aldrich; reference 14). The extracts were cleared by spinning at 15,000 rpm at 4° for 15 min and then diluted with the lysis buffer to achieve ∼2 mg/ml protein concentration. The total cell extracts were resolved by SDS-PAGE, and proteins were detected by immunoblotting as described 14 23 30. Anti-STAT1 (E-23), anti-STAT3 (C-20), anti-STAT5 (C17), and anti-STAT6 (M-20) were purchased from Santa Cruz Biotechnology, Inc. Tyrosine phosphorylation of STATs was detected by using anti–phospho-STAT–specific antibodies (anti-PY-STATs; New England BioLabs, Inc./Upstate Biotechnology) as described 31. Rabbit polyclonal and mouse monoclonal anti-CIS3 antibodies against COOH-terminal peptide (204–225) have been described previously 23 30 and were obtained from Immuno-Biological Laboratories Co., Ltd.
Immunohistochemistry.
Colon tissues were fixed with 10% formalin, paraffin embedded, and sectioned. Slides were incubated with either a 1:100 dilution of anti–phospho-STAT3–specific antibodies (1:1 mixture of anti-PS-STAT3 and anti-PY-STAT3; New England BioLabs, Inc.) or a 1:100 dilution of polyclonal anti-JAB/SOCS1 (Immuno-Biological Laboratories) and stained with LSAB kit according to the manufacturer's instructions (Dako). The samples were then lightly stained with hematoxylin and examined.
Northern Hybridization.
Total RNA from various kinds of tissue was prepared with Trizol (GIBCO BRL) according to the manufacturer's instructions. Total RNA (5 μg) was separated on 1.0% agarose gels containing 2.4% formaldehyde and then transferred to positively charged nylon membranes. After fixation under calibrated ultraviolet irradiation, the membranes were hybridized with digoxigenin (DIG)-labeled riboprobes and visualized using alkaline phosphatase–labeled anti-DIG antibody according to the manufacturer's instructions (Boehringer). The probe cDNA for CIS3, JAB, and G3PDH has been described previously 31.
In Situ Hybridization.
A PstI fragment of mouse CIS3 cDNA was cloned into Bluescript (Stratagene). DIG-labeled sense and antisense riboprobes were synthesized by using a DIG-RNA labeling kit (Boehringer) according to the manufacturer's instructions. Colon tissue obtained from Balb/c mice treated with 4% DSS for 7 d was cryostat sectioned to 7-μm thickness. Fixation in 4% formaldehyde, hybridization, and detection with DIG-labeled probe were performed according to Boehringer's protocol.
RT-PCR.
IL1β and control G3PDH mRNAs were detected by standard RT-PCR methods using Standard GeneAmp RNA PCR kit (PE Biosystems) according to the manufacturer's instructions. Primer sets were CAGAGTTCCCCAACTGGTACAT and AAGGAGGAAACACAGGCTCTCT (IL-1β) and ACCACAGTCCATGCCATCAC and TCCACCACCCTGTTGCTGTA (G3PDH).
Luciferase Assay.
The STAT3 responsive promoter-luciferase reporter gene and LIF-dependent luciferase assay in 293 cells transfected with Myc-tagged wild-type (WT) or mutant JAB cDNA have been described 12.
Results
STAT3 Is Activated in Colitis.
First, we examined which STAT is activated in colitis. Substantial amounts of STAT1, STAT3, STAT5, and STAT6 were detected in the colon of human UC and CD patients as well as DSS-treated mice. DSS-induced colitis is an experimental acute inflammatory bowel disease that is not dependent on T or B cells, as it occurs in SCID mice that lack lymphocytes 28. However, inflammatory cytokine production by macrophages and/or epithelial cells has been shown to contribute to the pathophysiology of colitis 5. Among STAT1, STAT3, STAT5, and STAT6, we could repeatedly detect the phosphorylation of STAT3 in the colon of DSS-treated mice as well as UC and CD patients (Fig. 1 A; PY-STAT), whereas the phosphorylation of STAT1, STAT5, or STAT6 was undetectable or very low by our assay. We also examined STAT3 activation in other types of intestinal inflammation. STAT3 phosphorylation was evident in a non-inflammatory bowel disease (IBD) infectious colitis patient (Fig. 1 B, lane 1). STAT3 activation was also detected in both Th1-dependent colitis in IL-10−/− mice (Fig. 1 B, lines 5 and 6) and macrophage- and neutrophil-specific STAT3 conditional knockout (Mφ-STAT3−/−) mice (reference 25; lines 7 and 8) as well as in IL-4–dependent colitis in TCR-α chain knockout (TCR−/−) mice (reference 32; lane 9). STAT3 activation was also observed in other T cell–dependent colitis induced by the haptenating reagent, TNBS (lanes 11 and 12) as well as by transplantation of CD45RBhighCD4+ T cells into SCID mice (line 13). Thus, STAT3 was activated not only in acute T cell–independent colitis but also in chronic T cell–dependent colitis. Clinical signs such as persistent diarrhea and the macroscopic examination of colon tissues indicated that the IL-10−/− mice suffered from much more severe colitis than the Mφ-STAT3−/− mice, probably due to the difference of age (IL-10−/− mice were 16 wk old, whereas Mφ-STAT3−/− mice were 9 wk old). Higher levels of STAT3 phosphorylation were observed in the IL-10−/− mice than in the Mφ-STAT3−/− mice (cf. lanes 5 and 6 and lanes 7 and 8). Furthermore, in the TNBS-induced colitis, one of the two mice (TNBS-1) showed much more severe symptoms along with stronger STAT3 phosphorylation than those of the other (TNBS-2; cf. lanes 11 and 12). The severity of colitis in the CD4+ T cell–transplanted SCID mouse was rather mild compared with other colitis that we examined in this study, and STAT3 phosphorylation in the colon was weak (lane 13). These data suggest that the levels of STAT3 phosphorylation were well correlated to the severity of colitis.
Figure 1.



Expression and activation of STATs in human and mouse colon tissues. (A) Colon tissue extracts from the normal region (NR) of colon cancer patients (lane 1), those from UC or CD patients (lanes 2–4), or those from Balb/c mice treated without (day 0) or with 4% DSS for 7 d (lanes 5 and 6) were immunoblotted with antiphospho-STATs (PY-STAT) or anti-STAT antibodies. As a positive control (pos), IFN-γ–treated NIH-3T3 cells (for STAT1), LIF-treated CMT cells (STAT3), IL-3–treated Ba/F3 cells (STAT5), and IL-4–treated 32D cells (STAT6) are shown in lane 7. We analyzed more than seven patients and three DSS-treated mice, obtaining similar results. (B) STAT3 activation in UC and IC patients (lanes 1 and 2) as well as several mouse models (lanes 3–13). Colon samples were obtained from 10–15-wk-old WT C57BL6 mice (lanes 3 and 4), 16-wk-old IL-10−/− mice (lanes 5 and 6), 9-wk-old Mφ-STAT3−/− mice (lanes 7 and 8), 15-wk-old TCR−/− mouse (lane 9), TNBS-treated (lanes 11 and 12) or untreated (lane 10) mice, and CD4+ T cell–transplanted SCID mouse (lane 13). (C) Immunohistochemical detection of activated STAT3 in DSS-induced colitis. C57BL6 mice were treated with (b and d) or without (a and c) 2% DSS for 5 d, and colon tissues were fixed with 10% formalin. Slides with sectioned tissues of 7-μm thickness were immunostained with anti–phospho-STAT3–specific antibodies (c and d) or control IgG (a and b) and were lightly stained with hematoxylin.
We examined in which cell types STAT3 was being activated by using immunohistochemistry with anti–phospho-STAT3–specific antibodies (Fig. 1 C). Nuclear accumulation of activated STAT3 was observed in epithelial cells as well as lamina propria cells in DSS-induced mouse colitis but not in untreated colon, indicating that STAT3 is activated not only in infiltrated cells but also in epithelial cells in colitis.
STAT3 has been shown to be mainly activated by IL-6–related cytokines, and the elevation of the IL-6 level has been reported in DSS-induced colitis. On the other hand, IFN-γ has been shown to activate mainly STAT1. For this reason, we investigated the role of STAT3 in the pathology of DSS-induced colitis using IL-6−/− and IFN-γ−/− mice in a C57BL/6 background (Fig. 2). All IL-6+/+ mice developed severe colitis characterized by loss of weight, marked epithelial hyperplasia, extensive leukocyte infiltration, and goblet cell depletion. Inflammatory infiltrates, which consisted mainly of mononuclear cells, were observed in the lamina propria in WT mice. These changes were not very obvious in IL-6−/− mice (Fig. 2a and Fig. b). On the other hand, IFN-γ–deficient mice developed colitis as severely as WT mice. Then, we compared STAT3 activation in WT and IL-6−/− mice. As shown in Fig. 2 C, STAT3 phosphorylation in the colon of WT mice was much more intensive than that in IL-6−/− mice. These data suggest that STAT3 activation in intestinal cells contributes to the development of colitis and constitutive activation of STAT3 may prevent healing of colitis.
Figure 2.


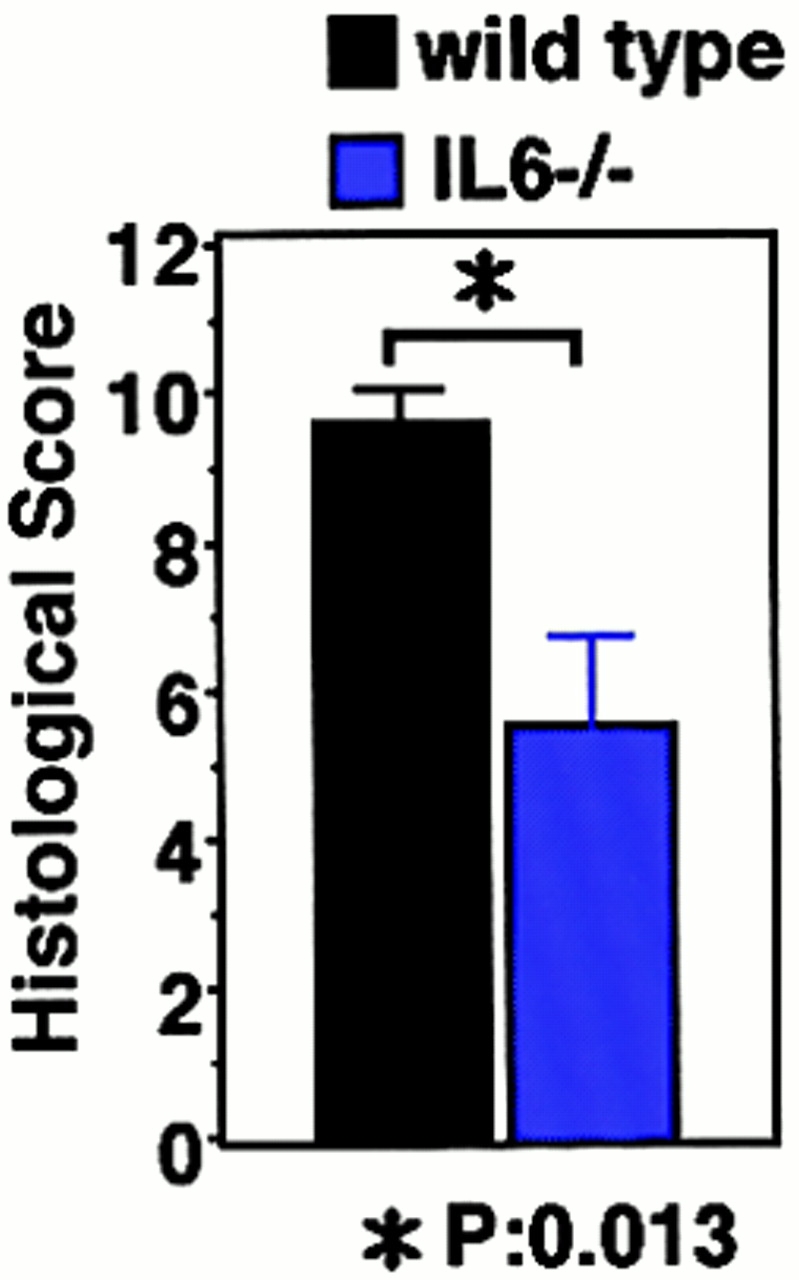

DSS-induced colitis in IL-6−/− and IFN-γ−/− mice. (A) Time course of body weight loss. WT C57BL6 mice or IL-6−/− or IFN-γ−/− mice (n = 3 for each knockout mice) were treated with 2% DSS for indicated periods, and body weight was measured daily. Relative body weight (%) compared with that of day 0 is plotted with a standard error. Results were analyzed using the t test. A P value of <0.05 was considered to be statistically significant. (B) Histological section of an ulcer with hematoxylin and eosin staining. Pathology was graded on a scale of 0–12. Data represent the average with SEM for the group. The average score for IL-6−/− mice, but not IFN-γ−/− mice, was significantly different from that for the WT mice (P = 0.013). Statistical analysis was performed using the t test. (C) STAT3 phosphorylation in the colon of WT and IL-6−/− mice treated with DSS. Colon tissue extracts from three independent mice of each strain were immunoblotted with anti–phospho-STAT3 (αPY-STAT3) and anti-STAT3 (αSTAT3) antibodies.
Correlation between CIS3 Induction and STAT3 Activation in Colitis.
As CIS3/SOCS-3 and JAB/SOCS-1 have been suggested to be involved in the negative regulation of inflammatory cytokine signaling, including IL-6 and IFN-γ, we investigated how CIS3 and JAB are implicated in colitis. We did not observe a drastic increase in JAB message in DSS-induced colitis (see Fig. 4) or human colitis patients (data not shown). Total RNA was isolated from colon samples, and CIS3 mRNA expression was detected using Northern hybridization (Fig. 3 A). An elevated CIS3 mRNA level was observed in the colon of Balb/c mice treated with 4% DSS for 7 d (lane 4). CIS3 expression was also elevated in the colon of TCR−/− mice (lane 6) compared with that of WT syngenic mice (lane 5). Elevated expression of CIS3 was also observed in all of the chronic colitis models described in Fig. 1 B including IL-10−/− mice (lines 9 and 10), Mφ-STAT3−/− mice (lines 11 and 12), TNBS-induced colitis (lanes 13 and 14), and CD45RBhighCD4+ T cell–mediated colitis (lane 15). Interestingly, the expression levels of CIS3 mRNA were not directly correlated to the extent of STAT3 phosphorylation in these model mice (cf. Fig. 1 B). CIS3 levels in Mφ-STAT3−/− mice were higher than those in IL-10−/− mice (cf. Fig. 3 A, lanes 9 and 10 and 11 and 12), whereas STAT3 phosphorylation in IL10−/− mice was stronger than that in Mφ-STAT3−/− mice (Fig. 1 B, lanes 5 and 6 and 7 and 8). Furthermore, TNBS-2 mouse (Fig. 3 A, lane 14) exhibited higher level of CIS3 expression but lower level of STAT3 activation (Fig. 1 B, lane 12) compared with the TNBS-1 (Fig. 1 B, lane 11, and Fig. 3 A, lane 13). These data suggest that STAT3 activation and CIS3 induction are strongly correlated, but these are not simple parallel events.
Figure 4.
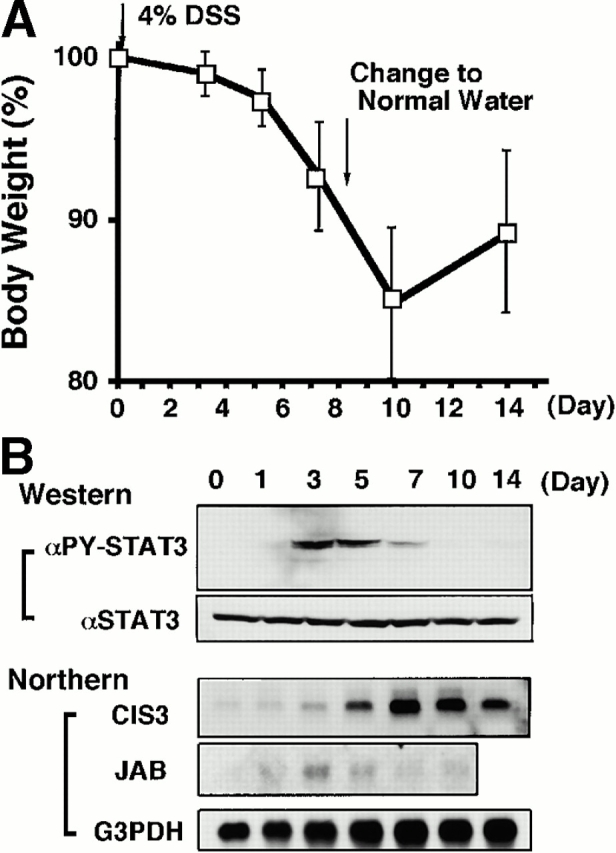
Time course of DSS-induced body weight loss (A), STAT3 activation, and induction of CIS3 and JAB (B) in mouse colon after DSS treatment. Balb/c mice were treated with 4% DSS for indicated periods. (A) Mice (n = 3 for each DSS concentration) were treated with indicated concentrations of DSS, and body weight was measured daily. The relative body weight (%) compared with that of day 0 is plotted with a standard error. (B, Western) Colon tissue extracts from mice treated with DSS for indicated periods were immunoblotted with anti–phospho-STAT3 (αPY-STAT3) and anti-STAT3 (αSTAT3) antibodies. (B, Northern) Total RNA was extracted from the colon. Northern hybridization using cDNA probes for mouse CIS3, JAB, and control G3PDH was performed.
Figure 3.



Expression of CIS3 in colon tissue obtained from several colitis model mice and human patients. (A; Northern) Mucosal samples were taken from intestinal resection. Total RNA was extracted from NIH-3T3 cells treated without (lane 1) or with 1,000 U/ml IFN-γ for 30 min (lane 2), colon tissue obtained from Balb/c mice without (lane 3) or with DSS-induced colitis (lane 4) for 7 d, TCR−/− mice (lane 6), or control syngenic mice (lane 5). RNA samples were also obtained from colon tissues from the same mice described in the legend to Fig. 1; WT C57BL6 mice (lanes 7 and 8), IL-10−/− mice (lanes 9 and 10), Mφ-STAT3−/− mice (lanes 11 and 12), TNBS-treated (lanes 13 and 14), and CD4+ T cell–transplanted SCID mice (lane 15). Total RNA was also extracted from human mucosal samples taken from intestinal resections from non-IBD control patients (lanes 16 and 17), or UC (lanes 18 and 19), CD (lanes 21 and 22), or IC (lane 23) patients. The human cell lines Raji (lane 24) and HEL (lane 25) were used as negative and positive controls for CIS3 mRNA, respectively. Northern hybridization was performed with cDNA probes for coding regions of human and murine CIS3 and control G3PDH. We analyzed more than nine UC or CD patients and obtained similar results. Representative data are shown. (B; Western) Expression of CIS3 protein in colitis. Cell extracts were prepared from HCT cells treated without (lane 1) or with (lane 2) 2 ng/ml LIF for 3 h, colon tissues of Balb/c mice treated without (lane 3) or with (lane 4) 4% DSS for 7 d, or colon tissues from non-IBD control (lane 5) or UC patients (lane 6). CIS3 protein was immunoprecipitated with a rabbit anti-CIS3 polyclonal antibody and then immunoblotted with a mouse anti-CIS3 monoclonal antibody. (C) Localization of CIS3 expression examined by in situ hybridization. Balb/c mice were treated with 4% DSS in the drinking water for 7 d. Colon tissues from DSS-treated (a, b, and d) or untreated (c) mice were cryostat sectioned to 7-μm thickness. After fixation of samples with 4% formaldehyde, hybridization and detection were performed with DIG-labeled CIS3 antisense (b–d) and sense (a) probes. The samples were lightly stained with 1% methyl green. Original magnification: ×20 (a and b), ×40 (c), and ×100 (d).
We also examined CIS3 levels in colon samples from UC, CD, and IC patients. In these cases, CIS3 expression was higher than normal in the colon samples (Fig. 3 A, lanes 18–23; representative data from seven patients are shown). As shown in Fig. 3 B, the expression of CIS3 in protein level was confirmed by immunoblotting with anti-CIS3 monoclonal antibody. CIS3 protein was induced by treatment with LIF in HCT, a human colon carcinoma cell line (lanes 1 and 2). A high level of CIS3 protein was detected in the colon of DSS-treated mice (lane 4) as well as in a UC patient (lane 6), but not in normal colons (lanes 3 and 5).
To determine which cells express CIS3, we performed in situ hybridization in a DSS-treated colon (Fig. 3 C). CIS3 mRNA was mainly detected in hyperplastic epithelial cells and lamina propria cells (Fig. 3 C, panels b and d) but also weakly in lymphoid cells (Fig. 3 C, panel b). Sense oligonucleotides did not hybridize at all (Fig. 3 C, panel a). CIS3 expression was not very obvious in a normal colon (Fig. 3 C, panel c). These data suggested that STAT3 phosphorylation and CIS3 expression occurred at the same region of DSS-induced colitis.
Time Course of STAT3 Activation and CIS3 Induction in DSS-induced Colitis.
We postulated that CIS3 is involved in the pathological deterioration of colitis. To explore this possibility further, we examined the time course of STAT3 activation and CIS3 expression in DSS-induced colitis. The colon inflammation became severe at day 7, and mice recovered from colitis on day 14, after DSS had been removed from the drinking water at day 8 (Fig. 4 A). As shown in Fig. 4 B, STAT3 phosphorylation was transiently observed in DSS-treated mice, reached a plateau on days 3–5, and decreased thereafter. JAB was not induced very intensively compared with CIS3 (Fig. 4 B, Northern). On the other hand, CIS3 mRNA levels reached a plateau on day 7 and maintained a high level until day 10, gradually decreasing thereafter as mice recovered from their severe pathological condition. CIS3 mRNA was not elevated in DSS-treated IL-6−/− mice (data not shown), suggesting that CIS3 expression is regulated by the IL-6/STAT3 system. This is consistent with our previous observation that CIS3 is induced by IL-6 via STAT3 in hematopoietic cells 31. These data suggest that STAT3 is activated as a result of an increase in the IL-6 level and that the CIS3 level increases thereafter. Subsequently, CIS3 may reduce the severity of colitis through the suppression of STAT3 activation.
We confirmed the induction of CIS3 in colon epithelial cells and the effect of forced expression of CIS3 on STAT3 activity using a cultured intestinal epithelial cell line, CMT-93 24. As shown in Fig. 5 A, STAT3 activation is also ahead of CIS3 induction in CMT cells, and the level of CIS3 was maintained even after STAT3 phosphorylation disappeared. It has been demonstrated that expression of the proinflammatory cytokine IL-1β is regulated by STATs 33. As shown in Fig. 5B and Fig. C, forced expression of CIS3 suppressed LIF-induced STAT3 phosphorylation as well as IL-1β production. These data suggest that CIS3 is induced via STAT3 and that CIS3 suppresses STAT3 activation in a colon-derived cell line. Repression of STAT3 activity by CIS3 in epithelial cells may contribute to suppression of the severity of colitis by reducing proinflammatory cytokine production.
Figure 5.
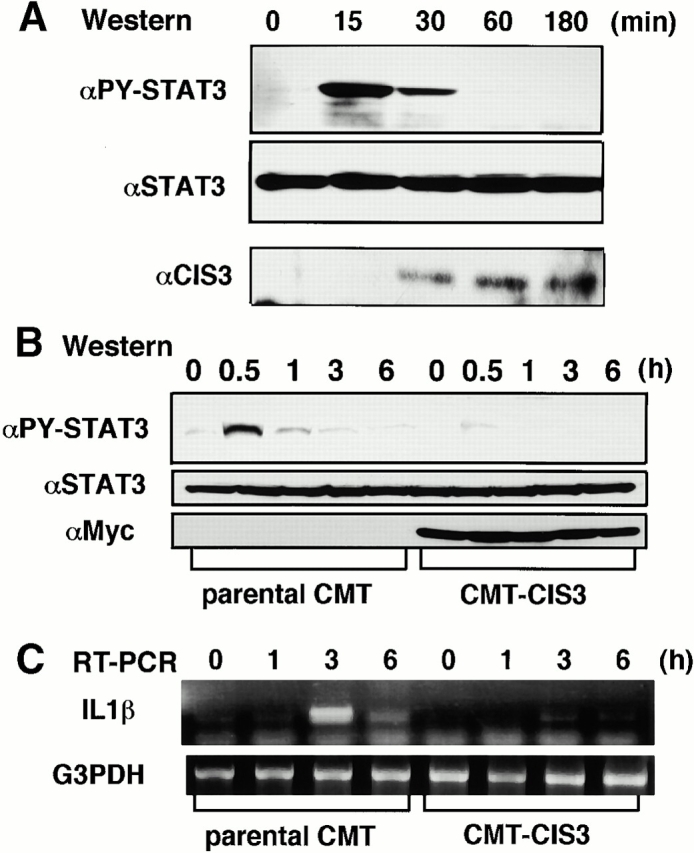
Activation of STAT3, induction of CIS3 protein, and the effect of forced expression of CIS3 on LIF-induced STAT3 activation and IL-1β production in a mouse colon cell line, CMT. (A, Western) Cells grown in six-well dishes were stimulated with 10 ng/ml LIF for indicated periods, and total cell extracts were then immunoblotted with anti–phospho-STAT3, anti-STAT3, and rabbit anti-CIS3 polyclonal antibodies. In B and C, CMT cells stably transformed with CIS3 (CMT-CIS3) were stimulated with 10 ng/ml LIF for indicated periods, and then STAT3 phosphorylation and IL-1β production were measured by immunoblotting (B, Western) and RT-PCR analysis (C, RT-PCR), respectively. Three independent CMT transformants were examined and similar results were obtained.
A Mutant Form of JAB/SOCS1 Reversed the Negative Effect of JAB and CIS3 and Induced More Severe Colitis in Tg Mice.
To investigate the physiological meaning of STAT3 activation and CIS3 expression more precisely in DSS-induced colitis, we developed a dominant negative form of CIS3 and JAB. We have previously shown that both JAB and CIS3 possess two JAK binding sites, kinase inhibitory region (KIR), and an SH2 domain 15. We found that a point mutation either in KIR or the SH2 domain (F59D-JAB or R105E-JAB) strongly augmented LIF-induced STAT3 activation (data not shown). We examined the effect of F59D-JAB on JAB- and CIS3-mediated suppression of STAT3 activation. As shown in Fig. 6A and Fig. B, overexpression of F59D-JAB reverted the negative effect of both WT JAB and CIS3 on LIF-induced STAT3 reporter gene activation (Fig. 6 A) as well as STAT3 phosphorylation (Fig. 6 B) in 293 cells. Although the precise molecular mechanism is under investigation and will be published elsewhere, we speculate that F59D-JAB masks the phosphorylated activation loop of JAKs, thereby preventing the binding of JAB or CIS3 to the catalytic pocket of the JAK tyrosine kinase domain.
Figure 6.
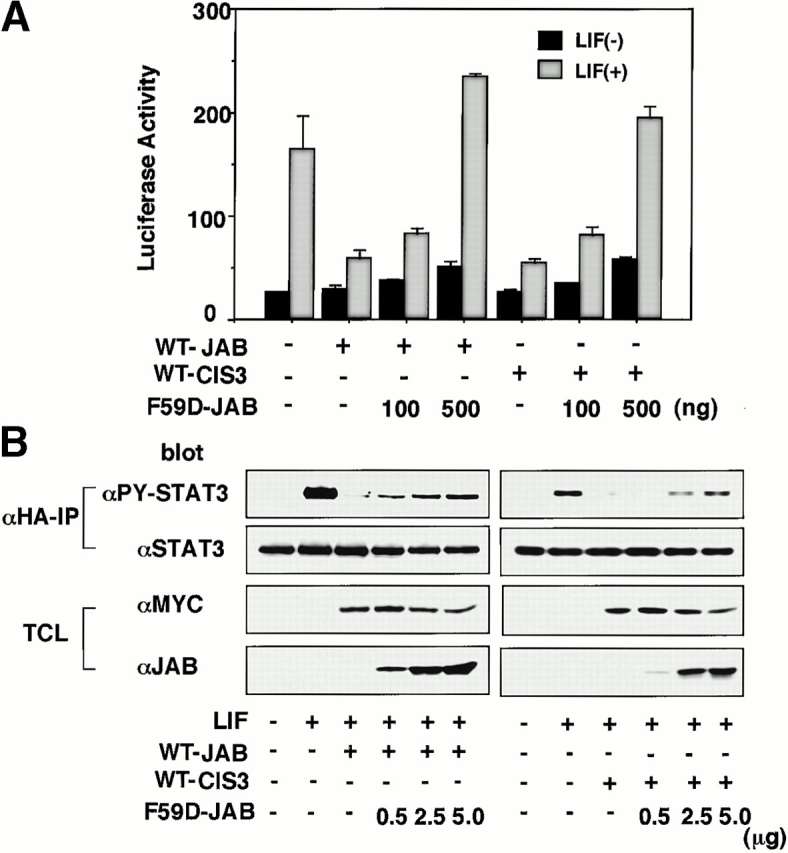
The effect of F59D-JAB on STAT3 repression by WT JAB and CIS3 in 293 cells. (A) 293 cells were transfected with 10 ng WT JAB or CIS3, APRE-luciferase reporter gene, and indicated amounts of F59D-JAB plasmid (ng). 2 d after transfection, LIF-induced STAT3 activation was measured by luciferase assay. (B) 293 cells were transfected with 100 ng Myc-tagged WT JAB or CIS3 and hemagglutinin (HA)-tagged STAT3 with indicated amounts of F59D-JAB. After stimulation with 100 ng/ml LIF for 30 min, cell extracts were immunoprecipitated with anti-HA, and then the immunoprecipitates (αHA-IP) were blotted with anti-phosphorylated STAT3 (αPY-STAT3) or anti-STAT3 (αSTAT3). Total cell extracts (TCL) were blotted with anti-Myc (αMyc) and anti-JAB (αJAB) antibodies to confirm the expression of Myc-JAB, Myc-CIS3, and F59D-JAB.
To examine the effect of F59D-JAB expression in DSS-induced colitis, we created four F59D-JAB Tg mice using a β-actin promoter 14. Expression was observed in most tissues except for liver and brain in a typical Tg line (Fig. 7 A). In the colon, F59D-JAB protein was expressed in most cells including goblet, epithelial, and lamina propria cells (Fig. 7 B). As shown in Fig. 8 A, Tg lines exhibited stronger STAT3 activation in the colon of DSS-treated mice than in that of their WT littermates. Our WT littermates were slightly resistant against DSS-induced colitis compared with C57BL/6 mice. As shown in Fig. 8B and Fig. c, all F59D-JAB Tg mice exhibited a more extensive loss of weight (Fig. 8 B) and a more severe epithelial hyperplasia and goblet cell depletion than their WT littermates (Fig. 8 C) when they received appropriate concentrations of DSS in water. These data suggest that expression of F59D-JAB resulted in hyperactivation of STAT3 and a more severe pathological profile in DSS-induced colitis.
Figure 7.


Expression of F59D-JAB in Tg mice. (A) RT-PCR analysis for tissues from F59D-JAB Tg mouse. 1 μg total RNA from each tissue was used as a template to detect F59D-JAB and control G3PDH mRNAs. Similar levels of expression were observed in the other three Tg lines. (B) Immunohistochemical detection of F59D-JAB in the colon of WT littermate or F59D-JAB Tg mouse (Tg).
Figure 8.
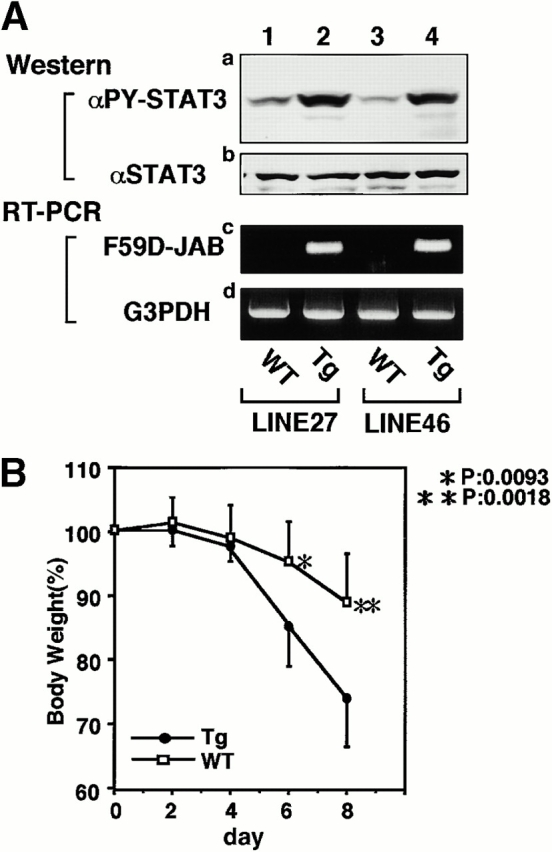

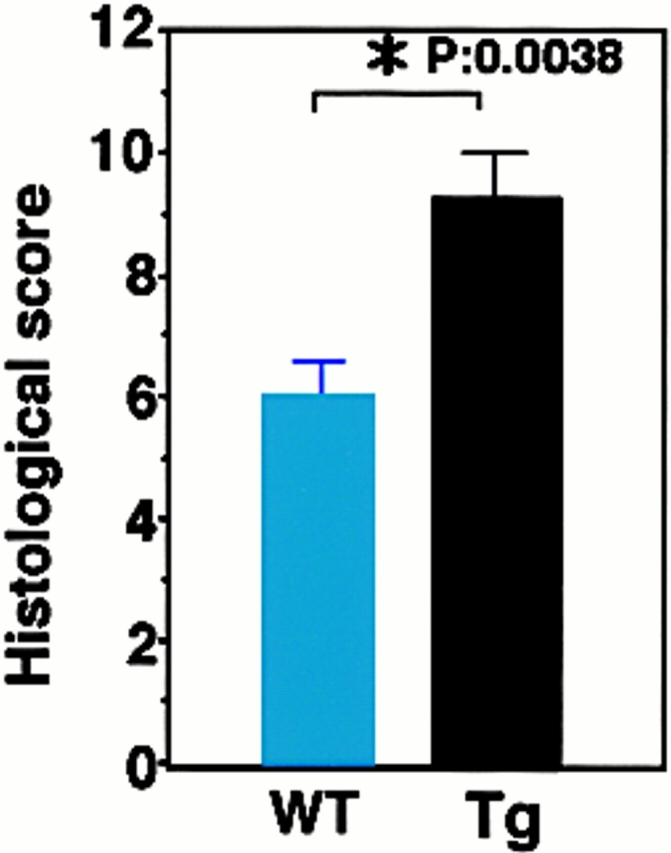
DSS-induced STAT3 activation and colitis in F59D-JAB Tg mice. (A, panels a and b; Western) colon tissue extracts from two lines of WT littermate or Tg mice treated with 2% DSS for 7 d mice were immunoblotted with anti–phospho-STAT3 (αPY-STAT3) and anti-STAT3 (αSTAT3) antibodies. (A, panels c and d; RT-PCR) F59D-JAB expression in the colon sample was confirmed by RT-PCR analysis. Results from two representative independent Tg lines (line 27 and line 46) are shown. (B) Time course of DSS-induced body weight loss. WT littermates or F59D-JAB Tg mice were treated with 2% DSS for indicated periods, and body weight was measured daily. We examined four independent Tg lines (total n = 10). Relative body weight (%) compared with that of day 0 is plotted with a standard error. Results were analyzed using the t test. A P value of <0.05 was considered to be statistically significant. (C) Histological section of an ulcer with hematoxylin and eosin staining. Pathology was graded on a scale of 0–12. Data represent the average with SEM for the group.
Next, we measured IL-6 and IL-10 levels in colon organ culture from WT and Tg mice. As shown in Fig. 9, IL-6 and IL-10 levels were not significantly different, suggesting that the more severe colitis in Tg mice was not due to higher levels of IL-6 or lower levels of IL-10. Taken together, our data suggest that sustained activation of STAT3 perpetuated the development of DSS-induced colitis and that CIS3 induction by STAT3 may play an important role in STAT3 repression and in the termination or healing of colitis.
Figure 9.
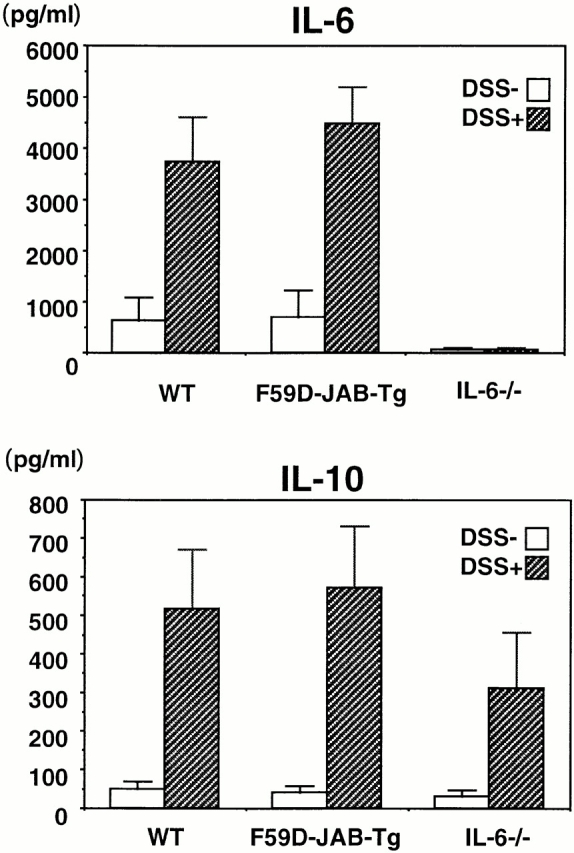
IL-6 and IL-10 production in organ cultures from colons of DSS-treated mice. WT, F59D-JAB-Tg, or IL-6−/− mice were treated with 2% DSS for 7 d, and cytokine levels were measured by standard enzyme immunoassay in 24-h organ culture of the distal region of the colon samples (n = 2).
Discussion
In this study, we focused on the role of STAT3 activation and CIS3/SOCS3 expression in colitis using model mice. STAT3 activation was found in almost all colitis models examined in this study including DSS treatment, TCR-α gene knockout, CD4+ T cell transfer, IL-10 knockout, macrophage- and neutrophil-specific STAT3 conditional knockout, as well as TNBS immunization. Conditional knockout of STAT3 in macrophages and neutrophils resulted in chronic enterocolitis with age 25. This is probably due to the enhancement of the Th1 response by the block of antiinflammatory cytokine IL-10 signaling. Colitis in TCR-α knockout is IL-4 dependent 32 and both Th1- and Th2-type cytokines are involved in the development of distinct forms of TNBS-induced colitis 29. Therefore, STAT3 is activated in many type of colitis irrespective of the cause, suggesting that STAT3 activation is related to the progression or development of the disease rather than the initiation. We frequently observed that the levels of STAT3 phosphorylation correlated to the severity of colitis, which supports this hypothesis. Therefore, STAT3 activation may more likely be involved in the process of controlled inflammation rather than colitis itself.
As IL-6 is a major activator of STAT3 in inflammation, and overproduction of IL-6 has been found in many type of colitis, IL-6 may play an important role in the development of colitis. Recently, Yamamoto et al. 6 and Atreya et al. 7 reported that administration of anti–IL-6 receptor monoclonal antibodies suppressed colitis induced by transplantation of CD45RBhighCD4+ T cells into SCID mice. These studies demonstrated that IL-6 perpetuates colitis by protecting lamina propria T cells from apoptosis. We also showed that IL-6–deficient mice developed less severe colitis induced by DSS, suggesting that IL-6 also play a promotive role in T cell–independent, acute colitis. Although IL-6 also activates the Ras–mitogen-activated protein (MAP) kinase pathway, phosphorylation of MAP kinase (Erk2) was not often observed in DSS-induced colitis. Therefore, we focused on STAT3 activation in IL-6 signaling in colitis. The mechanism whereby IL-6/STAT3 increases the severity of colitis is not clear at present. It has been suggested that IL-6 protects the apoptosis of CD4+ T cells in developing colitis 6 7, leading to the deterioration of the pathological condition. This may also be possible in epithelial cells, resulting in the hyperproliferation of both epithelial and lamina propria cells in response to IL-6. IL-6–activated macrophages and colon epithelial cells have been shown to secrete inflammatory cytokines, including TNF-α and IL-1β, which contribute to the development of colitis. These cytokines activate nuclear factor (NF)-κB pathway. TNF-α has been shown to play an important role in the development of inflammation, including colitis 2 5. However, as IL-6−/− mice developed less severe DSS-induced colitis as well as antigen- or collagen-induced arthritis 8 9 10, STAT3 pathway is also important for inflammation. Further study is necessary to clarify the relationship between TNF-α/NF-κB and IL-6/STAT3 pathways.
STAT3 probably plays both positive and negative roles in the development of inflammation. To assess the role of STAT3 in vivo more precisely, the STAT3 gene was disrupted in a tissue- or cell-specific manner by the Cre-loxP recombination system. In STAT3-deficient T cells, IL-6–induced T cell proliferation was impaired due to the lack of IL-6–mediated prevention of apoptosis 34, which is consistent with the protective effect of anti–IL-6 receptor monoclonal antibody against T cell–mediated colitis 6 7. Suppression of epithelial apoptosis and delayed mammary gland involution was observed in mice with a conditional knockout of STAT3 in the mammary gland 35. Thus, hyperactivation of STAT3 may induce apoptosis of epithelial cells in certain conditions. These data suggest that STAT3 plays a different, sometimes opposite, role in the apoptosis, proliferation, and cell functions of different cell types. However, as DSS-induced colitis is, at least, T cell independent, STAT3 activation may have a prominent role to promote inflammation by enhancing inflammatory cytokine production from epithelial cells or inducing proliferation of epithelial cells in DSS-treated mice. Therefore, suppression of STAT3 activity may be necessary for the elimination of inflammation in the colon.
We found that CIS3 was highly expressed in epithelial and lamina propria cells in the colon of DSS-treated mice but not in normal mice. A high level of expression of CIS3 was also observed in human UC and CD as well as in TCR-deficient mice. These data suggest that CIS3 expression is observed not only in acute colitis but also in other kinds of chronic colitis in which activated T cells are involved. However, STAT3 activation and CIS3 expression were not simply parallel events. A time course experiment indicated that STAT3 activation was 1 d ahead of CIS3 induction; STAT3 activation became apparent during days 3–5 and decreased thereafter, whereas CIS3 expression was induced at day 5 and maintained high levels thereafter. Based on the evidence that forced expression of CIS3 can inhibit IL-6–mediated STAT3 activation, CIS3, which is induced by STAT3 activation, acts as a negative feedback regulator of STAT3. The negative effect of CIS3 on STAT3 activation in colon epithelial cells is confirmed by the forced expression of CIS3 in CMT-93 cells (Fig. 5). These data raise the possibility that CIS3 expression is one, if not the only one, mechanism that negatively regulates inflammatory reaction in colitis. This is consistent with our observation in F59D-JAB Tg mice, which exhibited a severe inflammatory reaction along with the hyperactivation of STAT3. Induction of CIS3 may terminate further activation of STAT3, which contributes to the healing of colitis.
However, we observed both strong activation of STAT3 and high level of CIS3 in CD and UC patients. This may be explained by the possibility that the CIS3 expression in chronic symptoms may not reach a high enough level to shut off STAT3 activation completely because of an extremely high IL-6 level in chronic patients. Alternatively, CIS3 function may be suppressed by posttranslational modification or by the altered expression of interacting proteins. CIS3/SOCS3 has been shown to be phosphorylated and probably ubiquitinated in response to cytokine stimulation 30 36. These modifications and rapid degradation by proteasome could be reasons for the incomplete suppression of STAT3 by CIS3 in chronic inflammation. As CIS3 is induced not only by IL-6 but also by many other cytokines and growth factors, induction of CIS3 by antiinflammatory cytokines or growth factors may also be important for the downregulation of STAT3 activation and lead to the healing of colitis.
We found that an F59D-JAB mutant could overcome the negative effect of WT CIS3 and JAB in cultured cells. A detailed analysis of the mechanism of this “dominant negative” effect is under way. This mutant will be a useful tool to investigate the role of CIS3 and JAB in diseases. Our F59D-JAB Tg mice grew normally and did not exhibited any significant differences, from their WT littermates, without stimulation. This is very different from JAB or CIS3 knockout mice, which exhibit neonatal or embryonic lethality, respectively. This discrepancy may be due to a low level of expression of F59D-JAB in our Tg mice, which can only partially overcome the negative effect of JAB and CIS3. A high level of F59D-JAB expression may be lethal in Tg mice, because we obtained no Tg pups when we injected other tagged constructs that ensure a higher level of F59D-JAB expression into zygotes. To confirm the role of CIS3 or JAB in intestinal inflammation, a study of the conditional knockout of these genes is being undertaken.
Acknowledgments
We thank Ms. H. Ohgusu, Ms. M. Sasaki, and Ms. I. Ueno for their excellent technical assistance.
This work was supported in part by grants from the Ministry of Science, Education, and Culture of Japan, the TORAY Research Foundation, the Welfide Medicina Research Foundation, Foundation for Advancement of International Science, and the Mitsubishi Foundation.
Footnotes
Abbreviations used in this paper: CD, Crohn's disease; CIS, cytokine-inducible SH2 protein; DIG, digoxigenin; DSS, dextran sulfate sodium; IBD, inflammatory bowel disease; IC, infectious colitis; JAK, Janus kinase; LIF, leukemia inhibitory factor; RT, reverse transcription; STAT, signal transducer and activator of transcription; Tg, transgenic; UC, ulcerative colitis; WT, wild-type.
References
- Podolsky D.K. Inflammatory bowel disease. N. Engl. J. Med. 1991;325:928–937. doi: 10.1056/NEJM199109263251306. [DOI] [PubMed] [Google Scholar]
- Strober W., Neurath M.F. Immunological diseases of the gastrointestinal tract. In: Rich R.R., editor. Clinical Immunology. Mosby; St. Louis, MO: 1995. pp. 1401–1428. [Google Scholar]
- Gross V., Andus T., Caesar I., Roth M., Schölmerich J. Evidence for continuous stimulation of IL-6 production in Crohn's disease. Gastroenterology. 1992;102:514–519. doi: 10.1016/0016-5085(92)90098-j. [DOI] [PubMed] [Google Scholar]
- Fuss I.J., Neurath M., Boirivant M., Klein J.S., de la Motte C., Strong S.A., Fiocchi C., Strober W. Desperate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. J. Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- Rogler G., Andus T. Cytokines in inflammatory bowel disease. World J. Surg. 1998;22:382–389. doi: 10.1007/s002689900401. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Yoshizaki K., Kishimoto T., Ito H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J. Immunol. 2000;164:4878–4882. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- Atreya R., Mudter J., Finotto S., Mullberg J., Jostock T., Wirtz S., Schutz M., Bartsch B., Holtmann M., Becker C. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammationevidence in Crohn disease and experimental colitis in vivo. Nat. Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- Mendel I., Katz A., Kozak N., Ben-Nun A., Revel M. Interleukin-6 functions in autoimmune encephalomyelitisa study in gene-targeted mice. Eur. J. Immunol. 1998;28:1727–1737. doi: 10.1002/(SICI)1521-4141(199805)28:05<1727::AID-IMMU1727>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Ohshima S., Saeki Y., Mima T., Sasai M., Nishioka K., Nomura S., Kopf M., Katada Y., Tanaka T., Suemura M., Kishimoto T. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc. Natl. Acad. Sci. USA. 1998;95:8222–8226. doi: 10.1073/pnas.95.14.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzi T., Fattori E., Lazzaro D., Costa P., Probert L., Kollias G., De Benedetti F., Poli V., Ciliberto G. Interleukin 6 is required for the development of collagen-induced arthritis. J. Exp. Med. 1998;187:461–468. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Farrell A.M., Liu Y., Moore K.W., Mui A.L. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanismsevidence for Stat3-dependent and -independent pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1006–1018. doi: 10.1093/emboj/17.4.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H., Sasaki A., Yoshimura A. Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol. 2000;18:143–164. doi: 10.1146/annurev.immunol.18.1.143. [DOI] [PubMed] [Google Scholar]
- Yoshimura A., Ohkubo T., Kiguchi T., Jenkins N.A., Gilbert D.J., Copeland N.G., Hara T., Miyajima A. A novel cytokine-inducible gene CIS encodes an SH2 containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:2816–2826. doi: 10.1002/j.1460-2075.1995.tb07281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A., Seki Y., Kubo M., Ohtsuka S., Suzuki A., Hayashi I., Tsuji K., Nakahata T., Okabe M., Yamada S., Yoshimura A. Suppression of STAT5 functions in liver, mammary glands, and T cells in cytokine-inducible SH2-containing protein 1 transgenic mice. Mol. Cell. Biol. 1999;19:6396–6407. doi: 10.1128/mcb.19.9.6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H., Misawa H., Sakamoto H., Masuhara M., Sasaki A., Wakioka T., Ohtsuka A., Imaizumi T., Matsuda T., Ihle J.N., Yoshimura A. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1309–1320. doi: 10.1093/emboj/18.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuhara M., Sakamoto H., Matsumoto A., Suzuki R., Yasukawa H., Mitsui K., Wakioka T., Tanimura S., Sasaki A., Misawa H. Cloning and characterization of novel CIS family genes. Biochem. Biophys. Res. Commun. 1997;239:439–446. doi: 10.1006/bbrc.1997.7484. [DOI] [PubMed] [Google Scholar]
- Starr R., Willson T.A., Viney E.M., Murray L.J., Rayner J.R., Jenkins B.J., Gonda T.J., Alexander W.S., Metcalf D., Nicola N.A., Hilton D.J. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Naka T., Narazaki M., Hirata M., Matsumoto T., Minamoto S., Aono A., Nishimoto N., Kajita T., Taga T., Yoshizaki K. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Naka T., Matsumoto T., Narazaki M., Fujimoto M., Morita Y., Ohsawa Y., Saito H., Nagasawa T., Uchiyama Y., Kishimoto T. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc. Natl. Acad. Sci. USA. 1998;95:15577–15582. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R., Metcalf D., Elefanty A.G., Brysha M., Willson T.A., Nicola N.A., Hilton D.J., Alexander W.S. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc. Natl. Acad. Sci. USA. 1998;95:14395–14399. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander W.S., Starr R., Fenner J.E., Scott C.L., Handman E., Sprigg N.S., Corbin J.E., Cornish A.L., Darwiche R., Owczarek C.M. SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Marine J.C., Topham D.J., McKay C., Wang D., Parganas E., Stravopodis D., Yoshimura A., Ihle J.N. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/s0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Marine J.C., McKay C., Wang D., Topham D.J., Parganas E., Nakajima H., Pendeville H., Yasukawa H., Sasaki A., Yoshimura A., Ihle J.N. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999;98:617–627. doi: 10.1016/s0092-8674(00)80049-5. [DOI] [PubMed] [Google Scholar]
- Nguyen H.H., Boyaka P.N., Moldoveanu Z., Novak M.J., Kiyono H., McGhee J.T., Mestecky J. Influenza virus-infected epithelial cells present viral antigens to antigen-specific CD8+ cytotoxic T lymphocytes. J. Virol. 1998;72:4534–4536. doi: 10.1128/jvi.72.5.4534-4536.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Clausen B.E., Kaisho T., Tsujimura T., Terada N., Forster I., Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Kanauchi O., Nakamura T., Agata K., Mitsuyama K., Iwanaga T. Effects of germinated barley foodstuff on dextran sulfate sodium-induced colitis in rats. J. Gastroenterol. 1998;33:179–188. doi: 10.1007/s005350050067. [DOI] [PubMed] [Google Scholar]
- Cooper H.S., Murthy S.N., Shah R.S., Sedergran D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- Dieleman L.A., Ridwan B.U., Tennyson G.S., Beagley K.W., Bucy R.P., Elson C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- Dohi T., Fujihashi K., Kiyono H., Elson C.O., McGhee J.R. Mice deficient in Th1- and Th2-type cytokines develop distinct forms of hapten-induced colitis. Gastroenterology. 2000;119:724–733. doi: 10.1053/gast.2000.16500. [DOI] [PubMed] [Google Scholar]
- Sasaki A., Yasukawa H., Suzuki A., Kamizono S., Syoda T., Kinjyo I., Sasaki M., Johnston J.A., Yoshimura A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells. 1999;4:339–351. doi: 10.1046/j.1365-2443.1999.00263.x. [DOI] [PubMed] [Google Scholar]
- Suzuki R., Sakamoto H., Yasukawa H., Masuhara M., Wakioka T., Sasaki A., Yuge K., Komiya S., Inoue A., Yoshimura A. CIS3 and JAB have different regulatory roles in interleukin-6 mediated differentiation and STAT3 activation in M1 leukemia cells. Oncogene. 1998;17:2271–2278. doi: 10.1038/sj.onc.1202143. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A., Mizoguchi E., Bhan A.K. The critical role of interleukin 4 but not interferon gamma in the pathogenesis of colitis in T-cell receptor alpha mutant mice. Gastroenterology. 1999;116:320–326. doi: 10.1016/s0016-5085(99)70128-9. [DOI] [PubMed] [Google Scholar]
- Tsukada J., Waterman W.R., Koyama Y., Webb A.C., Auron P.E. A novel STAT-like factor mediates lipopolysaccharide, interleukin 1 (IL-1), and IL-6 signaling and recognizes a gamma interferon activation site-like element in the IL1B gene. Mol. Cell. Biol. 1996;16:2183–2194. doi: 10.1128/mcb.16.5.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Kaisho T., Yoshida N., Takeda J., Kishimoto T., Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosisgeneration and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- Chapman R.S., Lourenco P.C., Tonner E., Flint D.J., Selbert S., Takeda K., Akira S., Clarke A.R., Watson C.J. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of stat3. Genes Dev. 1999;13:2604–2616. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.G., Farley A., Nicholson S.E., Willson T.A., Zugaro L.M., Simpson R.J., Moritz R.L., Cary D., Richardson R., Hausmann G. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]