

Abstract
Colorectal cancer (CRC) is among the most prevalent cancers worldwide and represents a major public health challenge in the developed world. From the perspective of translational investigation, scientists have enormous opportunity to elucidate the molecular genetic mechanisms contributing to CRC pathogenesis since the majority of cancers arise from adenomatous precursor lesions. The process of adenoma growth and transformation is accompanied by cumulative mutations in dominant genetic pathways that confer a growth advantage. While this developmental process permits interrogation of informative pathways prior to the development of cancer, only a minority of adenomas progress to CRC. Accordingly, a major challenge for clinical translational investigators is to identify the molecular signatures that indicate increased likelihood for adenoma progression. By corollary, these molecular signatures include mutations in high penetrance alleles, including the Adenomatous Polyposis Coli (APC) gene as well as other alleles in the Wnt/β-catenin signaling pathway that specify increased genetic susceptibility to CRC. Interactions between these high penetrance alleles and other modifier genes as well as with environmental factors are of particular importance in understanding the complex network of events leading to CRC. This brief review will highlight three areas where important questions concerning genetic and environmental risk factors have fueled translational investigation into possible pathways leading to CRC.
There have been substantial advances in the last two decades in our understanding of the molecular pathways that lead to colorectal cancer (CRC), the results of research that demonstrated the role of mutational activation of oncogenes coupled with loss of function of tumor suppressor genes in a model that advanced the concept of finite, but cumulative mutational events (summarized in (1)). These studies in preclinical, animal models as well as cell-based models, clinical observational and randomized studies in humans have led to a more complete understanding of the role of both environmental and genetic factors in CRC pathogenesis.
One tangible result of this expanded scientific foundation is an emerging consensus that familial or inherited factors play an increasingly relevant role in our approach to patients with CRC(2). Examples of dominant genetic pathways include those governed by Adenomatous Polyposis Coli (APC) tumor suppressor gene, and also the microsatellite instability pathway whose tumor phenotype results from hereditary or acquired mutations in mismatch repair genes that lead to replication error (2). Inherited mutations in the APC tumor suppressor gene (ie familial adenomatous polyposis) account for a very small proportion (less than 1%) of all CRC, yet somatic mutations are found in over 80% of sporadic cases of CRC, suggesting that this is a key genetic event in most tumors (2). Similarly, inherited germline mutations in mismatch repair genes (ie Lynch syndrome) account for perhaps 2–3% of all cases of CRC, yet the functional somatic signature of this repair defect, microsatellite instability, is found in approximately 15% of cases of sporadic CRC (2).
However, despite these seminal advances in our understanding of the aberrant pathways that contribute to CRC pathogenesis, there is growing awareness of the complexity in mutational signatures that accompany CRC. For example, sequence information from the consensus coding sequence database demonstrated that individual CRC tumors accumulated approximately 90 mutant genes, but only a subset were felt to participate in tumorigenesis, the remainder representing “passenger” mutations (3). This apparent redundancy highlights a major challenge in translational research, namely understanding how complex genetic traits and environmental factors interact and how to design experimentally testable models that might reveal unanticipated interactions between dominant and recessive pathways.
Cyclooxygenases, aspirin and the genetics of CRC chemoprevention
There has been an exponential increase in the last two decades in our understanding of the pathways involved in cyclooxygenase (cox) signaling and their relationship to CRC pathogenesis, driven largely by studies demonstrating that sustained aspirin use or chronic intake of non-steroidal anti-inflammatory drugs decreases both adenoma formation and recurrence and also decreases the incidence of CRC (4–6). The mechanisms and pathways by which cox-2 inhibition in particular mediates chemoprevention in human subjects as well as in relevant preclinical animal models has been the focus of intense interest.
PGE2, the dominant prostanoid produced through the cox-2 pathway, is synthesized and secreted by stromal fibroblasts as well as by epithelial cells, and transduces signals in epithelial cells through interactions mediated through one or more of four receptors, principally (at least in the colonic epithelium) by endoprostanoid (EP) receptor subtypes 2 and 4. Activation of EP receptors leads to an array of downstream events, including activation of the epidermal growth factor receptor (EGFR), activation of the phosphatidyl-3-kinase (PI3K) and Akt pathways, which together result in the release of glycogen synthase kinase 3β from its stable complex with axin and β-catenin (7). Inactivation of glycogen synthase kinase 3β stabilizes β-catenin and permits its nuclear translocation, which in turn results in growth stimulation through transcriptional activation of TCF/LEF family members (7). There is additional complexity however, since PGE2 signaling through EP2 and EP4 leads to a feed-forward loop in which PGE2 itself stimulates cox-2 expression and thereby further enhances prostaglandin production (8, 9). This summary overview illustrates some of the complexity in dissecting functional intersections between cox-2 dependent prostaglandin production and the Wnt/β-catenin/TCF signaling pathways of CRC pathogenesis.
Several features of the cox-2/CRC pathogenesis pathway interactions have been validated in preclinical animal models and in cell lines, and serve as a foundation for translational research initiatives in understanding the relevant pathways in human CRC. These include the observations that cox-2 knockout mice crossed into the Apcmin background (a relevant model of intestinal adenomatous polyposis) demonstrated a dramatic reduction in polyposis, along with decreased production of PGE2 (10). In addition, EP2 knockout mice also demonstrate reduced intestinal polyposis when crossed into the Apcmin background (8). Pharmacologic inhibitors of EP4 suppress polyposis in the Apcmin model (9), findings consistent with numerous other reports that selective cox-2 inhibitors inhibit polyp development, while administration of PGE2 accelerates their appearance and progression (8, 9). These findings in animal models have been extended to studies in human colorectal cancer cell lines, where many of the downstream events of cox-2 inhibition and PGE2 signaling are more readily dissected (11). Taken together, the findings from preclinical models, cell culture and human clinical investigational studies provide a substantial foundation for exploring cox-2 dependent pathways and their intersection with other dominant genetic pathways for growth regulation in chemoprevention of CRC.
With this general background in mind, there is considerable translational research interest emerging from the recent report that chronic aspirin use is associated with prevention of CRC particularly in the subgroup (about two thirds) where cox-2 immunostaining was moderately or strongly positive (4). These authors found that sustained (over several years) intake of at least five aspirin tablets per week was associated with an age-standardized CRC incidence rate of 37 per 100,000 person-years in subjects with cox-2 positive tumors, compared with 56 per 100,000 person-years for subjects not using aspirin (4). These findings raise important questions for new translational research initiatives, including the importance of cox-2 independent pathways in aspirin users whose tumors are cox-2 positive as well as in those subjects whose tumors are cox-2 negative. Are there alternative pathways that become dominant following cox-2 inhibition in certain subjects and how can these be identified in human subjects? Are there fundamental differences in CRC pathogenesis in cox-2 negative versus positive tumors? Are cox-2 positive tumors responsive to aspirin use via cox-2 independent pathways? Can we identify individuals who are most likely to show benefit from cox-2 inhibition as an approach to optimizing the risk-reward benefit from long-term aspirin use? There are also questions of a more basic nature that will require further exploration. These include understanding the cell-specific regulation of cox-2 expression and the epithelial mesenchymal/stromal cell dialog that occurs during the course of CRC development. How does PGE2-mediated signaling occur between cells located at a distance as opposed to adjacent locations? What are the implications of PGE2 signaling via different EP receptors and their downstream pathways and are these relevant considerations in the adenoma to carcinoma progression?
Estrogen, Hormone Replacement Therapy and Colorectal Cancer Risk
Significant epidemiologic data exists regarding an association between estrogen supplementation and CRC risk. The bulk of the data consists of case control and cohort studies, with the Women’s Health Initiative (WHI) being the only randomized placebo-controlled trial addressing the issue of estrogen supplementation and CRC risk. The WHI examined a cohort of postmenopausal women who were enrolled in a set of clinical trials, two of which involved randomized, placebo-controlled treatment with combined estrogen plus progestin or estrogen alone. Results of the trial comparing combination HRT versus placebo were reported in 2002 (12). The trial enrolled 16,608 post menopausal women who received conjugated equine estrogen (0.625 mg daily) plus medroxyprogesterone acetate 92.5mg daily) in a single tablet, or placebo. The cohort was followed over an average of 5.2 years, with the primary endpoints of the study being coronary heart disease and invasive breast cancer; CRC was one of several secondary endpoints. At the conclusion of the study, the hazard ratio for CRC was 0.63 (0.43–0.92), for endometrial cancer 0.83 (0.47–1.47) and for invasive breast cancer 1.26 (1.00–1.59). The authors concluded that combined HRT produced a 37% reduction in CRC incidence, and noted a benefit starting at 3 years of therapy.
In 2004, the WHI reported specifically on CRC risk in study subjects (13) with a total of 122 CRC cases confirmed; 48 in the HRT group and 74 in the placebo group (HR 0.61, 0.42–0.87). There were 43 invasive cancers in the HRT group compared to 72 in the placebo group (HR 0.56. 0.38–0.81). Despite a 44% reduction in invasive CRC in the HRT group, the cancers that developed in the HRT group were more likely to be lymph node positive (3.2% vs 0.8%, p=0.002) and advanced (76.2% vs. 48.5%, p=0.004). This unexpected difference was not explained by screening rates or symptoms.
In parallel with the above study the WHI also conducted a randomized placebo controlled trial of conjugated equine estrogen (0.625 mg daily) versus placebo with the same primary and secondary endpoints of the combined HRT study (14). The trial enrolled 10,739 post-menopausal women, and followed them for an average of 6.8 years. The hazard ratio for CRC was 1.08 (0.75–1.55) and for invasive breast cancer 0.77 (0.59–1.01). The authors concluded that estrogen-only supplementation did not protect against CRC risk (14).
On a practical note, an important obstacle surrounds the implementation of HRT as a chemopreventive agent. The WHI study was halted prematurely due to an overall increased risk for disease in the subjects taking HRT versus placebo. In particular, there was an increase in invasive breast cancer and cardiovascular disease in the HRT group, and although the HRT group had fewer CRCs, the cancers that developed in this group were more advanced at diagnosis (13). Of interest, a similar reduction in CRC was not seen in the estrogen-only arm of the WHI, (14) a finding that remains unexplained. As a consequence, although HRT is unattractive as a chemopreventive agent for CRC, there remains intense interest with respect to estrogen and its effect on CRC risk and the pathways by which such interactions might be tested.
Biological data provides insight into several possible mechanisms by which estrogen may impact CRC risk. Data support a role for estrogen-induced proliferation of colorectal neoplasia and also for induction of apoptosis and suppression of colorectal neoplasia. For example, certain estrogen metabolites have been shown to induce apoptosis in both tissues and CRC cell lines (15, 16). One such metabolite, 2-methoxyestradiol (2-MeOE2) induced apoptosis in CRC cell lines and cells cultured with 2-MeOE2 at increasing doses showed a dose dependent increase in p53 and p21WAF1CIP1 expression (17). 2-MeOE2 binds weakly to estrogen receptors (ER), suggesting that induction of apoptosis in this setting may be an ER independent event.
Considerable attention has focused on how estrogens may impact CRC pathogenesis via ER-related mechanisms. Estrogen receptor β (ERβ) expression was demonstrated in normal colon tissue, with a progressive decline in ERβ expression in CRC accompanying loss of differentiation (18). ERα, by contrast is minimally expressed in colorectal tissue, but 17β-estradiol (E2) induced apoptosis in LoVo colon cancer cells following transfection of ERα (19). In another study, E2 induced apoptosis in COLO 205 colon cancer cells via an ERβ-dependent pathway in which there was decreased vascular endothelial growth factor (VEGF) mRNA and protein secretion, suggesting inhibition of angiogenesis as a possible downstream event (20). VEGF has been linked to induction and maintenance of the neovasculature in human CRC (21), suggesting a possible mechanistic link between E2 and CRC prevention.
Reconciling the expansive yet conflicting data from colon cancer cell lines in conjunction with the epidemiologic data discussed above is challenging and thus the role of estrogen and HRT in CRC remains uncertain. The bulk of epidemiologic data clearly points to a protective effect of HRT, but a limitation to these studies is that they identify an association but provide no evidence of causation. Estrogens and estrogen receptors have been shown to both promote and repress growth in colorectal cancer cell lines, while HRT (although not estrogen alone) has been consistently associated with reduction in CRC risk. One plausible mechanism to account for the discrepancy is that HRT generates estrogen metabolites in-vivo with accompanying ER-dependent effects that are unique and distinct from those demonstrated in colon cancer cell lines. The role of estrogen and HRT in CRC is intriguing and is an area of ongoing investigation. Ideally, future investigation will reconcile the evidence from tissue and cancer cell lines with the robust epidemiologic data already available. In particular, breakthroughs might include differentiating which estrogen metabolites infer CRC protection and how, and why only combined HRT appears to decrease CRC risk in women.
Obesity and colorectal cancer risk
There is consensus agreement that maintaining an appropriate body weight, through strategies including regulated calorie intake coupled with physical activity, represents one of the most effective approaches to cancer prevention, second only to smoking cessation. This statement applies broadly also to CRC prevention. In regard to the genetic-environmental factors that influence CRC, there are several lines of evidence that suggest a uniquely informative role for obesity, increased calorie intake and increased disease susceptibility. First, studies of migrants moving from a low- to a high-risk area for CRC have shown that these migrant families acquire the cancer pattern of the host country within a single generation (22). Secondly, the shift in dietary behaviors accompanying cultural trends towards Westernization has produced a striking increase in CRC within populations previously considered to have a low prevalence rate. A specific example of this phenomenon has emerged from observational studies for the last 40 years in Japan (a country with one of the world’s lowest incidence of CRC at the beginning of the century), where the age-standardized incidence rate of colon cancer has increased 9.4 times for males and 4.7 times for females (23). By way of emphasizing the importance of environmental modifiers of genetic risk, studies show that the chances of identical twins developing cancer at the same site are generally less than 10% (24). Finally, data from epidemiological studies strongly suggest that increased calorie consumption, decreased physical activity, and excessive adiposity, are key players in the pathogenesis of some types of cancer, in particular CRC (25).
Data from large epidemiological studies indicate that excessive adiposity, physical inactivity and malnutrition are associated with increased incidence and/or death from CRC (26, 27). In particular, there is a clear association between abdominal obesity, as reflected by a higher waist circumference, and colon cancer and advanced adenoma risk in both men and women (28), findings confirmed in studies where visceral fat—measured by CT scanning—was strongly associated with colorectal adenoma detection and inversely associated with circulating adiponectin levels (29). Accumulating evidence in experimental animals support these data and indicate that normal colonic epithelial cells as well as CRC cells proliferate more rapidly in obese animals and in mice fed a hypercaloric diet (30, 31). Moreover, tumor development and size is increased after subcutaneous injection of CRC cells in obese compared to non-obese mice (32). By contrast, animal studies have demonstrated that calorie restriction strongly inhibits cancer and slows tumor growth (33). In particular, calorie restriction inhibits spontaneous, transplanted, and carcinogen-induced colon cancer in mice and rats (34-36). This said, the underlying mechanisms by which excessive caloric intake/adiposity promotes and calorie restriction prevents CRC remain incompletely understood (37).
Excessive adiposity is associated with insulin resistance, dyslipidemia, low-grade inflammation, and changes in hormone and growth factor levels that likely play a role in the pathogenesis of CRC (25, 38). Chronic positive energy balance, for example, promotes adipose tissue hypertrophy, adipokine-mediated insulin resistance, compensatory hyperinsulinemia and increased sex hormone availability (39, 40). By contrast, calorie restriction, which is the most effective and reproducible intervention for preventing cancer, improves insulin sensitivity, and reduces circulating levels of insulin, leptin, sex hormones, and increases circulating levels of adiponectin and sex hormone binding globulin (37).
Calorie restriction may have additional beneficial effects on cancer prevention independent of adiposity, including a reduction in insulin-like growth factor 1 (IGF-1), reduced inflammatory cytokine levels, reduced oxidative stress, and enhanced repair of DNA damage (41, 42). Insulin is a recognized growth factor, promotes proliferation of colon cancer cells in vitro and promotes colonic tumor growth in experimental animals (43–45). Hyperinsulinemia has been hypothesized to promote CRC both directly and indirectly through increases in insulin-like growth factor-1 (IGF-1), which itself is a potent mitogen and inhibitor of apoptosis (46, 47). Independent lines of evidence suggest that increased circulating IGF-1 may promote CRC pathogenesis through up-regulation of Akt, p53 and NF-kB pathways (48–50). Moreover, excessive adiposity, insulin resistance and high serum IGF-1 levels are also associated with higher oxidative stress and free radical mediated-DNA damage, which are key players in the pathogenesis of cancer (51, 52). The adipokine leptin also has been shown to stimulate the growth of colon cancer cells (53, 54). Further support for the possible role of leptin in promoting CRC growth comes from the demonstration that both CRC cell lines and tissue express functional leptin receptors (55) and that leptin promotes mitogenesis and inhibits apoptotis in several different CRC cell lines (56). Several features of the proposed pathways linking excess adiposity and insulin resistance to CRC pathogenesis are summarized in Figure 1.
Figure 1.
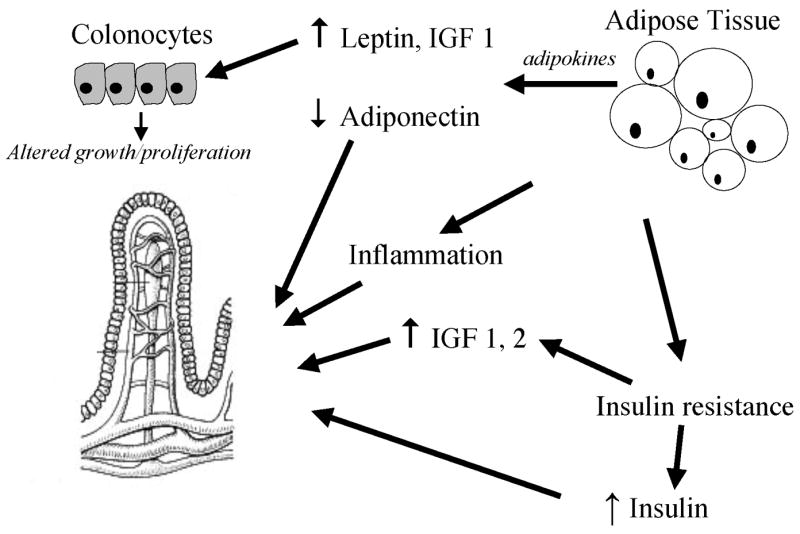
Excess adiposity is associated with insulin resistance, compensatory hyperinsulinemia, increased inflammation and increased production of adipokines including leptin. Insulin resistance also leads to upregulation of IGF1 and IGF2 production. Inflammatory cytokines, insulin, IGFs and leptin stimulate colorectal cancer cell proliferation, both via systemic as well as local paracrine pathways. Abdominal obesity is inversely associated with circulating adiponectin levels.
It has been postulated that changes in hormone metabolism, specifically insulin and sex hormones, may be a common pathway by which environmental risk factors promote CRC development (57, 58). This proposal raises the possibility that interactions between estrogen-signaling and insulin resistance particularly in a permissive genetic setting may explain some aspects of CRC tumorigenesis. A key question emerging from this particular suggestion is to understand the mechanisms whereby obesity is associated with a greater increase in CRC risk for men than for women (59, 60). As with the other areas examined in this review, unraveling the importance of these individual components is complex and advances will come from a combination of basic and translational approaches. Given the array of data that suggest calorie restriction in humans is effective in replicating the metabolic adaptations and benefits (including extending life expectancy and reduced manifestations of aging) seen in calorie restricted animals, it seems intuitive that the same pathways should be reasonable candidates for CRC reduction. However, much work will need to be undertaken to identify appropriate biomarkers for CRC risk reduction in obese individuals and to validate their corresponding utility in controlled studies. Among the questions to be addressed will be an exploration of potential pathways that link the regulation of cox-2 gene expression to excess adiposity or caloric intake.
Current and future research efforts will focus on identifying subsets of patients in whom specific genetic pathways can be targeted with appropriate interventions. With the array of preclinical animal models now available, coupled with the widespread dissemination of reagents with which to interrogate specific genetic pathways, it is now feasible to tailor translational studies to a more refined analysis of genetic-environmental interactions at play in CRC pathogenesis.
Acknowledgments
Work from the authors’ laboratories was supported by grants HL-38180, DK-56260, DK-52574 (to NOD), Clinical Nutrition Research Unit Grant DK56351, NIH General Clinical Research Center RR00036, and the Longer Life Foundation (an RGA/Washington University Partnership) (to LF).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 2.de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004;4:769–780. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- 3.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 4.Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med. 2007;356:2131–2142. doi: 10.1056/NEJMoa067208. [DOI] [PubMed] [Google Scholar]
- 5.Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp DD, Loprinzi CL, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348:883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 6.Turini ME, DuBois RN. Cyclooxygenase-2: a therapeutic target. Annu Rev Med. 2002;53:35–57. doi: 10.1146/annurev.med.53.082901.103952. [DOI] [PubMed] [Google Scholar]
- 7.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 8.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 9.Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi K, et al. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 2002;62:28–32. [PubMed] [Google Scholar]
- 10.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 11.Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, Morrow J, Beauchamp RD, DuBois RN. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest. 1997;99:2254–2259. doi: 10.1172/JCI119400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Writing group for the Women’s Health Initiative Investigators. Risks and benefits of hormone replacement therapy in healthy postmenapausal women. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 13.Chlebowski RT, Wactawski-Wende J, Ritenbaugh C, Hubbell FA, Ascensao J, Rodabough RJ, Rosenberg CA, Taylor VM, Harris R, Chen C, et al. Estrogen plus progestin and colorectal cancer in postmenopausal women. N Engl J Med. 2004;350:991–1004. doi: 10.1056/NEJMoa032071. [DOI] [PubMed] [Google Scholar]
- 14.Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [see comment] [DOI] [PubMed] [Google Scholar]
- 15.Mukhopadhyay T, Roth JA. Induction of apoptosis in human lung cancer cells after wild-type p53 activation by methoxyestradiol. Oncogene. 1997;14:379–384. doi: 10.1038/sj.onc.1200835. [DOI] [PubMed] [Google Scholar]
- 16.Schumacher G, Kataoka M, Roth JA, Mukhopadhyay T. Potent antitumor activity of 2-methoxyestradiol in human pancreatic cancer cell lines. Clin Cancer Res. 1999;5:493–499. [PubMed] [Google Scholar]
- 17.Carothers AM, Hughes SA, Ortega D, Bertagnolli MM. 2-Methoxyestradiol induces p53-associated apoptosis of colorectal cancer cells. Cancer Lett. 2002;187:77–86. doi: 10.1016/s0304-3835(02)00409-3. [DOI] [PubMed] [Google Scholar]
- 18.Konstantinopoulos PA, Kominea A, Vandoros G, Sykiotis GP, Andricopoulos P, Varakis I, Sotiropoulou-Bonikou G, Papavassiliou AG. Oestrogen receptor beta (ERbeta) is abundantly expressed in normal colonic mucosa, but declines in colon adenocarcinoma paralleling the tumour’s dedifferentiation. Eur J Cancer. 2003;39:1251–1258. doi: 10.1016/s0959-8049(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 19.Hsu HH, Cheng SF, Chen LM, Liu JY, Chu CH, Weng YJ, Li ZY, Lin CS, Lee SD, Kuo WW, et al. Over-expressed estrogen receptor-alpha up-regulates hTNF-alpha gene expression and down-regulates beta-catenin signaling activity to induce the apoptosis and inhibit proliferation of LoVo colon cancer cells. Mol Cell Biochem. 2006;289:101–109. doi: 10.1007/s11010-006-9153-3. [DOI] [PubMed] [Google Scholar]
- 20.Qiu Y, Langman MJ, Eggo MC. Targets of 17beta-oestradiol-induced apoptosis in colon cancer cells: a mechanism for the protective effects of hormone replacement therapy? J Endocrinol. 2004;181:327–337. doi: 10.1677/joe.0.1810327. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995;55:3964–3968. [PubMed] [Google Scholar]
- 22.Stammermann GN, Nomura AM, Chyou PH, Kato I, Kuroishi T. Cancer incidence in Hawaiian Japanese: migrants from Okinawa compared with those from other prefectures. Jpn J Cancer Res. 1991;82:1366–1370. doi: 10.1111/j.1349-7006.1991.tb01807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minami Y, Nishino Y, Tsubono Y, Tsuji I, Hisamichi S. Increase of colon and rectal cancer incidence rates in Japan: trends in incidence rates in Miyagi Prefecture. J Epidemiol. 2006;16:240–248. doi: 10.2188/jea.16.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminiki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 25.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 26.Giovannucci E, Ascherio A, Rimm EB, Colditz GA, Stampfer MJ, Willett WC. Physical activity, obesity, and risk of colon cancer and adenoma in men. Ann Intern Med. 1995;122:327–334. doi: 10.7326/0003-4819-122-5-199503010-00002. [DOI] [PubMed] [Google Scholar]
- 27.Giovannucci E, Colditz GA, Stampfer MJ, Willett WC. Physical activity, obesity, and risk of colorectal adenoma in women (United States) Cancer Causes Control. 1996;7:253–263. doi: 10.1007/BF00051301. [DOI] [PubMed] [Google Scholar]
- 28.Russo A, Franceschi S, La Vecchia C, Dal Maso L, Montella M, Conti E, Giacosa A, Falcini F, Negri E. Body size and colorectal cancer risk. Int J Cancer. 1998;78:161–165. doi: 10.1002/(sici)1097-0215(19981005)78:2<161::aid-ijc7>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 29.Otake S, Takeda H, Suzuki Y, Fukui T, Watanabe S, Ishihama K, Saito T, Togashi H, Nakamura T, Matsuzawa Y, et al. Associateion of visceral fat accumulation and plasma adiponectin with colorectal adenoma: evidence for particpation of insulin resistance. Clin Cancer Res. 2005;22:3642–3646. doi: 10.1158/1078-0432.CCR-04-1868. [DOI] [PubMed] [Google Scholar]
- 30.Newmark HL, Lipkin M, Maheshwari N. Colonic hyperplasia and hyperproliferation induced by a nutritional stress diet with four components of Western-style diet. J Natl Cancer Inst. 1990;82:491–496. doi: 10.1093/jnci/82.6.491. [DOI] [PubMed] [Google Scholar]
- 31.Newmark HL, Yang K, Lipkin M, Kopelovich L, Liu Y, Fan K, Shinozaki H. A Western-style diet induces benign and malignant neoplasms in the colon of normal C57BL/6 mice. Carcinogenesis. 2001;22:1871–1875. doi: 10.1093/carcin/22.11.1871. [DOI] [PubMed] [Google Scholar]
- 32.Yakar S, Nunez NP, Pennisi PPB, Sun H, Fallavollita L, Zhao H, Scavo L, Novosyadlyy R, Kurshan N, et al. Increased tumor growth in mice with diet-induced obesity: impact of ovarian hormones. Endocrinology. 2006;147:5826–5834. doi: 10.1210/en.2006-0311. [DOI] [PubMed] [Google Scholar]
- 33.Hursting SD, Lavigne JA, Berrigan D, Perkins SN, Barrett JC. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med. 2003;54:131–152. doi: 10.1146/annurev.med.54.101601.152156. [DOI] [PubMed] [Google Scholar]
- 34.Giovanella BC, Shepard RC, Stehlin JS, Venditti JM, Abbott BJ. Calorie restriction: effect on growth of human tumors heterotransplanted in nude mice. J Natl Cancer Inst. 1982;68:249–257. [PubMed] [Google Scholar]
- 35.Reddy BS, Wang CX, Maruyama H. Effect of restricted caloric intake on azoxymethane-induced color tumor incidence in male F344 rats. Cancer Res. 1987;47:1226–1228. [PubMed] [Google Scholar]
- 36.Lasko CM, Good CK, Adam J, Bird RP. Energy restriction modulates the development of advanced preneoplastic lesions depending on the level of fat in the diet. Nutr Cancer. 1999;33:69–75. doi: 10.1080/01635589909514750. [DOI] [PubMed] [Google Scholar]
- 37.Fontana L, Klein S. Aging, adiposity, and calorie restriction. JAMA. 2007;297:986–994. doi: 10.1001/jama.297.9.986. [DOI] [PubMed] [Google Scholar]
- 38.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 40.Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Visceral fat adipokine secretion is associated wity systemic inflammation in obese humans. Diabetes. 2007;56:1010–1013. doi: 10.2337/db06-1656. [DOI] [PubMed] [Google Scholar]
- 41.Dunn SE, Kari FW, French J, et al. Dietary restriction reduces insulin-like growth factor I levels, which modulates apoptosis, cell proliferation, and tumor progression in p53-deficient mice. Cancer Res. 1997;57:4667–4672. [PubMed] [Google Scholar]
- 42.Matsuzaki J, Kuwamura M, Yamaji R, Inui H, Nakano Y. Inflammatory responses to lipopolysaccharide are suppressed in 40% energy-restricted mice. J Nutr. 2001;131:2139–2144. doi: 10.1093/jn/131.8.2139. [DOI] [PubMed] [Google Scholar]
- 43.Koenuma M, Yamori T, Tsuruo T. Insulin and insulin-like growth factor 1 stimulate proliferation of metastatic variants of colon carcinoma. Jpn J Cancer Res. 1989;80:51–58. doi: 10.1111/j.1349-7006.1989.tb02244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koohestani N, Tran T, Lee W, et al. Insulin resistance and promotion of aberrant crypt foci in the colons or rats on a high-fat diet. Nutrition Cancer. 1997;29:69–76. doi: 10.1080/01635589709514604. [DOI] [PubMed] [Google Scholar]
- 45.Tran T, Medline A, Bruce W. Insulin promotion of colon tumors in rats. Cancer, Epidemiology, Biomarkers and Prevention. 1996;5:1013–1015. [PubMed] [Google Scholar]
- 46.Giovannucci E. Insulin, insulin-like growth factors and colon cancer. J Nutr. 2001;131:3109S–3120S. doi: 10.1093/jn/131.11.3109S. [DOI] [PubMed] [Google Scholar]
- 47.Giovannucci E. Insulin-like growth factor-I and binding protein-3 and risk of cancer. Hormone Research. 1999;51:34–41. doi: 10.1159/000053160. [DOI] [PubMed] [Google Scholar]
- 48.LeRoith D, Baserga R, Helman L, Roberts CT., Jr Insulin-like growth factors and cancer. Ann Intern Med. 1995;122:54–59. doi: 10.7326/0003-4819-122-1-199501010-00009. [DOI] [PubMed] [Google Scholar]
- 49.Edinger AL. Growth factors regulate cell survival by controlling nutrient transporter expression. Biochem Soc Trans. 2005;33:225–227. doi: 10.1042/BST0330225. [DOI] [PubMed] [Google Scholar]
- 50.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Keaney JFJ, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol. 2003;23:434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 52.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 53.Liu Z, Uesaka T, Watanabe H, Kato N. High fat diet enhances colonic cell proliferation and carcinogenesis in rats by elevating serum leptin. Int J Oncol. 2001;19:1009–1014. doi: 10.3892/ijo.19.5.1009. [DOI] [PubMed] [Google Scholar]
- 54.Attoub S, Noe V, Pirola L, Bruyneel E, Chastre E, Mareel M, Wymann MP, Gespach C. Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3-kinase-, rho-, and rac-dependent signaling pathways. FASEB J. 2000;14:2329–2338. doi: 10.1096/fj.00-0162. [DOI] [PubMed] [Google Scholar]
- 55.Hardwick JC, Van Den Brink GR, Offerhaus GJ, Van Deventer SJ, Peppelenbosch MP. Leptin is a growth factor for colonic epithelial cells. Gastroenterology. 2001;121:79–90. doi: 10.1053/gast.2001.25490. [DOI] [PubMed] [Google Scholar]
- 56.Hoda MR, Keely SJ, Bertelsen LS, Junger WG, Dharmasena D, Barrett KE. Leptin acts as a mitogenic and antiapoptotic factor for colonic cancer cells. Br J Surg. 2007;94:346–354. doi: 10.1002/bjs.5530. [DOI] [PubMed] [Google Scholar]
- 57.Kaaks RTP, Akhmedkhanov A, et al. Serum C-peptide, insulin-like growth factor (IGF)-I, IGF-binding proteins, and colorectal cancer risk in women. J Nat Cancer Inst. 2000;92:1592–1600. doi: 10.1093/jnci/92.19.1592. [DOI] [PubMed] [Google Scholar]
- 58.Giovannucci E. Insulin and colon cancer. Cancer Causes & Control. 1995;6:164–179. doi: 10.1007/BF00052777. [DOI] [PubMed] [Google Scholar]
- 59.Murphy T, Calle E, Rodriguez C, Khan H, Thun M. Body mass index and colon cancer mortality in a large prospective study. Am J Epidemiol. 2000;152:847–854. doi: 10.1093/aje/152.9.847. [DOI] [PubMed] [Google Scholar]
- 60.Caan B, Coates A, Slattery ML, Potter JD, Quesenberry C, Edwards S. Body size and colon cancer in a large case-control study. International J Obesity Relat Metab Disord. 1998;22:178–184. doi: 10.1038/sj.ijo.0800561. [DOI] [PubMed] [Google Scholar]