

Abstract
B7H/B7RP (hereby called B7H) is a new member of the B7 family of costimulatory molecules and interacts with inducible costimulatory molecule (ICOS). Its function for CD8 T cells has not been reported. We report here that expression of B7H on the tumor cells reduced tumorigenicity and induced immunity to subsequent challenge with parental tumor cells. The immune protection correlates with an enhanced cytotoxic T lymphocyte (CTL) response against P1A, the major tumor antigen expressed in the J558 tumor. To understand the mechanism of immune protection, we adoptively transferred transgenic T cells specific for tumor antigen P1A into mice that bore P1A-expressing tumors. We found that while the transgenic T cells divided faster in mice bearing the B7H+ tumors, optimal B7H-induced clonal expansion of P1CTL required costimulation by B7–1 and B7–2 on the endogenous host antigen-presenting cells (APCs). Interestingly, when B7H+ and B7H− tumors were coinjected, P1CTL selectively eliminated the B7H+ tumor cells. Moreover, B7H expressed on the tumor cells made them highly susceptible to destruction by CTL in vivo, even if the CTL was administrated into mice with large tumor burdens. Tumors that recurred in the P1CTL-treated mice lost transfected B7H and/or H-2Ld, the class I molecule that presents the P1A peptide. Taken together, our results reveal that B7H costimulates clonal expansion of, and cognate destruction by CD8+ T lymphocytes in vivo.
Keywords: cytotoxic T lymphocytes, tumor immunity, B7H, effector function, clonal expansion
Introduction
An important feature of the immune response is the multitude of regulation. An important issue is whether an immune response is regulated at both the inductive and effector phases. While multiple factors, such as antigen (1), costimulation (2–5), CD4 T cell help (6), and APCs (7) regulate the induction of CTL responses, the mechanisms responsible for regulating the effector function of T cells have not been fully elucidated.
Perhaps the most striking demonstration of controls at the effector phase is that tumor-reactive CTLs coexist with the cancer cells in vivo. Thus, we have demonstrated that P1A-expressing plasmocytoma J558 grew in the transgenic mice that express a P1A-specific TCR in an overwhelming number of T cells (8). Moreover, despite a lack of overt immune suppression on antigen-specific CTLs, established cancers are highly resistant to transgenic tumor-specific CTLs (9). Corresponding to the observations in experimental animals, several groups reported that melanomas progressed in many patients despite the high number of CTL-specific for melanoma (10, 11). While some have observed clear functional defects among these tumor-reactive T cells (10), others have failed to do so (11). These observations raise an intriguing possibility that the effector function of tumor-reactive T cells is normally restrained in vivo.
We (8, 12) and others (13, 14) have reported that expression of costimulatory molecule B7–1 on tumors promoted T cell effector function. For instance, when B7–1+ and B7–1− tumors were injected into the same mouse, tumor-specific T cells preferentially eliminated B7–1+ tumors (8, 12). Corresponding to this, we reported that downregulation of either transfected B7–1 or MHC class I molecules is sufficient to allow tumor recurrence in the mice that had originally rejected the tumor (15). Thus, the costimulatory molecule B7–1 played a significant role in the destruction of tumor cells by CTLs in vivo. Similarly, in the autoimmune nonobese diabetic (NOD) model, when B7–1 was expressed in some, but not all pancreatic islet cells, the T cells preferentially destroyed the B7–1+ β-cells (16). The selective destruction of the B7–1+ cells over the surrounding B7–1− cells suggests that costimulatory molecule B7–1 can promote the effector function of autoreactive CTLs.
A fundamental issue raised by these studies is whether the CTL effector function is controlled by the costimulatory molecules, much as is the induction. A major obstacle to this notion is that B7–1 is neither present nor considered inducible on the nonhematopoietic tissues which CD8 T cells can efficiently survey. In this regard, the recently uncovered members of the B7 family, B7H1 (17) or PDL1 (18) and B7H (19) (20), B7H3 (21), and PD-L2 (22) or B7-DC (22) have considerably wider tissue distribution. Northern blot analysis of RNA expression suggested that B7H is expressed in the heart, lung, and kidney at levels comparable to those in the spleen and thymus; and that B7-H1 mRNA is abundant in heart, skeletal muscle, placenta, and lung tissues, but is weak in thymus, spleen, kidney, and liver tissues (17, 23). Moreover, B7H is inducible by proinflammatory cytokines such as interferon and tumor necrosis factor (19, 24, 25), and its receptor inducible costimulatory molecule (ICOS)* is expressed on activated, but not resting, T cells (26). It is thus plausible that B7H may act as a costimulator late in the immune response when T cells have reached target tissues. To address this important issue, we expressed B7H on a P1A-expressing tumor cell line J558, and used P1A-specific transgenic T cells to analyze the role for B7H in antitumor CTL response in vivo. Here we report that, in addition to promoting CD8 T cell clonal expansion, B7H expressed on the tumor cells drastically enhanced cognate destruction of cancer cells by CD8 T cells in vivo. Our study provides direct evidence that B7H costimulates CD8 T cells in vivo. Moreover, our data support the notion that costimulation is an important parameter in the effector function of T cells.
Materials and Methods
Experimental Animals
Transgenic mice expressing TCR specific for tumor antigen P1A35–43:Ld complex have been described (8). The TCR transgenes were backcrossed with BALB/cByJ for at least eight generations before they were used for this study. Wild-type BALB/c mice were purchased from National Cancer Institute. BALB/c mice with a targeted mutation of RAG-2 gene were purchased from Taconic.
Construction of B7H-Green Fluorescent Protein Fusion Gene and Production of B7H-transfected J558 Cells
Total RNA was isolated from the mouse spleen cells by TRIzol (GIBCO BRL) and 2 μg of total RNA were used for first strand cDNA synthesis using the random hexamers (GIBCO BRL). The mouse B7H cDNA was amplified by PCR using Pfu DNA polymerase (forward primer: ATGCAGCTAAAGTGTCCCTGT and reverse primer: GGCGTGGTCTGTAAGTTCAAG). The PCR amplified B7H fragment was cloned into the BamHI and EcoRI sites of pEGFP-N3 (CLONTECH Laboratories, Inc.). The positive clones were sequenced using a primer in the pCMV IE region. The mB7H–green fluorescent protein (GFP) fusion DNA was transfected into the J558 cell line using DMRIE reagent (Life Technologies) and the presence of B7H and GFP was verified by reverse transcription (RT)-PCR and flow cytometry.
Cell Lines
Plasmocytomas J558 transfected with either vector alone (J558-Neo), or B7–1 (J558-B7–1) have been described (12). Three independent clones of J558-B7H were produced as described above and used for the current study.
Antibodies and Fusion Protein
Anti–B7–1 (3A12) (27) and anti–B7–2 (GL-1) mAbs (28) were purified from the hybridoma supernatants using protein G affinity column. For in vitro analysis, the antibodies were biotinylated. Cy-chrome-conjugated anti-CD8, PE-conjugated anti-Vα8, and biotinylated anti–H2-Ld mAb HB27 were purchased from BD PharMingen.
The extracellular portion of murine ICOS was amplified by RT-PCR using cDNA from mouse spleen as template, 5′-ATGAAGCCGTACTTCTGCCATG as forward primer, 5′-TCTTCAGCTGGCAGCAGAGCTGGG as the reverse primer. The PCR product was cloned into the pIg vector (Novagen). ICOSIg fusion gene was then inserted into HindIII/KpnI site of pcDNA3.1/Hygro+ (Invitrogen). The construct was transfected into CHO cells using Fugene 6 transfection system (Boehringer). The Hygromycin B resistant clones were expanded. The ICOSIg protein was purified using protein G column.
Adoptive Transfer of Purified Transgenic T Cells
Pools of spleen and lymph node cells from the recombination activating gene (RAG)-2+/+ or RAG-2−/− P1CTL-transgenic mice were incubated with a cocktail of mAbs (anti-CD4 mAb GK1.5, anti-FcR mAb 2.4G2, anti-CD11c mAb N418). After removal of unbound mAbs, the cells were incubated with anti-Ig–coated magnetic beads. The antibody-coated cells were removed by a magnet. The unbound cells consisted of more than 90% CD8 T cells, with no detectable CD4 T cells. The purified T cells were adoptively transferred into RAG-2−/− mice that either had established tumors, or had received tumor cells on the same day of adoptive transfer. In some experiments, the CD8 T cells were labeled with CFSE before their adoptive transfer as described (29).
Tumorigenicity Assay
5 × 106 J558 cells were injected in the flanks as described (12). The tumor size and incidence were determined by physical examination.
CTL Assays
A 6-h 51Cr release assay was used to measure CTL activity. The effector T cells were either the P1CTL activated for 96 h with 0.1 μg/ml of P1A peptide (8), or tumor-infiltrating lymphocytes isolated from either J558-B7H or J558-Neo tumors, as described (12).
Results
B7H-expressing Tumors Induce Stronger Antitumor CTL Response in Wild-Type Mice
As no anti-B7H mAb is available at this point, we have produced a fusion gene encoding a B7H-GFP chimera protein which can be easily quantitated by flow cytometry. The plasmid containing B7H-GFP was therefore used to transfect the J558 cells. After selection with G418, the drug resistant clones were screened by flow cytometry. Three independent clones were selected for the current studies. As shown in Fig. 1 A, all three clones expressed significant levels of B7H-GFP. Fusion with GFP did not abrogate the function of B7H as the J558-B7H-GFP can bind to ICOSIg (Fig. 1 B). Moreover, the B7H-GFP is expressed on the cell surface as revealed by confocal microscopy (data not shown). To avoid variation of individual clones, we maintained the clones individually, but mixed equal numbers of cells from the three clones for all the experiments described in the current study. As shown in Fig. 1 C, both J558-Neo and J558-B7H expressed and presented the tumor antigen P1A as they were readily killed by P1A-specific transgenic T cells. While B7–1 significantly increased lysis of J558 cells by CTL as we have described (12), B7H did not substantially enhance the lysis by P1CTL in vitro.
Figure 1.


Generation of tumor cell lines expressing B7H-GFP fusion protein. (A) Intensity of green fluorescence in J558 cells transfected with vector alone (J558-Neo) or B7H-GFP. Profiles of three independent clones were presented. These clones were cultured separately, mixed at an 1:1:1 ratio, and used in all subsequent experiments. (B) ICOSIg binds to J558-B7H, but not to J558-Neo. The J558-transfectants were incubated with either human IgG1Fc or ICOSIg (50 μg/ml), and the binding was measured by flow cytometry. (C) Susceptibility of J558-B7H, J558-Neo, and J558-B7 to transgenic P1CTL, which had been stimulated in vitro for 4 d with P1A peptide (0.1 μg/ml). Two independent experiments were presented. Unpulsed or P1A-pulsed P388D1(H-2d) target cells were used as negative and positive controls, respectively.
Expression of GFP as a cytosolic protein drastically reduced the growth rate of the J558 cells in vitro and the tumorigencity of J558 even in RAG-2−/− mice that do not have T and B lymphocytes (data not shown). This is not the case for B7H-GFP, which was expressed on the cell surface and did not affect cell growth rate and tumorigenicity in the absence of T and B lymphocytes (Fig. 2 B; see also Figs. 7 B and 8 C). We therefore used J558 cells transfected with vector alone as control for this study. We tested the tumorigenicity of the J558-B7H and J558-Neo in syngeneic BALB/c mice. As shown in Fig. 2, all mice that received the J558-Neo tumor cells developed palpable tumors within 11 d, while only 10% of the mice inoculated with the J558-B7H cells developed tumors during the same period. Although 60% of the mice did develop tumors within a 6-wk period, the remaining 40% of the mice never developed tumors. In contrast, in the immune deficient RAG-2−/− mice, the two tumor cell lines induced tumors at a similar rate. Thus, while nonspecific immunity may cause a minor reduction of the tumorigenicity of the B7H-transfected J558 cells, the decreased tumorigenicity of the B7H-transfected tumor cells is primarily due to specific immune response. To confirm that the immunity was not solely specific for any part of the B7H-GFP fusion protein, we challenged the mice that had rejected the J558-B7H tumor with the parental J558 cells. As shown in Fig. 2, C and D, while 100% of the naive mice developed the J558 tumors at the site of injection, none of the mice that rejected the J558-B7H tumors develop the J558 tumors.
Figure 2.

B7H expression reduced tumorigenicity of the J558 cells in immune competent mice and induced protection to subsequent challenge with the parental tumors. (A) Tumor incidence of J558-Neo (n = 6) and J558-B7H (n = 10) in immune competent mice. Data are representative of four independent experiments. In total, 24/26 mice receiving J558-Neo cells developed tumors, while 23/39 mice receiving the J558-B7H cells developed tumors. (B) J558-B7H and J558-Neo tumors grew at a similar rate in syngeneic RAG-2−/− mice (n = 7). Data shown are representative of four independent experiments, which in total involved 23 mice per group. All mice developed tumors within 2 wk. Syngeneic BALB/c or BALB/crag2−/− mice received 5 × 106 tumor cells in the flank, and the tumor incidences were determined by physical examination. (C and D) Mice that rejected J558-B7H tumors were immune to subsequent challenge with parental J558 cells. Syngeneic BALB/c mice that had rejected J558-B7H tumors and naive control mice were challenged with 5 × 106 J558 cells at the opposite flank. The tumor incidence (C) and growth kinetics (D) were measured by physical examination. This experiment was repeated twice with similar results.
Figure 7.

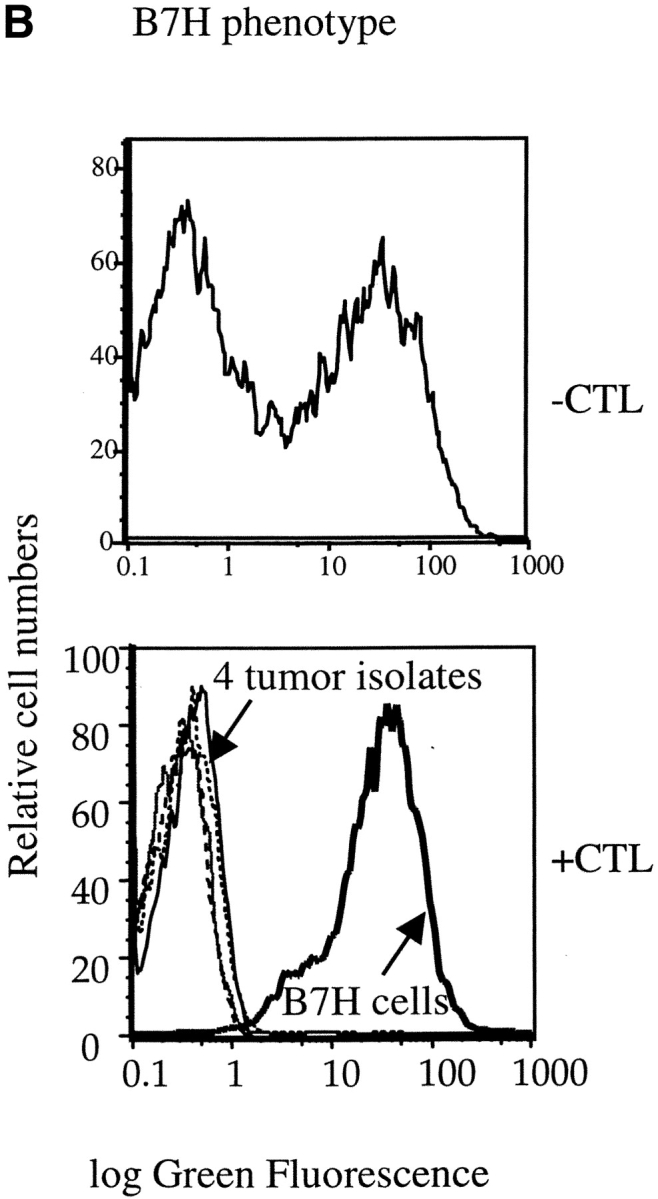
Selective elimination of B7H-expressing tumor cells by transgenic T cells in vivo. J558-Neo and J558-B7H were mixed at 1:1 ratio and injected into the flank of the BALB/c RAG-2−/− mice. Half of the mice received 5 × 106 of P1CTL intraperitoneally at the same day. (A) P1CTL delayed the development of tumor cells. Data presented were percentage of mice with palpable tumors at different times after injection of tumor cells. (B) Selective elimination of B7H cells by P1CTL. Top, substantially comparable representation of J558-B7H and J558-Neo tumor cells. Mice were killed when the tumor reached ∼5% body weight (3 wk after injection), and the freshly isolated tumor cells were analyzed for both GFP intensity and for tumor markers, the plasma cell antigen PC1 that is present on tumor cells, but not on host cells in the RAG-2−/− host. Data presented are the histograms of gated PC1+ tumor cells. Note that about half of the tumor cells expressed B7H. Bottom, tumor cells isolated by biopsies were all devoid of GFP. At 7 wk after injection, the tumor cells were isolated by needle biopsies from at least three locations in the tumors. After a short-term culture (<1 wk), their GFP intensity was analyzed by flow cytometry. Note that all ex vivo tumor cells (plain lines) were devoid of B7H. A J558-B7H cell line was used as control.
A major tumor antigen in the J558 tumor is P1A (30). To verify that expression of B7H on the tumor cells enhances antitumor immune response, we evaluated anti-P1A CTL response in tumor-infiltrating lymphocytes (TILs) from the J558-Neo and J558-B7H tumors. As shown in Fig. 3 A, J558-B7H was infiltrated a large number of CD8+ T cells, and essentially all CD8+ T cells expressed high levels of CD44. While J558-Neo was also infiltrated with activated CD8 T cells, its frequency was ∼20-fold lower. Moreover, strong P1A-specific CTL activity was observed in the TIL of the J558-B7H tumors, but not in those of the J558-Neo tumors (Fig. 3 B). TIL from J558-B7H kills both J558-B7H and J558-Neo targets (Fig. 3 C). Thus, B7H enhanced antitumor CTL response, including the anti-P1A CTL response in vivo.
Figure 3.
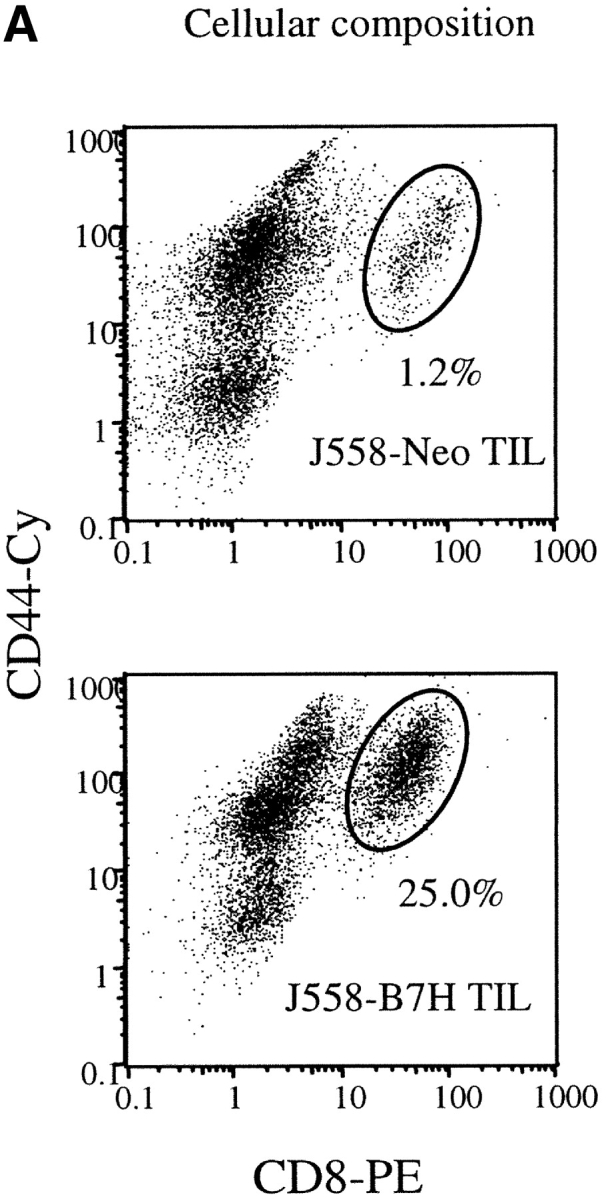
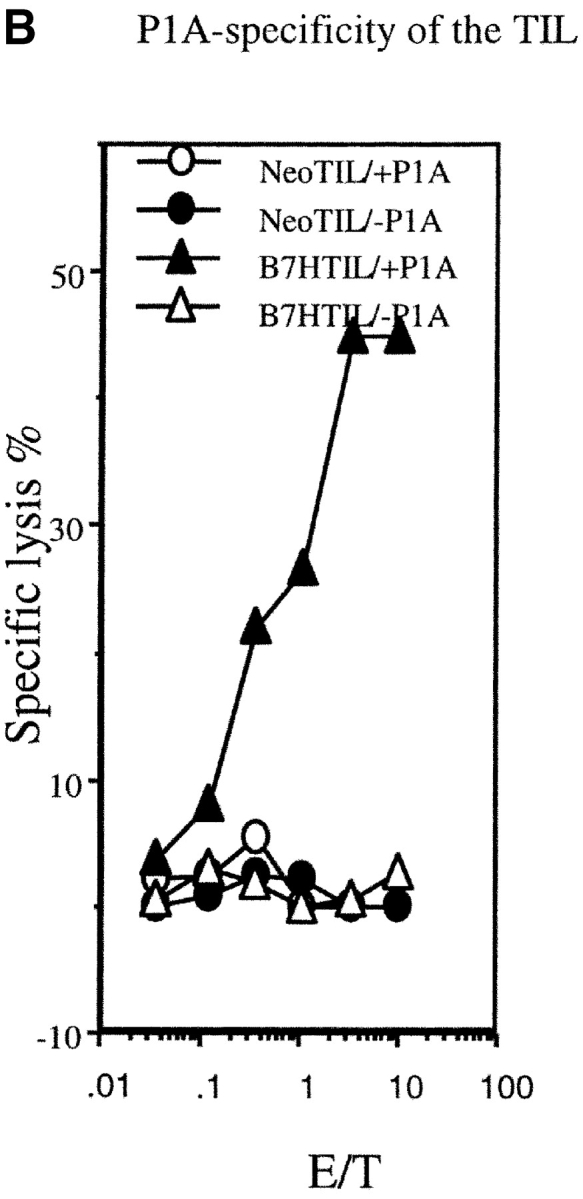

B7H increases P1A-specific CTL in the TIL from wild-type BALB/c mice. The TIL were enriched by depletion tumor cells as described (reference 12). The cytotoxicity of the freshly isolated TIL were determined using either P1A-pulsed or unpulsed P388D1, or the J558-Neo and J558-B7H cells as targets. (A) Substantial increase of activated CD8 T cells among the TIL. The TIL were stained with PE-conjugated anti-CD8 and Cy-conjugated anti-CD44 mAbs and analyzed by flow cytometry. (B) P1A-specific CTL in the TIL from the J558-B7H, but not the J558-Neo tumors. Freshly isolated TIL were used as effectors while the P1A-pulsed and unpulsed P388D1 were used as targets. (C) TIL from J558-B7H (right panel), but not those from J558-Neo (left panel), lysed both J558-B7H and J558-Neo tumors. As in B, except that the J558-Neo and J558-B7H were also used as targets. Data shown are representative of at least three independent experiments. TIL used in A and B were isolated at day 24 of tumor injection, while those used in C were isolated on day 20 of tumor cell injection.
B7H Costimulates Clonal Expansion of Tumor-specific T Cells, Although Optimal In Vivo Division of P1CTL Depends on B7–1/2 on Host APCs
We have recently produced a transgenic mouse line that expresses TCR-specific for P1A, which we called P1CTL (8). To analyze if the B7H promotes the induction of T cell clonal expansion, we labeled the purified transgenic CD8 T cells with CFSE and stimulated them with either J558-Neo or J558-B7H cells. As shown in Fig. 4 , regardless of the tumor cells used, insignificant T cell division was found at 48 h of coculture. By 72 h, however, many of the J558-B7H–stimulated T cells had mounted up to five divisions, while T cells cocultured with J558-Neo barely divided. Both groups divided at 96 h after stimulation (data not shown), although there were approximately fivefold more nondividing P1CTL in the J558-Neo stimulated culture (∼10%) than the J558-B7H–stimulated culture (∼2%). Thus, B7H accelerated T cell clonal expansion in vitro.
Figure 4.

B7H expressed on the tumor cells promotes clonal expansion of purified P1CTL. Purified CD8 T cells from P1CTL transgenic mice were labeled with CFSE and stimulated with irradiated J558-Neo or J558-B7H for either 48 or 72 h. The viable cells were stained with PE-anti-Vα8 and Cy-anti-CD8 mAbs. The data shown are histograms of gated CD8+Vα8+ T cells. This experiment was repeated twice with similar results.
To test the function of B7H for CD8 T cell activation in vivo, we labeled P1CTL with CFSE and adoptively transferred them into syngeneic RAG-2−/− mice that were either tumor-free or bore either J558-Neo or J558-B7H tumors. At 36, 60, and 84 h after the T cell transfer, spleen cells were harvested, and the number of divisions that the transgenic T cells had undergone in vivo was analyzed by flow cytometry. As shown in Fig. 5 , left panels, P1CTL divided slowly in the tumor-free mice, with a significant number of T cell dividing only at 84 h of adoptive transfer. In the mice that bore the J558-Neo tumors, few cells divided at 36 h of transfer. By 60 h, most of the cells in the spleen were products of one to six divisions, and an overwhelming majority of the cells accumulated at 84 h were products of six or more divisions. Interestingly, the P1CTL had divided significantly faster in the mice that bore the J558-B7H tumors. Thus, significant divisions of T cells were already detected at 36 h of transfer in the J558-B7H tumor-bearing mice. By 60 h, an overwhelming majority of the T cells accumulated in the spleen had divided more than four times. Corresponding to this, substantially more T cells had accumulated in the spleen of J558-B7H tumor bearing mice at 60 and 84 h after adoptive transfer (Fig. 5).
Figure 5.

Expression of B7H on the tumor cells promotes T cell clonal expansion. Purified CD8 T cells from P1CTL transgenic mice were labeled with CFSE and adoptively transferred into RAG-2−/− BALB/c mice bearing either J558-Neo or J558-B7H tumors, or mice that received no tumor cells as control. At 36, 60, and 84 h after the adoptive transfer, T cells were isolated from either spleen or tumors and were analyzed for the number of divisions that they had undergone. FACS® profiles of CFSE intensity of gated CD8+Vα8+ T cells isolated from spleen were presented. The percent of the CD8+Va8+ cells were shown in the panels.
To test if costimulation by B7–1 and B7–2 on the host APCs is required for clonal expansion of T cells, we injected anti–B7–1 and anti–B7–2 mAbs in mice that bore J558-B7H tumors and analyzed the division of T cells in the spleens. As the J558 tumor cells did not express B7–1 and B7–2, even during an ongoing immune response in vivo (31), the anti-B7 mAbs must have blocked the costimulatory function of B7–1 and B7–2 on the host APCs. As shown in Fig. 6 , anti–B7–1 and anti–B7–2 substantially reduced the division rate of T cells. Thus, despite B7H expression on the tumor cells, optimal activation of tumor-specific T cells requires costimulation by B7–1 and/or B7–2 expressed on the host APCs.
Figure 6.
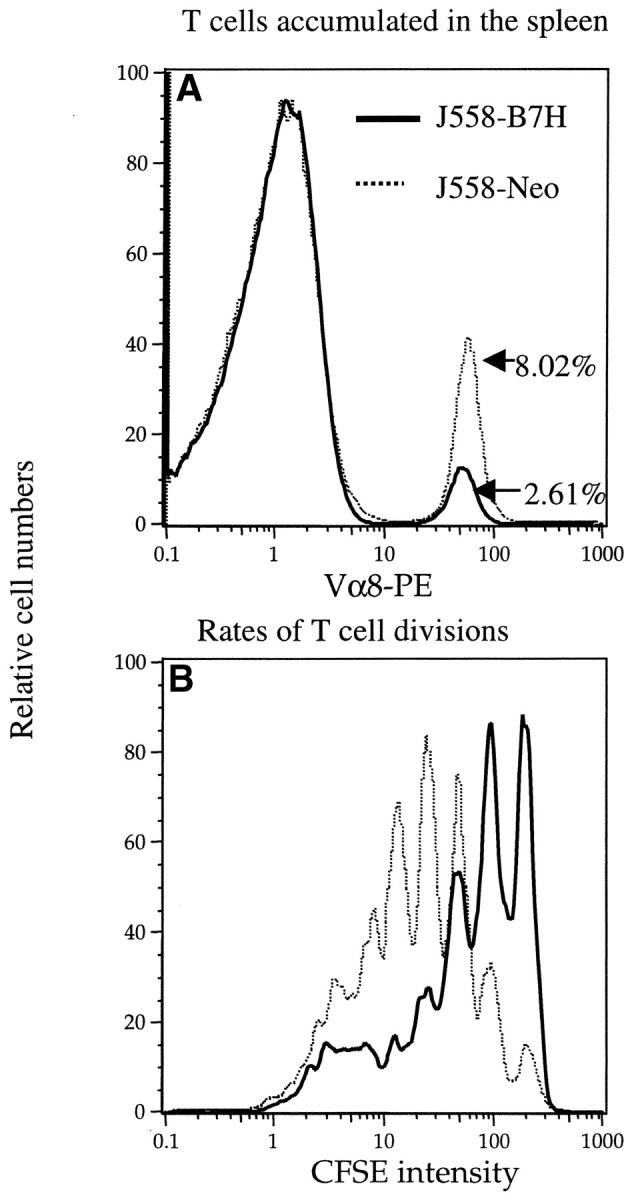
B7–1 and/or B7–2 on host cells were required for optimal T cell division in vivo. (A) The amount of Vα8+ T cells recovered from the spleen, the percentages of the P1CTL recovered were given in the panel. (B) Distributions of CFSE intensities among the Vα8+ T cells. Naive P1CTL were labeled with CFSE and adoptively transferred into the RAG-2−/− J558-B7H tumor-bearing mice that had received either control (a mixture of rat and hamster) IgG or anti–B7–1 and B7–2 mAbs on days 0, 1, and 2 of T cell adoptive transfer. Spleen cells were harvested and analyzed on day 3 of the adoptive transfer. This experiment was repeated twice with similar results.
B7H Promotes the Cognate Destruction of Cancer Cells In Vivo
To test if B7H may play a role subsequent to the initial T cell activation, such as the effector function of T cells, we injected a mixture of J558-B7H and J558-Neo into syngeneic RAG-2−/− mice in the presence or absence of tumor-specific T cells. Despite of the fact that P1CTL successfully delayed the growth of tumors, the majority of the mice eventually developed tumors at the site of injection (Fig. 7 A). To determine the origin of the tumor cells, we isolated the tumor cells and analyzed the expression of B7H by flow cytometry. As shown in Fig. 7 B, top panel, tumor cells recovered from mice that received no T cells consisted of a mixture of both B7H+ and B7H− subpopulations. The nearly equal contribution of the two types of tumor cells indicated that B7H expression did not convey a significant growth disadvantage in the absence of P1CTL. In contrast, in mice that received P1CTL, all tumor cells that survived in vivo were devoid of B7H. As the B7H+ and B7H− cells were injected as a mixture, selective elimination of B7H+ tumor cells indicates that B7H promotes the cognate destruction of tumor cells.
A major caveat of the above experiment is that T cells that mediate the rejection can be specific for the GFP fusion protein, as the T cells used in the above experiments were from transgenic mice that may have had rearrangement of the endogenous TCR. To rule out this possibility, we bred the P1CTL into the syngeneic RAG-2−/− background to produce the RAG-2−/− P1CTL. As shown in Fig. 8 , RAG-2−/− P1CTL was at least as efficient as the RAG+/+ P1CTL (Fig. 7) in rejecting a mixture of the J558-B7H and J558-Neo tumors. In addition, the RAG-2−/− P1CTL divided faster in mice that bore the J558-B7H tumor (Fig. 8 B), much as the RAG-+/+ P1CTL (Fig. 5). Moreover, the tumor cells that survived the RAG-2−/− P1CTL were predominantly B7H− (Fig. 8 C). These results confirmed that the function of B7H described here was not due to the antigenicity of the GFP.
Figure 8.

RAG-2−/− P1CTL proliferate more rapidly in mice bearing J558-B7H tumors than in the J588-Neo tumor-bearing mice, and preferentially reject B7H+ tumors. (A) Tumor rejection. RAG-2−/− BALB/c mice were inoculated with a mixture (1:1) of J558-Neo and J558-B7H cells. One group of mice also received 5 × 106 RAG-2−/− P1CTL. Tumor incidence was determined by physical examination. (B) T cell division in vivo. RAG-2−/− P1CTL were labeled with CFSE and adoptively transferred into mice that bore J558-B7H (dotted line) or J558-Neo (bold line) tumors of comparable sizes. Spleen cells were harvested at 60 h after adoptive transfer, and stained with PE-anti-Vα8 and Cy-anti-CD8 mAbs. Data shown are the histograms of CFSE intensity of the gated CD8+Vα8+ T cells (14.8% of spleen cells in the J558-B7H group and 2.2% in the J558-Neo group). (C) Preferential rejection of the B7H+ tumors. Tumors cells were isolated from mice that received 1:1 ratio of J558-B7H and J558-Neo cells with or without RAG-2−/− P1CTL. The presence of B7H+ tumor cells was measured based on the GFP+ cells. Top panels depict two representative tumors from mice that did not receive T cells, while the two bottom panels depict those that received T cells. The percentage of GFP+ cells are indicated in the panels.
Established B7H-expressing Tumor Cells Are Highly Sensitive to T Cell Therapy
A major challenge in tumor immunotherapy is to eliminate the established tumors. As B7H promoted cognate destruction of J558 tumors, we tested if expression of B7H allowed T cells to eliminate large burdens of tumors. We injected RAG-2−/− mice with either J558-Neo or J558-B7H tumor cells. At 3 wk after tumor inoculation when the tumors had reached a diameter ∼1.5 cm, we adoptively transferred 5 × 106 P1CTL. As shown in Fig. 9 A, P1CTL caused a temporary reduction in the size of the J558-Neo tumors. However, after a short pause, the J558-Neo tumor progressed rapidly and all mice succumbed to the tumor shortly thereafter. Interestingly, in the two mice with the J558-B7H tumor and P1CTL, the tumor rapidly shrank to either impalpable (J558-B7H-A), or into a small debris of <2 mm with no tumor when examined by biopsies (J558-B7H-B). These results suggest that expression of B7H enhanced tumor susceptibility to T cell therapy.
Figure 9.


B7H expression increased susceptibility of established tumors to T cell therapy. J558-Neo and J558-B7H tumors were allowed to grow in the BALB/crag-2−/− mice for three weeks until they reached ∼1.5 cm in diameter. Transgenic T cells (5 × 106/mouse) were then injected intraperitoneally. (A) Tumor size after T cell injection. Note the complete disappearance and recurrence of the J558-B7H tumors. *, found dead; ** moribund and killed. (B). Mechanism of tumor recurrence. Analysis of cell surface expression of MHC class I H-2Ld, and B7H. Left, a J558-B7H cell line as control. Middle, phenotype of the J558-B7H-A tumor cells. Right, phenotype of the J558-B7H-B tumor cells.
Eventually, the tumors originated from the B7H+ cells recurred and grew progressively. As mice with recurrent tumors had large numbers of P1CTL (data not shown), we isolated the tumor cells and analyzed the cell surface expression of H-2Ld, the restricting element for the P1A peptide, and B7H. As shown in Fig. 9 B, in comparison to the parental J558-B7H tumor cells, almost all cells from the J558-B7H-A had reduced cell surface H-2Ld by 10-fold and the majority of the tumor cells also downregulated the B7H. This is consistent with our previous reports (15, 32). Interestingly, the cells from J558-B7H-B maintained a substantial level of H-2Ld, but lost all B7H. Selective loss of B7H in the recurrent tumors reaffirms the notion that B7H promotes CTL recognition of tumor cells in vivo.
Discussion
An important advance in the area of T cell costimulation is the identification of the members of the extended B7 family and their receptors (17, 19–21, 26, 33). Until now, most studies have focused on the roles of new B7 family members in CD4 T cells responses. Although the function of other members is still controversial (17, 18, 21, 22, 33, 34), accumulating evidence supports a critical role for B7H–ICOS interaction in the induction and the effector function of Th2 cells, including the induction of germinal center formation, regulation of Th1 response, and development of allergy and autoimmune disease (33, 35, 36). Essentially all of these studies have focused on CD4 T cells, and the function of the new B7 family members for CD8 T cells has not been reported.
The results presented in this study showed that B7H promoted both clonal expansion and effector function of tumor-specific CD8 T cells in vivo. Thus, in syngeneic mice, transfection with the B7H-GFP gene induced rejection of J558-B7H-GFP and subsequent immunity to unmodified J558 cells. Corresponding to this, we observed that the TIL isolated from J558-B7H, but not J558-Neo, had strong ex vivo cytotoxicity specific for P1A, the major tumor antigen expressed on the J558 tumors. As adoptive transfer of RAG-2−/− P1CTL conferred protection in the absence of other antigen-specific T cells, it is most likely that the enhanced P1A-specific CTL response contributed to the rejection of J558 tumors in the syngeneic nontransgenic mice.
B7H-expressing J558 tumor cells are substantially more potent in inducing clonal expansion of tumor-specific T cells. Interestingly, clonal expansion of P1CTL in vivo requires costimulation by B7–1 and B7–2. As B7–1 and B7–2 are neither expressed, nor induced on the J558 tumor cells (31), it is likely that anti–B7–1/2 blocked cross-presentation of P1A antigen by host APCs. Consistent with this, we have recently demonstrated that both direct and cross-presentation of P1A antigen to T cells leads to activation of P1A-specific CTL in vivo (37). The fact B7H enhance clonal expansion of tumor-specific T cells in vivo explains the increased number of tumor-specific CTLs among the B7H TIL, and the increased immunity to subsequent challenge with parental tumor cells. However, when P1CTL were adoptively transferred to mice that had a mixture of both B7H+ and B7H− cells, the T cells preferentially rejected the B7H+ tumor cells. More strikingly, the increased susceptibility of the B7H+ tumors could be observed even when the tumors were allowed to grow to 1–2 cm in diameter before T cell transfer. The mechanism by which B7H promotes cognate destruction of J558-tumor in vivo is not fully understood at the present. As the effect of B7H on tumor cell susceptibility is modest at best, the simplest interpretation, that B7H promote cytolysis of tumor cells to CTL lysis, while not excluded, may not explain the enhanced cognate destruction. Moreover, we have not observed induction of cytokine gene expression by B7H, as measured by RNase protection (data not shown).
We and others have observed that expression of B7–1 on the tumor cells promotes destruction of tumor cells in vivo. While Wu et al. (13) suggested that enhanced lysis by NK cells is responsible, our results demonstrated that B7–1 enhanced CTL-mediated rejection of J558 cells in vivo, and enhanced cytolysis in vitro (12). It is worth mentioning that for at least three lineages of P1A-expressing tumors, J558, Meth A, and P815, B7–1 expressed on the tumor cells promoted cognate destruction of tumor cells (38). As expression of B7–1 did not enhance lysis of P815 and Meth A by P1CTL (data not shown), it is unlikely that enhanced lysis as measured by the in vitro CTL was essential for the function of B7–1 at the effector phase. Regardless of the mechanism, the role for B7–1 at the effector phase indicated that the effector function of antitumor CTL can be regulated in vivo. Even though B7H is not unique in its intrinsic ability to promote the cognate destruction of tumor cells, the expression pattern of B7H and its receptor ICOS suggests an important implication of our results. Unlike B7–1 and B7–2, which appear restricted to the hematopoietic cells in vivo, the B7H gene is expressed in essentially all nonhematopoietic tissues tested, such as the heart, lung, brain, kidney, and liver (23). In addition, B7H is highly inducible by proinflammatory cytokines such as tumor necrosis factor and interferon γ (19, 24, 25). As activated, but not resting, T cells express ICOS (26), it is possible that the effector function of activated T cells is regulated by B7H, and that inflammation may ultimately determine target susceptibility to CTL recognition. Finally, as B7H enhances tumor susceptibility to T cell therapy, one may be able to tap into the potential of the large number of tumor-specific T cells in the melanoma patients (10, 11) by activating B7H-mediated signal transduction within the tumor milieu.
Acknowledgments
We thank Jennifer Kiel for editorial assistance, and Dr. Peisheng Du for typing the transgenic mice.
This study is supported by grants from National Institute of Health (CA58033, CA69091, and AI32981 to Y. Liu and CA82355 to P. Zheng).
Note Added in Proof: A recent study by Wallin et al. ( J. Immunol. 2001. 167:132–139) demonstrated that expression of B7H on tumors promoted their rejection, primarily by enhancing recall CD8 T cell response.
Footnotes
Abbreviations used in this paper: GFP, green fluorescence protein; ICOS, inducible costimulatory molecule; RAG, recombination activating genes; TIL, tumor-infiltrating lymphocyte.
References
- 1.Zinkernagel, R.M. 1996. Immunology taught by viruses. Science. 271:173–178. [DOI] [PubMed] [Google Scholar]
- 2.Lafferty, K.J., S.J. Prowse, C.J. Simeonovic, and H.S. Warren. 1983. Immunobiology of tissue transplantation: a return to the passenger leukocyte concept. Annu. Rev. Immunol. 1:143–173. [DOI] [PubMed] [Google Scholar]
- 3.Janeway, C. 1989. Immunogenicity signals 1, 2, 3… and 0. Immunol. Today. 10:283–286. [DOI] [PubMed] [Google Scholar]
- 4.Liu, Y., and P.S. Linsley. 1992. Costimulation of T-cell growth. Curr. Opin. Immunol. 4:265–270. [DOI] [PubMed] [Google Scholar]
- 5.Mueller, D.L., M.K. Jenkins, and R.H. Schwartz. 1989. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol. 7:445–480. [DOI] [PubMed] [Google Scholar]
- 6.Cantor, H., and E.A. Boyse. 1975. Functional subclasses of T lymphocytes bearing different Ly antigens. II. Cooperation between subclasses of Ly+ cells in the generation of killer activity. J. Exp. Med. 141:1390–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steinman, R.M., J.C. Adams, and Z.A. Cohn. 1975. Identification of a novel cell type in peripheral lymphoid organs of mice. IV. Identification and distribution in mouse spleen. J. Exp. Med. 141:804–820. [PMC free article] [PubMed] [Google Scholar]
- 8.Sarma, S., Y. Guo, Y. Guilloux, C. Lee, X.-F. Bai, and Y. Liu. 1999. Cytotoxic T lymphocytes to an unmutated tumor antigen P1A: normal development but restrained effector function. J. Exp. Med. 189:811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanson, H.L., D.L. Donermeyer, H. Ikeda, J.M. White, V. Shankaran, L.J. Old, H. Shiku, R.D. Schreiber, and P.M. Allen. 2000. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 13:265–276. [DOI] [PubMed] [Google Scholar]
- 10.Lee, P.P., C. Yee, P.A. Savage, L. Fong, D. Brockstedt, J.S. Weber, D. Johnson, S. Swetter, J. Thompson, P.D. Greenberg, et al. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5:677–685. [DOI] [PubMed] [Google Scholar]
- 11.Romero, P., P.R. Dunbar, D. Valmori, M. Pittet, G.S. Ogg, D. Rimoldi, J.L. Chen, D. Lienard, J.C. Cerottini, and V. Cerundolo. 1998. Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J. Exp. Med. 188:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramarathinam, L., M. Castle, Y. Wu, and Y. Liu. 1994. T cell costimulation by B7/BB1 induces CD8 T cell-dependent tumor rejection: an important role of B7/BB1 in the induction, recruitment, and effector function of antitumor T cells. J. Exp. Med. 179:1205–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu, T.C., A.Y. Huang, E.M. Jaffee, H.I. Levitsky, and D.M. Pardoll. 1995. A reassessment of the role of B7-1 expression in tumor rejection. J. Exp. Med. 182:1415–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chong, H., G. Hutchinson, I.R. Hart, and R.G. Vile. 1998. Expression of B7 co-stimulatory molecules by B16 melanoma results in a natural killer cell-dependent local anti-tumour response, but induces T-cell-dependent systemic immunity only against B7-expressing tumours. Br. J. Cancer. 78:1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng, P., S. Sarma, Y. Guo, and Y. Liu. 1999. Two mechanisms for tumor evasion of preexisting cytotoxic T-cell responses: lessons from recurrent tumors. Cancer Res. 59:3461–3467. [PubMed] [Google Scholar]
- 16.Allison, J., L.A. Stephens, T.W. Kay, C. Kurts, W.R. Heath, J.F. Miller, and M.F. Krummel. 1998. The threshold for autoimmune T cell killing is influenced by B7-1. Eur. J. Immunol. 28:949–960. [DOI] [PubMed] [Google Scholar]
- 17.Dong, H., G. Zhu, K. Tamada, and L. Chen. 1999. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 5:1365–1369. [DOI] [PubMed] [Google Scholar]
- 18.Freeman, G.J., A.J. Long, Y. Iwai, K. Bourque, T. Chernova, H. Nishimura, L.J. Fitz, N. Malenkovich, T. Okazaki, M.C. Byrne, et al. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swallow, M.M., J.J. Wallin, and W.C. Sha. 1999. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity. 11:423–432. [DOI] [PubMed] [Google Scholar]
- 20.Yoshinaga, S.K., J.S. Whoriskey, S.D. Khare, U. Sarmiento, J. Guo, T. Horan, G. Shih, M. Zhang, M.A. Coccia, T. Kohno, et al. 1999. T-cell co-stimulation through B7RP-1 and ICOS. Nature. 402:827–832. [DOI] [PubMed] [Google Scholar]
- 21.Chapoval, A.I., J. Ni, J.S. Lau, R.A. Wilcox, D.B. Flies, D. Liu, H. Dong, G.L. Sica, G. Zhu, K. Tamada, and L. Chen. 2001. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2:269–274. [DOI] [PubMed] [Google Scholar]
- 22.Latchman, Y., C.R. Wood, T. Chernova, D. Chaudhary, M. Borde, I. Chernova, Y. Iwai, A.J. Long, J.A. Brown, R. Nunes, et al. 2001. PD-L2 is a second ligand for PD-I and inhibits T cell activation. Nat. Immunol. 2:261–268. [DOI] [PubMed] [Google Scholar]
- 23.Ling, V., P.W. Wu, H.F. Finnerty, K.M. Bean, V. Spaulding, L.A. Fouser, J.P. Leonard, S.E. Hunter, R. Zollner, J.L. Thomas, et al. 2000. Cutting edge: identification of GL50, a novel B7-like protein that functionally binds to ICOS receptor. J. Immunol. 164:1653–1657. [DOI] [PubMed] [Google Scholar]
- 24.Aicher, A., M. Hayden-Ledbetter, W.A. Brady, A. Pezzutto, G. Richter, D. Magaletti, S. Buckwalter, J.A. Ledbetter, and E.A. Clark. 2000. Characterization of human inducible costimulator ligand expression and function. J. Immunol. 164:4689–4696. [DOI] [PubMed] [Google Scholar]
- 25.Brodie, D., A.V. Collins, A. Iaboni, J.A. Fennelly, L.M. Sparks, X.N. Xu, P.A. van der Merwe, and S.J. Davis. 2000. LICOS, a primordial costimulatory ligand? Curr. Biol. 10:333–336. [DOI] [PubMed] [Google Scholar]
- 26.Hutloff, A., A.M. Dittrich, K.C. Beier, B. Eljaschewitsch, R. Kraft, I. Anagnostopoulos, and R.A. Kroczek. 1999. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 397:263–266. [DOI] [PubMed] [Google Scholar]
- 27.Wu, Y., Y. Guo, and Y. Liu. 1993. A major costimulatory molecule on antigen-presenting cells, CTLA4 ligand A, is distinct from B7. J. Exp. Med. 178:1789–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hathcock, K.S., G. Laszlo, H.B. Dickler, J. Bradshaw, P. Linsley, and R.J. Hodes. 1993. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science. 262:905–907. [DOI] [PubMed] [Google Scholar]
- 29.Lyons, A.B., and C.R. Parish. 1994. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171:131–137. [DOI] [PubMed] [Google Scholar]
- 30.Ramarathinam, L., S. Sarma, M. Maric, M. Zhao, G. Yang, L. Chen, and Y. Liu. 1995. Multiple lineages of tumors express a common tumor antigen, P1A, but they are not cross-protected. J. Immunol. 155:5323–5329. [PubMed] [Google Scholar]
- 31.Maric, M., P. Zheng, S. Sarma, Y. Guo, and Y. Liu. 1998. Maturation of cytotoxic T lymphocytes against a B7-transfected nonmetastatic tumor: a critical role for costimulation by B7 on both tumor and host antigen-presenting cells. Cancer Res. 58:3376–3384. [PubMed] [Google Scholar]
- 32.Zheng, P., Y. Guo, Q. Niu, D.E. Levy, J.A. Dyck, S. Lu, L.A. Sheiman, and Y. Liu. 1998. Proto-oncogene PML controls genes devoted to MHC class I antigen presentation. Nature. 396:373–376. [DOI] [PubMed] [Google Scholar]
- 33.Dong, C., A.E. Juedes, U.A. Temann, S. Shresta, J.P. Allison, N.H. Ruddle, and R.A. Flavell. 2001. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 409:97–101. [DOI] [PubMed] [Google Scholar]
- 34.Tseng, S.Y., M. Otsuji, K. Gorski, X. Huang, J.E. Slansky, S.I. Pai, A. Shalabi, T. Shin, D.M. Pardoll, and H. Tsuchiya. 2001. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193:839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tafuri, A., A. Shahinian, F. Bladt, S.K. Yoshinaga, M. Jordana, A. Wakeham, L.M. Boucher, D. Bouchard, V.S. Chan, G. Duncan, et al. 2001. ICOS is essential for effective T-helper-cell responses. Nature. 409:105–109. [DOI] [PubMed] [Google Scholar]
- 36.McAdam, A.J., R.J. Greenwald, M.A. Levin, T. Chernova, N. Malenkovich, V. Ling, G.J. Freeman, and A.H. Sharpe. 2001. ICOS is critical for CD40-mediated antibody class switching. Nature. 409:102–105. [DOI] [PubMed] [Google Scholar]
- 37.Guilloux, Y., X.F. Bai, X. Liu, P. Zheng, and Y. Liu. 2001. Optimal induction of effector but not memory antitumor cytotoxic T lymphocytes involves direct antigen presentation by the tumor cells. Cancer Res. 61:1107–1112. [PubMed] [Google Scholar]
- 38.Bai, X.F., J. Bender, J. Liu, H. Zhang, Y. Wang, O. Li, P. Du, P. Zheng, and Y. Liu. 2001. Local costimulation reinvigorates tumor-specific cytolytic T lymphocytes for experimental therapy in mice with large tumor burdens. J. Immunol. 167:3936–3943. [DOI] [PubMed] [Google Scholar]