

Abstract
Recombinant streptococcal pyrogenic exotoxin C (SPE-C) is a potent superantigen that stimulates Vβ2-bearing human T cells, but is inactive in mice. SPE-C binds with high affinity to both human HLA-DR and murine I-E molecules, but not to murine I-A molecules in a zinc-dependent fashion. Competition binding studies with other recombinant toxins revealed that SPE-C lacks the generic low affinity major histocompatibility complex (MHC) class II α-chain binding site common to all other bacterial superantigens. Despite this, SPE-C cross-links MHC class II to induce homotypic aggregation of class II–bearing B cells. Nondenaturing sodium dodecyl sulfate electrophoresis and size exclusion chromatography revealed that both wild-type and recombinant SPE-C exist in a stable dimer at neutral or alkaline pH. These data support a recent crystal structure of SPE-C and reveal yet another mechanism by which bacterial superantigens ligate and cross-link MHC class II.
Bacterial superantigens (SAgs)1 produced by Staphylococcus aureus and Streptococcus pyogenes simultaneously bind to MHC class II antigens and T cell receptor molecules via the Vβ domain and stimulate large numbers of T cells (1–3). SAgs stimulate high systemic levels of the proinflammatory cytokines TNF-α and IL-1β and the T cell mediators IL-2 and INF-γ that cause hypertension, fever, and shock. All SAgs require initial presentation by MHC class II molecules for maximal activity (4–7), but the role of the MHC class II molecule appears to differ depending on the SAg. Crystal structures are now available for staphylococcal enterotoxin (SE)A (8), SEB (9), SEC2 (10), and toxic shock syndrome toxin (TSST; 11), and although there are significant differences in surface features, each molecule conforms to the same overall folding pattern, with a small N-terminal β-barrel globular domain and a larger COOH-terminal globular domain of the β-grasp motif. Despite conservation of three-dimensional structure, individual staphylococcal and streptococcal SAgs have developed different ways of binding to MHC class II. For example, SEB and toxic shock syndrome toxin TSST-1 both bind to the invariant MHC class II α chain, but SEB binds out to the side of the α chain leaving the top of the class II molecule exposed (12), whereas TSST binds over the top of the class II α chain and obscures the α chain α helix, the peptide, and some of the β chain α helix (13). This implies that TCRs, when ligated by TSST, do not contact the MHC class II molecule to which TSST is bound. SEB, on the other hand, appears to rely heavily on continued interaction between TCRs and MHC class II to stabilize a trimeric complex between SEB, MHC class II, and TCR (14–16).
Some SAgs, such as SEA, have an additional binding site for the polymorphic β chain of MHC class II (17–19) that is 100 times stronger than α-chain binding. This interaction results from a tetrameric zinc coordination complex between H187, H225, and D227 of SEA and the highly conserved H81 of the MHC class II β chain (17), and provides SEA with the ability to cooperatively bind and stabilize a second SEA molecule to the low affinity class II α-chain site, promoting cross-linking of MHC class II molecules on the surface of the APC (17–19). Cross-linking of MHC class II is an important component of SEA activity (18, 20), although the exact mechanism by which this enhances SAg stimulation is not yet known. Experimental evidence indicates that the low affinity α-chain interaction is more important to TCR ligation than MHC class II binding because mutations in this site affect T cell activation without significantly altering class II binding, whereas mutations in the β-chain site destroy all β- and α-chain binding, and subsequent T cell activation (18, 19).
Despite their role in human disease, the streptococcal toxins have not been as well studied as the staphylococcal toxins. There are three known streptococcal superantigens, streptococcal pyrogenic exotoxin (SPE)-A, SPE-C (21), and SSA (22). SPE-A and SSA are most similar in sequence to SEB, whereas SPE-C shares little similarity to any other member of the SAg family, and is least homologous with TSST. Nevertheless, there is sufficient sequence homology among all of the staphylococcal and streptococcal toxins to suggest that they have evolved from a common ancestral gene and are likely to fold in a similar manner.
This study has examined SPE-C in detail and reveals unique features that distinguish it from other superantigens. Like TSST, SPE-C is a powerful in vitro activator of human Vβ2-bearing T cells with a potency approaching that of SEA. However, recombinant SPE-C is totally inactive against murine T cells. Evidence is presented that shows that SPE-C is a dimer molecule that no longer uses the generic α-chain binding site, but instead binds only to the β chain of MHC class II by a zinc-mediated mechanism similar to SEA. The conclusions made from these functional data are supported by the recently resolved crystal structure of SPE-C (23).
Materials and Methods
Toxin Purification.
All toxins were expressed from the pGEX vector in Escherichia coli as glutathione S transferase (GST) fusion proteins and purified by glutathione chromatography as previously described (24). Mature toxins were cleaved from GST by trypsin digestion and purified by two rounds of cation exchange chromatography. The first round using carboxymethyl sepharose and the second on a POROS HS (Perceptive Systems, Cambridge, MA) HPLC column. All toxins were resistant to trypsin digestion except SEB which has a single cleavage site in the disulphide loop region. This does not affect SEB activity.
Toxin Proliferation Assays.
Human peripheral blood lymphocytes were purified by Ficoll-Hypaque and incubated for 3 d at 106 cells/ml in duplicate in 96-well microtiter plates in media containing varying dilutions of recombinant toxins. 0.1 μCi [3H]thymidine was added to each well, and cells were incubated a further 24 h. Plates were harvested and counted on a scintillation counter.
Radiolabeling and Cell-binding Experiments.
5 μg of purified toxin was radioiodinated by the chloramine T method to a specific activity of ∼50–100 μCi/μg. Labeled toxin was separated from free iodine on a 1 ml Sephadex G25 (Pharmacia, Piscataway, NJ) column and stored at 1 μg/ml in 50% glycerol–1% hemaglobin phosphate buffered saline. LG-2 cells are a human B lymphoblastoid cell line homozygous HLA-DR1. Cells were harvested in log phase, washed in RPMI/10% fetal calf serum, and suspended at 107/ml of the same medium. For duplicate binding studies using either SEA, TSST, or SPE-C, the 125I-tracer toxin was used at 2 ng/ml with the addition of 0.01–100 μg of unlabeled toxin. 106 cells (100 μl) were added and incubated for 1 h at 37°C. Cells were washed three times with ice-cold medium and centrifuged pellets were counted in a gamma counter. For SEB inhibition studies, 1 μg/ml of 125I-SEB was used as tracer due to the much lower affinity that SEB binds to LG-2 cells with, and samples were incubated for 4 h at 37°C before washing and counting. For binding studies to mouse B cells, single cell suspensions of splenocytes were pretreated at 5 × 106/ml for 4 d in vitro with 20 μg/ml lipopolysacharride (Sigma Chemical Co., St. Louis, MO) in 10% FCS-DMEM to generate B cell blasts that were harvested and used for cell binding at 107 cells/tube. Cells were incubated for 1 h at 37°C, and then washed three times in ice-cold medium before gamma counting.
Fluorescence Labeling of SEB.
1 mg (1 mg/ml) SEB in 0.1 M sodium carbonate, pH 8.5, was incubated with 20 μl of 5 mg/ml fluorescein isothiocyanate in DMSO (Sigma Chemical Co.) overnight in the dark at room temperature. Fluoresceinated SEB was separated from free FITC on a 1-ml G25 column and stored at 4°C with 0.02% NaN3. LG-2 cells (6 × 105 cells) were incubated for 2 h at 37°C with shaking with 1 μg/ml FITC SEB and a 10-fold serial dilution of unlabeled toxin starting at 0.1 mg/ml. After incubation, cells were washed with 2 × 0.5 ml ice-cold PBS and examined immediately on a FACSCAN® (Becton Dickinson, San Jose, CA).
TCR Vβ Analysis.
These were performed as previously described using the reverse dot-blot procedure (24). In brief, human peripheral blood lymphocytes were incubated at 105/ml with 1 ng/ml of recombinant toxins for 3 d. The cultures were expanded twofold with medium containing 20 ng/ml IL-2. Cells were harvested at 4 d and RNA made by standard procedures (25). TCR β-chain messenger was reverse transcribed using a set of primers specific for a conserved region in all β-chain genes. Amplification of a 500-bp Vβ probe was accomplished by an anchor primer to the 5′ end of the β-chain primers plus a single Cβ region primer. This probe was radiolabeled and reverse blotted to filters containing individual β-chain genes. Relative changes in individual β-chain mRNA were compared to unactivated PBL.
Anti-TCR mAb FACS® Staining.
Activated T cells were incubated for 1 h on ice with 25 ml of anti-TCR BV2 (MPB2/ C11; a gift from A.W. Boylston, Universtiy of Leeds, Leeds, UK), anti-BV551 (LC4; a gift from R. Levy, Stanford University Medical School, Stanford, CA), anti-BV5S3 (42/1C1; a gift from A.W. Boylston, Universtiy of Leeds, Leeds, UK), anti-BV8.1 (C305; a gift from A. Weiss, University of California, San Francisco, CA), and anti-BV12S (S511; a gift from D. Posnett, Cornell University Medical College, NY). Washed cells were then incubated with 1 ml FITC goat anti–mouse (Becton Dickinson) and incubated on ice for a further 30 min. After washing, cells were analyzed on a FACSCAN®.
Zinc Blots.
Recombinant toxins (10 μg) were incubated in triplicate with 10 μM EDTA followed by 100 μM 65ZnCl (New England Nuclear, Boston, MA) in 20 mM Tris, pH 8.0, 10 mM MgCl2, 0.15 M NaCl made zinc free by addition of chelex resin (Sigma Chemical Co.) as previously described (17). Samples were then dot blotted to nitrocellulose filters using a 96-well dot-blot apparatus. Filters were washed briefly three times with zinc-free buffer, and then autoradiographed. Spots were cut out and counted on gamma counter (Packard Instrs., Meriden CT) to quantify 65Zn bound to each toxin.
Nondenaturing SDS Electrophoresis.
Toxin samples were incubated in standard Laemmli reducing sample buffer (containing 1% SDS and 10 mM dithiothreitol), and then resolved as normal of a 12.5% acrylamide gel. For denaturing conditions, samples were heated to 100°C for 2 min before loading. To prevent dimers from dissociating during running, the power was maintained below 2 W (20 mA and 100 V). Some reduced samples were treated with 20 mM iodoacetic acid, pH 7.0, before loading. EDTA was added to some samples at 10 mM.
LG-2 Cell Aggregation Assay.
Healthy LG-2 were washed in RPMI–10% FCS, vigorously resuspended at 5 × 105 cells/ml in RPMI–10% FCS and aliquoted as a single-cell suspension to a 96-well flat-bottom plate in a final volume of 100 μl. Cells in duplicate wells were stimulated by the addition of toxin to ∼37 nM (1.0 μg/ml). Aggregation was scored at various time points after incubation at 37°C as the percentage of LG-2 cells in aggregates, and was determined by light microscopy.
Western Blotting of S. pyogenes Strain 2035–derived SPE-C.
S. pyogenes strain 2035 was grown under anaerobic conditions in brain heart fusion medium at 37°C for 24 h without shaking. Supernatant proteins were concentrated by sequential (NH4)2SO4 precipitations, with cuts of <40%, 40–60%, 60–80%, and 80% saturation, and resuspended in 50 mM Tris–1 mM EDTA, pH 7.4, at ∼500–1,000 times their original concentration. Recombinant SPE-C or 10 μl of the 60–80% (NH4)2SO4-precipitable fraction of 2035 supernatant were combined with an equal volume of nonreducing 2% SDS sample buffer and separated by 0.1% SDS–12% PAGE. Denatured samples were heated (95°C) for 2 min before analysis. Fractions were dialyzed extensively against 25 mM Tris–50 mM NaCl–1 mM EDTA, pH 7.4, to remove salt. After separation by SDS-PAGE, proteins were transferred to a nitrocellulose filter (Hybond-C; Amersham Corp., Arlington Heights, IL) in an electroblotting apparatus using Towbin buffer lacking SDS (25 mM Tris.HCl, pH 8.5–150 mM glycine– 10% methanol). The filter was blocked in PBS–0.05% Tween–5% nonfat dried milk powder–0.1% normal rabbit serum and stained with 1:6,000 peroxidase-labeled affinity-purified rabbit anti–SPE-C immunoglobulin. The peroxidase conjugate was detected on radiographic film by chemiluminescence (ECL; Amersham Corp.) according to the manufacturer's instructions.
Size Exclusion Chromatography.
Recombinant SPE-C (2 mg/ ml) was dialyzed at 4°C overnight in 20 mM BisTris-Tris, pH 6.0 or 9.0. 20 μl samples were diluted into 100 μl 50 mM BisTris, pH 6.0, 7.0, 8.0, or 9.0/0.1 M NaCl and incubated for 1 h at room temperature before separation at 1 ml/min (±0.05 ml/min) on a Superose12 (Pharmacia) high resolution HPLC column attached to a Biocad Sprint (Perceptive Systems) preequilibrated with the respective incubation buffer. Trace chromatograms monitoring Abs280nm, pH, and conductivity were all recorded directly and subsequently analyzed for retention times, peak integration, and peak assignment using the on-line Biocad software. Traces were grouped and printed using the stacked trace mode which automatically aligns each trace to the injection point.
Results
All toxins were expressed in E. coli as GST fusion proteins and purified by glutathione agarose affinity chromatography, cleaved with trypsin, and purified by HPLC to a point of apparent homogeneity by SDS-PAGE gel analysis. To ensure native conformation after expression in E. coli, each recombinant toxin was tested for its potency in the standard 3-d [3H]thymidine assay. All, including SPE-C, were active at concentrations <1 pg/ml on human T cells (Table 1). The most potent of the recombinant toxins was SPE-C, which gave a half-maximal response of 5 fg/ml and was still active at <0.1 fg/ml. In contrast, SPE-C completely lacked proliferative activity for mouse T cells in all strains tested. No activity was observed above background in any mouse strain, even at the highest concentration of SPE-C tested (100 μg/ml). By comparison, all other recombinant toxins were active on murine T cells at <1 ng/ ml (except for SEB on C57Bl/6). The proliferative activity varied greatly between strains and toxins (Table 1). The half-maximal activity of SEB on BALB/c T cells was 4 pg/ ml, but 10,000 pg/ml for C57Bl/6 mice, representing a 2,500-fold difference between the two strains. The most consistently potent toxin on mouse T cells was TSST, which varied in its half-maximal activity from 0.6 pg/ml for C57Bl/6 mice to 20 pg/ml for C3H mice.
Table 1.
Relative Potency of Recombinant SA on Human and Mouse T Cells
| Proliferation potential P50% | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BALB/c | SJL | B10.M | C57Bl/6 | C3H | Human PBL | |||||||
| pg/ml | ||||||||||||
| SEA | 40 | 0.1 | 80 | 6 | 20 | 0.01 | ||||||
| SEB | 4 | 900 | 200 | 10,000 | 300 | 0.04 | ||||||
| SPEC-C | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 | 0.005 | ||||||
| TSST | 2 | 20 | ND | 0.6 | 20 | 0.03 | ||||||
Values reflect the concentration of recombinant toxin required to produce 50% maximal proliferation. There was no detectable proliferation for SPE-C at the highest concentration tested on murine splenocytes.
Amino acid sequence alignment of SEA and SPE-C revealed that SPE-C shared two out of the three zinc coordinating residues present in SEA. Residues H201 and D203 in SPE-C are analogous to H225 and D227 in SEA that form the essential bidentate component found in all solvent exposed zinc sites (8, 26). This suggested that SPE-C might bind zinc in the same way as SEA. Identification of the third ligand was not obvious from direct sequence alignment because the corresponding residues to SEA-H187 is G161, which is unlikely to act as a zinc ligand. However, three other residues in close proximity to G161 could potentially serve as zinc ligands. These are E159, D164, or H167.
Recombinant SPE-C was tested for its ability to bind radioactive 65Zn in a zinc dot-blot assay alongside recombinant SEA, SEE, SEB, and SPE-A (Fig. 1). SPE-C bound 65Zn 100 times greater than background and 50-fold higher than either SEB or SPE-A (two toxins that do not require Zn2+ for superantigen function). SEA bound the highest levels of 65Zn in this assay, whereas both SPE-C and SEE bound approximately half this level. Given that bound 65Zn is determined at nonequilibrium conditions, these differences reflect a combination of both the equilibrium conditions, these differences reflect a combination of both the equilibrium affinity and the off-rate of zinc for the different toxin binding sites.
Figure 1.

65Zn binding to SPE-C. Recombinant toxins were incubated with 100 μM 65Zn and blotted in triplicate to nitrocellulose. After washing, the strips were autoradiographed and the individual dots counted.
The importance of zinc to SPE-C activity was examined with direct binding of 125I SPE-C to human and mouse cells expressing MHC class II (Fig. 2). SPE-C bound with high affinity to human LG-2 cells (which are homozygous HLA-DR1), but binding was completely abolished in the presence of 10 μM EDTA (Fig. 2 A). Equivalent binding was seen in MHC class II mice expressed on LPS-activated mouse spleen cells expressing both I-A and I-E (BALB/c I-Eb, C3H/HeJ I-Es). Strains such as C57Bl/6, B10M/Sn, and SJL/J B-cell blasts that lack expression of the I-Eβ chain bound 125I–SPE-C poorly (Fig. 2 B) demonstrating that the primary target for SPE-C was the I-E molecule. Maximum binding of SPE-C to both human and mouse cells was restored by a 10 μM excess of zinc over the EDTA.
Figure 2.
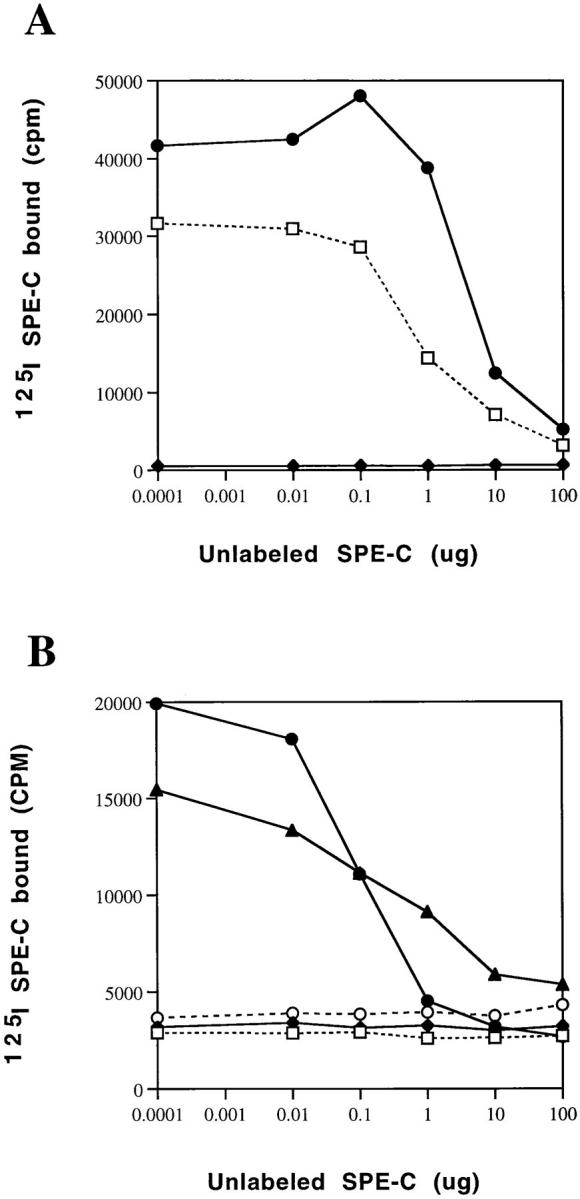
SPE-C binding to human and murine MHC class II. Radioiodinated SPE-C was incubated with (A) 106 LG-2 cells or (B) 106 LPS-activated mouse splenocytes with increasing amounts of unlabeled SPE-C for 1 h at 37°C, and then the cells were washed and counted. (A) □, Incubations in the media; ♦, media plus 10 mM EDTA; or •, media plus 10 mM EDTA and 20 mM ZnCl2. (B) □, B10M; ♦, SJL/J; •, BALB/c; ▴, C3H; ○, C57.
Competition binding experiments between SPE-C and other toxins were performed in an attempt to determine the orientation of SPE-C on MHC class II. The results of these binding experiments are shown in Fig. 3. SEA was the only toxin to inhibit 125I–SPE-C binding to LG-2 cells, and was only slightly less competitive than SPE-C itself (Fig. 3 A). Neither TSST nor SEB inhibited 125I–SPE-C, even in 10,000-fold molar excess. Both SEB and TSST bind exclusively to the MHC class II α chain (12, 13). In the reciprocal experiment, only SEA and SPE-C prevented 125I-SEA binding (Fig. 3 B). Neither SEB nor TSST inhibited. Although SEA and SPE-C appeared to bind to MHC class II in a similar fashion, competition experiments using 125I-SEB revealed important differences (Fig. 3 C). Even at the highest concentration tested (10 μg/ml), SPE-C did not inhibit 125I-SEB binding to LG-2 cells, whereas SEA remained an effective competitor, almost as efficient as SEB itself. The same experiment was performed using FITC-labeled SEB (Fig. 3 D) with identical results, confirming that SPE-C does not share with SEA the ability to prevent SEB binding and thus is unlikely to bind directly to the MHC class II α chain. The ability of SPE-C to inhibit TSST binding was also tested (Fig. 3 E). SPE-C was a strong inhibitor of 125I-TSST binding, a result contrary to the reverse experiment (Fig. 3 A) that showed that TSST did not prevent SPE-C binding.
Figure 3.

Competition binding studies. □, SPE-C; ♦, SEA; •, SEB; ▴, TSST. (A) 125I– SPE-C was incubated at 2 ng/ml with 106 LG-2 cells in the presence of increasing concentrations of unlabeled SPE-C, SEA, SEB, or TSST. Cells were incubated in duplicate for 1 h, and then washed and counted in a gamma counter. (B) Same as above except 125I-SEA was the tracer. (C) Same as above except that 125I-SEB was the tracer. (D) Fluoresceinated SEB (1 mg/ml) was incubated for 2 h at 37°C with LG-2 cells, and then washed briefly and analyzed by FACS® analysis. (E) Same as above except 125I-TSST was used as the tracer.
The human TCR Vβ specificity of recombinant SPE-C was determined by reverse PCR dot-blot analysis (24) and the relative Vβ enrichment compared with that induced by SPE-A or Con A. The results are presented in Table 2. SPE-C almost exclusively targeted Vβ2-bearing cells, enriching this particular Vβ mRNA over 30-fold (2–60%). The only other Vβs displaying significant enrichment over the Con A–activated control sample were Vβ4.1 (2-fold), Vβ12.5 (9-fold), and Vβ15 (1.6-fold), which increased from 0.2 to 1.7% (an 8-fold increase). The sum total of all Vβs stimulated by SPE-C was 94.4%, suggesting that all the Vβ species targeted were accounted for in the probe panel. This was in contrast to SPE-A where the panel of Vβ probes represented only 22.7% of the total enriched β-chain cDNA. Presumably, other Vβs absent from the probe panel were the major target of SPE-A action. All summed Vβs in the Con A–activated sample amounted to only 44.6% of the total β-chain cDNA, indicating that the Vβ panel represented about half of the total pool of transcribed TCR β chains. The PCR data were compared to surface TCR staining using a limited panel of anti–human TCR mAbs to Vβ2, 5.1, 5.3, 8.1, and 12. The results were mainly consistent, although the crucial Vβ staining of SPE-C–activated cells reflected a more modest increase in Vβ2-bearing cells from 2% in Con A–activated cells to 19% for SPE-C (10-fold increase) after 4 d of stimulation. The difference between PCR data and surface staining may simply reflect the more rapid and greater increase in intracellular TCR β-chain mRNA levels in activated T cells, compared to the much slower increase in cell number after activation.
Table 2.
Vβ Specificity of SPE-C on Human T Cells
| Vβ | Percent Vβ enrichment | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Resting | Con A | SPE-A | SPE-C | |||||||||||||
| PCR | mAb | PCR | mAb | PCR | mAb | PCR | mAb | |||||||||
| 1.1 | 0.0 | 0.0 | 0.0 | 0.5 | ||||||||||||
| 2.1 | 2.3 | 3 | 2.8 | 2 | 1.3 | <1 | 59.6 | 19 | ||||||||
| 3.2 | 6.0 | 15.7 | 10.4 | 18.8 | ||||||||||||
| 4.1 | 5.8 | 3.3 | 1.4 | 6.8 | ||||||||||||
| 5.1 | 4.2 | 2 | 3.3 | 1.7 | 1.1 | <1 | 1.4 | 3 | ||||||||
| 5.3 | 2.9 | 1.5 | 6.3 | 1.7 | 0.9 | <1 | 1.0 | <1 | ||||||||
| 6.3 | 0.7 | 0.9 | 0.3 | 0.6 | ||||||||||||
| 6.4 | 0.6 | 0.9 | 0.3 | 0.8 | ||||||||||||
| 6.9 | 1.2 | 2.4 | 0.0 | 1.0 | ||||||||||||
| 7.4 | 1.1 | 4.1 | 0.4 | 0.0 | ||||||||||||
| 8.1 | 0.5 | <1 | 3.0 | 2 | 0.4 | <1 | 0.2 | <1 | ||||||||
| 9.1 | 0.0 | 0.4 | 0.0 | 0.1 | ||||||||||||
| 12.5 | 0.3 | 1 | 0.2 | <1 | 1.5 | <1 | 1.7 | 1.5 | ||||||||
| 15.1 | 0.4 | 0.8 | 4.6 | 1.2 | ||||||||||||
| 23.1 | 1.1 | 0.3 | 0.2 | 0.7 | ||||||||||||
| Total | 27.1% | 44.6% | 22.7% | 94.4% | ||||||||||||
Human PBL were incubated with 1 ng/ml of recombinant toxins for 5 d. Relative enrichment of Vβ cDNAs was analyzed by PCR reverse dot blot (24). Cells were also stained with anti-Vβ mAbs at 5 d, after stimulation. PCR figures represent percentage of Vβ with respect to total Cβ. Antibody staining represents percent of total CD3 staining cells.
SEA and anti–MHC class II mAbs such as LB3.1 rapidly induce homotypic aggregation of LG-2 cells. In addition, both these MHC class II ligands induce the expression of proinflammatory cytokines such as IL-1 and TNF-α in MHC class II–bearing APCs (20, 27). This activity is a direct function of two separate binding sites for MHC class II molecules on SEA and presumably reflects the ability to cross-link MHC class II. Mutation of either site in SEA or papain cleavage of LB3.1 into F(ab) fragments abolishes the ability to induce homotypic aggregation. Purified recombinant SPE-C was tested in the LG-2 homotypic aggregation assay and found to be as potent as SEA (Fig. 4) suggesting that SPE-C was also capable of cross-linking MHC class II. In contrast, neither recombinant SEB nor SPE-A displayed any aggregation activity consistent with the presence of only a single MHC class II binding site on these molecules. Given that the previous experiments suggested the α-chain binding site was absent in SPE-C, two hypotheses were put forward to account for the presumed class II cross-linking activity of SPE-C. The first was that SPE-C binds to MHC class II via the zinc-mediated β-chain site and another unidentified site. The second hypothesis was that SPE-C might form dimers in solution with two identical β-chain binding sites. To test the second hypothesis, recombinant SPE-C was examined by nondenaturing SDS-PAGE in an attempt to observe SPE-C oligomers. Homodimers were clearly visible under both reducing and nonreducing conditions, representing >60% of the total amount of SPE-C loaded (Fig. 5). These dimers were absent when the sample was denatured in nonreducing conditions (not shown). One feature of these dimers was their discrete nature, even in the presence of 1% SDS and 10 mM dithiothreitol. No smearing between dimer and monomer was observed. Another feature of the SPE-C dimers was their dependence on neutral or alkaline pH for formation. At conditions below pH 6.0, virtually no dimers were detected, whereas at pH 7.0, >60% of the SPE-C was observed in dimer form. Neither EDTA nor iodoacetic acid (a sulfhydryl alkylating reagent) affected dimer stability, indicating the absence of covalently disulfide linked homodimers.
Figure 4.

SPE-C and SEA induce homotypic aggregation in LG-2 cells. Freshly split LG-2 cells were incubated with 1 μg/ml of recombinant toxin in 24-well culture plates and monitored for 24 h. Percent aggregation was determined by counting the number of cells not bound to an aggregated clump.
Figure 5.

Recombinant SPE-C forms stable dimers in solution. Recombinant SPE-C (5 mg/ml) was incubated in either 50 mM morpholine ethane sulphonic acid/Hepes/acetate buffer, pH 5.0, or 50 mM Tris-BisTris buffer, pH 7.0 or 9.0, with either 1 mM EDTA or 10 mM iodoacetamide. One volume of reducing SDS Laemmli sample buffer was added, and then samples were run on a 12.5% SDS gel without boling at a total of 0.5 W of power.
Because the SPE-C used for SDS nondenaturing studies was recombinant material stored at high concentration (5 mg/ml), protein dimerization might simply be an artifact of high protein concentration and/or recombinant expression in E. coli. Crude S. pyogenes–derived SPE-C was therefore examined by nondenaturing SDS-PAGE and Western blotting using an affinity-purified anti–SPE-C rabbit anti-serum (Fig. 6). Culture supernatant from an overnight growth of S. pyogenes strain 2035 was first concentrated 1,000-fold by ammonium sulfate precipitation because SPE-C was undetectable in the raw culture medium. The concentration of wild-type SPE-C was estimated at <10 ng/ml in the raw S. pyogenes culture supernatant and ∼10 μg/ml in the crude concentrated sample. Electroblotting of the SPE-C dimer was not as efficient as the monomer, so the relative monomer/dimer ratios were misleading in these experiments. Nevertheless, SPE-C dimer was readily detected in the crude S. pyogenes culture supernatant at approximately the same proportions as in the recombinant SPE-C, despite differences in both purity and concentration. To analyze the effect of pH on SPE-C under more physiological conditions (i.e., in the absence of SDS), SPE-C was dialyzed overnight at either pH 6.0 or 9.0, and then incubated at varying pHs before separation by size exclusion on a Superose 12 HPLC column (Fig. 7). The pH 6.0 dialyzed sample separated exclusively in the dimer form (95% by peak integration), irrespective of the pH of the separating buffer (Fig. 7 A). In contrast, 50% of the pH 9.0 dialyzed sample dissociated to the monomer form when the pH was lowered to 7.0 or less (Fig. 7 B). The apparent molecular weight of both the dimer and monomer peaks appeared to increase with increasing pH. This manifested in a consistent decrease in retention time proportional to increasing pH.
Figure 6.

Presence of SPE-C dimers in the culture supernatant of Streptococcus pyogenes. Concentrated streptococcal culture supernatant was run on a nondenaturing SDS gel and blotted to nitrocellulose. The Western blot was incubated with an affinity-purified rabbit anti–SPE-C antiserum (1:6,000) and detected by chemiluminescence. Den, boiled in SDS sample buffer; ND, nondenatured and reduced.
Figure 7.

Reversibility of SPE-C dimer formation. Recombinant SPE-C (2 mg/ml) was dialyzed overnight in (A) 10 mM BisTris-Tris, pH 6.0, or (B) 10 mM BisTris-Tris, pH 9.0. Samples (20 μl) from each dialysis were incubated for 1 h at room temperature in 100 μl 50 mM BisTris-Tris–0.1 M NaCl at either pH 6.0, 7.0, 8.0, or 9.0, and then analyzed by size exclusion chromatography. The exact retention time of each peak was determined with the Perceptive Biocad analysis software and is shown beside each peak. Each trace represents absorbance at 280 nm. The resulting figure was generated from the Perceptive Biocad program using the stacked trace mode. The small peak to the right of the pH 6.0 generated dimer is not monomer, but an SPE-C cleavage product also seen to the right of the pH 9.0 monomer peak.
This was unlikely to be due to decreasing charge interactions between the protein and Superose column media because 0.1 M NaCl was included as a counter ion to prevent such weak ionic interactions. Thus, under normal physiological conditions, the majority of recombinant SPE-C existed in dimer form that only dissociated to the monomer in the presence of an ionic detergent or after dialysis at elevated pH. This readily explained the apparent MHC class II cross-linking activity of SPE-C, and moreover, suggested that the dimer had two zinc-containing binding sites.
Discussion
The data presented in this paper demonstrate some unique features of SPE-C that set it apart from all other bacterial SAg. Most notable are (a) the apparent absence of the generic MHC class II α-chain binding site common to every other superantigen so far studied and (b) the formation of stable, noncovalent dimers under normal physiological conditions. The evidence for the absence of an MHC class II α-chain binding site in SPE-C is compelling. First, SPE-C does not compete for SEB binding to the MHC class II α chain, in contrast to SEA, which competes well. Second, amino acid comparison of SPE-C with SEA and SEB indicate major differences in the β1–β2 loop region that forms the hydrophobic ridge essential for α-chain binding site. SEB F44.L45.Y46.F47 that is central to MHC class II α-chain binding is replaced by T33.T34.H35.T36 in SPE-C. Third, in the crystal structure of SPE-C, the generic MHC class II α-chain binding site is structurally quite different from either SEA or SEB. As a consequence, SPE-C forms a symmetrical interaction with the same NH2-terminal region of another SPE-C molecule resulting in a dimer with two COOH-terminal domains pointing outwards with two potential zinc-containing β-chain binding sites located at opposite poles of the dimer (23). Significantly, many of the residues in SPE-C that are analogous to α-chain binding residues in SEA and SEB (12) are buried in the dimer interface.
Both recombinant and wild-type SPE-C form discrete, stable dimers in solution with functional properties consistent with the dimer observed in the SPE-C crystal structure (23). Although we have not formally proven that the solution dimer is exactly the same as the crystal dimer, it would seem surprising that SPE-C should undergo a significant change from one dimer structure to another during crystal formation, given the apparent stability of the solution form. Analysis of the dimer by nondenaturing SDS electrophoresis (in the presence of 1% SDS) revealed a pH dependence for dimer formation with a midpoint around pH 6–7 and a combination of both dimer and monomer forms above neutral pH. Histidine residues might form part of the solution dimer interface (pK of the free imidazole nitrogen is 6.5) and in the crystal structure, SPE-C H35 and E54 form a fourfold zinc coordination with H35 and E54 from a second SPE-C molecule. SPE-C mutants at these positions are currently being examined to confirm the importance of dimerization to SPE-C function. The role of zinc in dimer formation is not immediately obvious from our solution studies because the addition of EDTA had no effect on dimer stability. Notably, the crystal structure dimer was initially formed in the absence of zinc, indicating that it is not essential for dimer formation. Zinc was only seen in this position after soaking the crystal in 50 mM zinc (23). Currently, we believe that the zinc atom in the crystal dimer interface is merely opportunistic and does not serve any structural or functional role. Analysis of the solution dimer under more physiological conditions revealed that >95% of recombinat SPE-C was in the dimer form and required 1% SDS or mild alkaline treatment before it dissociated to the monomer. Evidence for an alkaline pH effect on the SPE-C structure was visible by size exclusion chromatography with a proportional decrease in column retention time (representing an higher apparent molecular weight) of both the dimer and monomer peaks with increasing pH. It is of special note that the dimer interface in the SPE-C crystal structure is large (>900Å2), and is formed from two regions, one made up predominantly of hydrophobic residues (the larger in surface area) and a second smaller hydrophilic region composed of residues in the β1–β2 loop (23). One possible explanation for the observed features of the SPE-C dimer might be that under normal physiological conditions, the hydrophobic interactions in the dimer interface predominate, but are disrupted by ionic detergent or mild alkaline conditions, thus placing a greater dependence for dimer stability on the pH-sensitive hydrophilic regions of the interface.
The zinc-containing β-chain binding site in the SPE-C crystal structure consists of residues H167, H201, and D203 (23). Although we have not formally confirmed by mutagenesis that these residues form the MHC class II β-chain binding site, its structural similarity to the zinc site of SEA (8), the finding that EDTA destroys SPE-C binding and the strong cross-competition between SEA and SPE-C suggest that this zinc site coordinates to the MHC class II β chain in a similar manner to SEA (17–19). In the SPE-C crystal dimer, two putative β-chain binding sites are at opposite ends of the dimer in an optimum position to ligate two MHC class II molecules (23). This structure is consistent with the potent APC activation activity observed, suggesting that SPE-C cross-links two independent MHC class II molecules via their β chains.
The ability of SPE-C to prevent TSST binding despite the absence of an α-chain binding site in the former toxin might best be explained by steric hindrance between the two toxins across the top of the MHC class II molecule rather than direct competition for α-chain sites. In support of this conclusion, SPE-C did not inhibit 125I-TSST binding in the presence of EDTA (not shown). The reciprocal competition experiment showed that TSST did not prevent SPE-C binding. One possible explanation for this might be that TSST binding to MHC class II is more restricted than SPE-C due to a greater dependence on peptide residues (28). This question will only be resolved by binding studies using a single soluble DR1/peptide combination.
The zinc-mediated binding site in the COOH-terminal domain of SEA, SEE, and SED is a remarkable addition to the generic low affinity MHC class II α-chain binding of other bacterial superantigens that allows these superantigens to bind to the MHC class II β chain despite the extensive polymorphism of this part of the MHC class II molecule. Nevertheless, SEA still requires additional binding to the MHC class II α-chain site for optimal activity. Multiple combined mutations in the SEA α-chain binding site (SEA-F47S.L48A.Y92A) reduce T cell activity by over 800-fold without significantly altering MHC class II binding affinity (19, 20, 29). The ability of SEA to cross-link MHC class II on the surface of the APC not only increases the rate of stability of binding, but also increases the effective surface concentration of this superantigen. In addition, by also activating the APC, bivalent superantigens such as SEA also promote cell adhesion (seen clearly as homotypic aggregation) and might also activate costimulations pathways that further enhance T cell responses. SPE-C appears to achieve the same end but by a different mechanism, dispensing with the generic α-chain binding site in favor of a preformed dimer molecule with two zinc-containing β-chain binding sites. The trade-off would appear to be a reduced TCR repertoire because, like TSST, only human Vβ2-bearing T cells are ligated by SPE-C (30). Nevertheless, this does not appear to affect the potency of SPE-C in vitro, at least towards human T cells. One major difference between SPE-C and TSST is that although TSST is extremely potent towards murine T cells, particularly murine Vβ15, SPE-C is completely inactive on murine T cells despite continued strong binding to the I-E molecule. The simplest explanation for this is that there are simply no mouse Vβs ligated by SPE-C. We find this surprising given the conservation of function of all other bacterial superantigens across species and the fact that SPE-C binding to murine class II (I-E) is conserved.
The location of the TCR binding site on SPE-C has not been identified. Given the similarities between SPE-C and TSST in both folding and Vβ specificity, it is likely that the TCR binding site on SPE-C is located in a similar position to TSST along the main COOH-terminal domain α helix (11, 13, 31) rather than in the shallow groove between the COOH- and NH2-terminal domains as has been mapped for SEA, SEB, or SEC3. Analogous residues in SPE-C to those that form the murine Vβ8.2 TCR binding site of SEC3 (32) partially overlap the SPE-C dimer interface and would not be available for TCR ligation in the dimer molecule (23).
Confirmation that the SPE-C dimer is required for optimal activity at normal toxins concentrations (i.e., in the picomolar range) must await mutational analysis, but in its absence, the functional and biochemical studies presented here combined with the recent crystal structure of SPE-C propose an intriging model of SPE-C as a preformed dimer with two MHC class II β-chain binding sites strategically placed at opposite poles to cross-link MHC class II, and potentially two TCR binding sites that might also induce cross-linking of the TCR. This study reveals yet another potential mechanism of bacterial superantigens and highlights the variation that has developed in this family of potent T cell mitogens, despite conservation around a similar protein fold and continued targeting of the MHC class II molecule as the primary cell surface receptor.
Acknowledgments
This work was supported by The Welcome Trust (UK), The Human Frontiers Science Programme, and the Health Research Council of New Zealand. J. Fraser is a Wellcome Trust Senior Research Fellow in Medical Science.
Footnotes
Abbreviations used in this paper: GST, glutathione S transferase; m, messenger; SAg, superantigen; SE, staphylococcal enterotoxin; SPE, streptococcal pyrogenic exotoxin; TSST, toxic shock syndrome toxin.
References
- 1.Janeway CJ, Yagi J, Conrad PJ, Katz ME, Jones B, Vroegop S, Buxser S. T-cell responses to Mls and to bacterial proteins that mimic its behavior. Immunol Rev. 1989;107:61–68. doi: 10.1111/j.1600-065x.1989.tb00003.x. [DOI] [PubMed] [Google Scholar]
- 2.Marrack P, Kappler J. The staphylococcal enterotoxins and their relatives. Science (Wash DC) 1990;248:705–711. doi: 10.1126/science.2185544. [DOI] [PubMed] [Google Scholar]
- 3.Herman A, Kappler JW, Marrack P, Pullen AM. Superantigens: mechanism of T-cell stimulation and role in immune responses. Annu Rev Immunol. 1991;9:745–772. doi: 10.1146/annurev.iy.09.040191.003525. [DOI] [PubMed] [Google Scholar]
- 4.Fischer H, Dohlsten M, Lindvall M, Sjogren HO, Carlsson R. Binding of staphylococcal enterotoxin A to HLA-DR on B cell lines. J Immunol. 1989;142:3151–3157. [PubMed] [Google Scholar]
- 5.Fleischer B, Schrezenmeier H. T cell stimulation by staphylococcal enterotoxins. Clonally variable response and requirement for major histocompatibility complex class II molecules on accessory or target cells. J Exp Med. 1988;167:1697–1707. doi: 10.1084/jem.167.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraser JD. High-affinity binding of staphylococcal enterotoxins A and B to HLA-DR. Nature (Lond) 1989;339:221–223. doi: 10.1038/339221a0. [DOI] [PubMed] [Google Scholar]
- 7.Mollick JA, Cook RG, Rich RR. Class II MHC molecules are specific receptors for staphylococcus enterotoxin A. Science (Wash DC) 1989;244:817–820. doi: 10.1126/science.2658055. [DOI] [PubMed] [Google Scholar]
- 8.Schad EM, Zaitseva I, Zaitsev VN, Dohlsten M, Kalland T, Schlievert PM, Ohlendorf DH, Svensson LA. Crystal structure of the superantigen staphylococcal enterotoxin type A. EMBO (Eur Mol Biol Organ) J. 1995;14:3292–3301. doi: 10.1002/j.1460-2075.1995.tb07336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swaminathan S, Furey W, Pletcher J, Sax M. Crystal structure of staphylococcal enterotoxin B, a superantigen. Nature (Lond) 1992;359:801–806. doi: 10.1038/359801a0. [DOI] [PubMed] [Google Scholar]
- 10.Papageorgiou AC, Acharya KR, Shapiro R, Passalacqua EF, Brehm RD, Tranter HS. Crystal structure of the superantigen enterotoxin C2 from staphylococcus aureus reveals a zinc-binding site. Structure (Lond) 1995;3:769–779. doi: 10.1016/s0969-2126(01)00212-x. [DOI] [PubMed] [Google Scholar]
- 11.Acharya KR, Passalacqua EF, Jones EY, Harlos K, Stuart DI, Brehm RD, Tranter HS. Structural basis of superantigen action inferred from crystal structure of toxic-shock syndrome toxin-1. Nature (Lond) 1994;367:94–97. doi: 10.1038/367094a0. [DOI] [PubMed] [Google Scholar]
- 12.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, Stauffacher C, Strominger JL, Wiley DC. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature (Lond) 1994;368:711–718. doi: 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]
- 13.Kim J, Urban R, Strominger J L, Wiley D. Toxic shock syndrome toxin-1 complexed with a major histocompatibility molecule HLA-DR1. Science (Wash DC) 1994;266:1870–1874. doi: 10.1126/science.7997880. [DOI] [PubMed] [Google Scholar]
- 14.Seth A, Stern LJ, Ottenhoff TH, Engel I, Owen MJ, Lamb JR, Klausner RD, Wiley DC. Binary and ternary complexes between T-cell receptor, class II MHC and superantigen in vitro. Nature (Lond) 1994;369:324–327. doi: 10.1038/369324a0. [DOI] [PubMed] [Google Scholar]
- 15.Wen R, Blackman MA, Woodland DL. Variable influence of MHC polymorphism on the recognition of bacterial superantigens by T cells. J Immunol. 1995;155:1884–1892. [PubMed] [Google Scholar]
- 16.Wen R, Cole GA, Surman S, Blackman MA, Woodland DL. Major histocompatibility complex class II– associated peptides control the presentation of bacterial superantigens to T cells. J Exp Med. 1996;183:1083–1092. doi: 10.1084/jem.183.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hudson KR, Tiedemann RE, Urban RG, Lowe SC, Strominger JL, Fraser JD. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J Exp Med. 1995;182:711–720. doi: 10.1084/jem.182.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abrahmsen L, Dohlsten M, Segren S, Bjork P, Jonsson E, Kalland T. Characterization of two distinct MHC class II binding sites in the superantigen staphylococcal enterotoxin A. EMBO (Eur Mol Biol Organ) J. 1995;14:2978–2986. doi: 10.1002/j.1460-2075.1995.tb07300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozono H, Parker D, White J, Marrack P, Kappler J. Multiple binding sites for bacterial superantigens on soluble class II MHC molecules. Immunity. 1995;3:187–196. doi: 10.1016/1074-7613(95)90088-8. [DOI] [PubMed] [Google Scholar]
- 20.Tiedemann RE, Fraser JD. Cross-linking of major histocompatibility complex class II by staphylococcal enterotoxin A is required for superantigen activity. J Immunol. 1996;157:3958–3966. [PubMed] [Google Scholar]
- 21.Wannamaker, L.W., and P.M. Schlievert. 1988. Exotoxins of group A Streptococci. In Bacterial Toxins. Vol. 4. C.M. Hardegree and A.T. Tu, editors. Marcel Dekker, New York. 267– 295.
- 22.Mollick JA, Miller GG, Musser JM, Cook RG, Grossman D, Rich RR. A novel superantigen isolated from pathogenic strains of Streptococcuspyogenes with amino terminal homology to staphylococcal enterotoxins B and C. J Clin Invest. 1993;92:710–719. doi: 10.1172/JCI116641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roussel, A., H.M. Baker, J.D. Fraser, and E.N. Baker. 1996. Crystal structure of the streptococcal superantigen SPE-C: dimerisation and zinc binding suggests a novel mode of interaction with MHC class II molecules. Nat. Struct. Biol. In press. [DOI] [PubMed]
- 24.Hudson KR, Robinson H, Fraser JD. Two adjacent residues in staphylococcal enterotoxins A and E determine T cell receptor V beta specificity. J Exp Med. 1993;177:175–184. doi: 10.1084/jem.177.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chomezynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocynate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–163. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Vallee BL, Auld DS. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry. 1990;29:5641–5659. doi: 10.1021/bi00476a001. [DOI] [PubMed] [Google Scholar]
- 27.Mehindate K, Thibodeau J, Dohlsten M, Kalland T, Sekaly RP, Mourad W. Cross-linking of major histocompatibility complex class II molecules by staphylococcal enterotoxin A superantigen is a requirement for inflammatory cytokine gene expression. J Exp Med. 1995;182:1573–1577. doi: 10.1084/jem.182.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thibodeau J, Cloutier I, Lavoie PM, Labrecque N, Mourad W, Jardetzky T, Sekaly RP. Subsets of HLA-DR1 molecules defined by SEB and TSST-1 binding. Science (Wash DC) 1994;266:1874–1878. doi: 10.1126/science.7997881. [DOI] [PubMed] [Google Scholar]
- 29.Tiedemann RE, Urban RJ, Strominger, Fraser JD. Isolation of HLA-DR1.(staphylococcal enterotoxin A)2 trimers in solution. Proc Natl Acad Sci USA. 1995;92:12156–12159. doi: 10.1073/pnas.92.26.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi Y, Kappler JW, Marrack P. A superantigen encoded in the open reading frame of the 3′ long terminal repeat of mouse mammary tumour virus. Nature (Lond) 1991;350:203–207. doi: 10.1038/350203a0. [DOI] [PubMed] [Google Scholar]
- 31.Hurley JM, Shimonkevitz R, Hanagan A, Enney K, Boen E, Malmstrom S, Kotzin BL, Matsumura M. Identification of class II major histocompatibility complex and T cell receptor binding sites in the superantigen toxic shock syndrome toxin 1. J Exp Med. 1995;181:2229–2235. doi: 10.1084/jem.181.6.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fields BA, Malchiodi EL, Li H, Ysern X, Stauffacher CV, Schlievert PM, Karjalainen K, Marriuzza RA. Crystal structure of a T cell receptor β-chain complexed with a superantigen. Science (Wash DC) 1996;384:188–192. doi: 10.1038/384188a0. [DOI] [PubMed] [Google Scholar]