

Abstract
Objective
To test the feasibility of applying a mimetic (specific for a patient-derived prothrombotic anticardiolipin antibody [aCL]) to study the homologous, disease-associated aCL in patients with antiphospholipid syndrome (APS).
Methods
We used the CL15 monoclonal aCL to screen 17 phage-display peptide libraries. Peptides (corresponding to recurrent peptide sequences) and their derivatives were synthesized and analyzed for binding to CL15 and for their abilities to inhibit CL15 from binding to cardiolipin. A peptide was chosen and used to study CL15-like IgG aCL in plasma samples from patients with APS, patients with systemic lupus erythematosus (SLE) but without APS, and normal healthy donors.
Results
Library screening with CL15 yielded 4 recurrent peptide sequences. Analyses of peptides showed that peptide CL154C reacted with antibody CL15 and inhibited binding of CL15 to cardiolipin, indicating that peptide CL154C may be a peptide mimetic for the CL15 aCL. Initial studies with plasma samples revealed that CL154C-reactive IgG was present (positivity defined as the mean + 3 SD optical density of the 25 normal controls) in 15 of 21 APS patients and 1 of 12 SLE patients.
Conclusion
These findings suggest that it is feasible to develop a specific enzyme-linked immunosorbent assay for each immunologically and functionally distinct disease-associated aCL. Additional testing of CL154C with a larger number of APS patients and SLE patients, as well as identification of peptide mimetics for each distinct aCL, will reveal the diagnostic potential of CL154C and other mimetics in identifying patients with aCL who are at risk of developing life-threatening thrombosis.
Recurrent thrombosis and/or pregnancy loss occurs in some patients with systemic lupus erythematosus (SLE) (1–3) and may be associated with the presence of antiphospholipid antibodies (aPL). These antibodies include anticardiolipin antibodies (aCL) and/or the lupus anticoagulants (LAC). The association of thrombosis and/or fetal loss with aPL is recognized as the antiphospholipid syndrome (APS) (2,4). To date, accumulated studies show that aPL represent a heterogeneous group of immunologically and functionally distinct antibodies that recognize various phospholipids, phospholipid-binding plasma proteins, and/or phospholipid–protein complexes (2,5–11).
Although some investigators reported that the clinical features of APS were more closely associated with the autoantibodies detected in the enzyme-linked immunosorbent assay (ELISA) for anti–β2-glycoprotein I (anti-β2GPI) than in the conventional ELISA for aCL (7,12–15), the roles of various aPL in thrombosis and/or fetal loss remain unclear (2). Thus, current qualitative diagnostic tests are limited in predictive value for the identification of patients with aPL who have or will develop a clotting disorder. Quantitative assays have shown that patients with high quantities of aCL have a significantly increased risk of clotting (2). Since only a minority of patients with aPL will have a significant thrombotic event, and most treatments for clotting are potentially hazardous, patients usually are not treated until after life-threatening events occur. Clearly, better predictive tests are needed, so that patients who are truly at risk for clotting can be treated earlier.
Considering that many SLE patients with aPL do not manifest the features of APS, we hypothesized that only certain aPL in patients with APS are prothrombotic, and that such aPL are likely to be necessary, but not sufficient, for causing thrombosis, since patients with APS do not always develop thrombosis. The putative prothrombotic aPL may have certain unique binding and functional properties. In accordance with these hypotheses, our strategy for developing better predictive tests was to develop a specific ELISA for each single species of disease-associated aPL.
Several studies have identified peptide mimetics that can differentiate related antibody molecules with different functional properties (16,17). On the other hand, the CL15 patient-derived IgG3κ monoclonal aCL were found to be prothrombotic in both an in vivo pinch-induced thrombosis model (18) and an in vivo stasis-induced thrombosis model (Roubey RAS: personal communication). The former model assesses the ability of IgG aCL to increase the sizes of thrombi induced by pinch injury to the vessel. The latter model evaluates the combined effects of IgG aCL and a local venous stasis; neither of these factors alone is sufficient to induce thrombosis (19). CL15 binds to cardiolipin in the presence of bovine serum or human β2GPI, but not to β2GPI alone (20). Accordingly, we used the CL15 prothrombotic monoclonal IgG aCL to screen 17 phage-display peptide libraries, identified a peptide mimetic for monoclonal antibody (mAb) CL15, and then studied the feasibility of using the mimetic peptide to detect CL15-like disease-associated antibodies in the plasma of APS patients.
MATERIALS AND METHODS
Phage-display peptide libraries
Two sets of peptide libraries were used in this study. The first, a set of 14 libraries, was constructed with the f88-4 phage vector (21) and included 2 linear libraries (X6 and X15, in which X represents a randomized amino acid comprising any of the 20 amino acids), 5 libraries comprising cysteine (Cys)–constrained cyclic peptides flanked by a single randomized residue (XCX4CX, XCX6CX, XCX8CX, XCX10CX, and XCX12CX, in which C stands for Cys), 4 libraries in which 4 cyclic peptides are flanked by 4 or 5 randomized residues (X5CX3CX4, X4CX4CX4, X4CX5CX4, and X4CX6CX4; these libraries are abbreviated as Cys3, Cys4, Cys5, and Cys6), and 3 half-Cys libraries (X15CX, XCX15, and X8CX8). Under nonreducing conditions, Cys typically forms a disulfide link, resulting in the display of cyclized peptides on the phage coat; thus, the first 5 cyclic libraries are abbreviated as LX4, LX6, LX8, LX10, and LX12, in which L denotes the disulfide-bridged loop. The characteristics of these libraries have been described previously (22). The second set of libraries included the linear 7-mer and 12-mer libraries and the cyclic 7-mer library from New England Biolabs (NEB; Beverly, MA).
Enrichment of CL15 binding phage
Two different library screening protocols were used. The libraries were screened either in pools of 2–3 libraries or individually. First, all 17 libraries were screened in 6 pools. Pool 1 contained cyclic libraries LX8, LX10, and LX12, pool 2 contained 3 half-Cys libraries, pool 3 contained cyclic libraries Cys3 and Cys4 and LX4, pool 4 contained cyclic libraries Cys5 and Cys6 and LX6, pool 5 contained linear libraries X6 and X15, and pool 6 contained the 3 NEB libraries. Panning (affinity selection) was performed according to published protocols, with minor modification (22). Briefly, phage were incubated with biotinylated CL15 (100 nM) overnight at 4°C, and then transferred to round-bottom microwells precoated with streptavidin for 4 hours at 4°C. The numbers of phage particles screened were 1012 virions for the f88-4–based libraries and 1011 virions for the M13-based libraries. After washing with Tris buffered saline (TBS), bound phage in the wells were eluted with 35 μl elution buffer (0.1M HCl adjusted to pH 2.2 with glycine, and containing 1 mg/ml bovine serum albumin [BSA]; fraction V; Sigma, St. Louis, MO) per well, followed by incubation for 10 minutes at room temperature (RT).
The eluted phage were neutralized with 1M Tris HCl (pH 9.1), and then were amplified by the addition of K91 Escherichia coli cells. The second to fourth rounds of panning were performed similarly, except that the input phage were reduced by 100-fold (1010 for the f88-4–based libraries) and wells were washed with TBS containing 0.5% Tween 20. Each sample of amplified, enriched phage obtained after every round of panning was gel-analyzed to quantitate the phage yield, and to determine the presence of fast-growing, non–tetracycline-resistant contaminants (genome size ~6 kb) that sporadically appear in our f88-4 cultures. Samples containing this contaminant were discarded and the panning was repeated, starting with the appropriate phage libraries.
Second, the NEB linear 12-mer and cyclic 7-mer libraries were screened individually according to the manufacturer’s instructions. Briefly, wells of a 96-well high-binding ELISA plate were coated with 100 μg/ml of CL15 overnight at 4°C. The wells were blocked (0.1M NaHCO3, pH 8.6, 5 mg/ml BSA) for 1 hour at 4°C and washed with TBS/0.1% (volume/volume) Tween 20 (TBS/T). Phage at 1.5 × 1011 plaque-forming units (PFU) in 100 μl of TBS/T were added to wells, and the plate was incubated for 1 hour at RT, with gentle rocking. After washing, bound phage were eluted with 100 μl of elution buffer (0.2M glycine-HCl, pH 2.2, 1 mg/ml of BSA). The eluted phage were neutralized, amplified, and then used in the subsequent rounds of panning. After the third and fourth rounds of panning, 10–20 clones were chosen at random and analyzed for reactivity with CL15 by ELISA (as described below).
Phage ELISA
The reactivities of pooled phage and chosen phage clones with the panning CL15 were analyzed by either the direct phage ELISA or the reversed phage ELISA. In the direct phage ELISA (22), wells were coated with anti-phage antibodies and blocked with BSA. Then, 1 × 1010 virions of the indicated pools or the f88-4 vector phage (as the negative control) were distributed to wells. After incubation and washing, CL15-reactive phage were detected with biotinylated CL15 and streptavidin-conjugated horseradish peroxidase (HRP).
For the reversed phage ELISA, wells were coated with 5 μg/ml of CL15 overnight at 4°C. After blocking, test phage or the negative control vector clone (M13 or f88-4) were added at 2 × 1010 PFU in the TBS/T. After a 2-hour incubation at RT, wells were washed with TBS/T and bound phage were detected with HRP-labeled anti-M13 antibodies (Pharmacia, Piscataway, NJ). The substrate tetramethylbenzidine (Kirkegaard and Perry Laboratories, Gaithersburg, MD) was added to wells and the optical density (OD) was measured at a wavelength of 450 nm, with a reference wavelength of 650 nm. The signal from the vector clone was used as background and was subtracted from the signals produced by the test clones.
Sequence determination of displayed peptides
After the third or fourth panning, chosen phage clones were analyzed by 2 different protocols. First, for libraries screened in pools, DNA of 5–10 clones from each selected library pool was purified and sequenced as described previously (22). Second, for the NEB libraries screened individually, single-stranded phage DNA (ssDNA) of 10–20 clones was purified and sequenced in accordance with the manufacturer’s instructions. Briefly, purified ssDNA was sequenced by polymerase chain reaction and purified further by passage through gel filtration cartridges (Edge Biosystems, Gaithersburg, MD). Amplified samples were analyzed by Laragen (Santa Monica, CA). Resultant sequences were analyzed using the Genetics Computer Group (GCG) programs to deduce the insert amino acid sequences.
Peptide synthesis
Peptides and their derivatives (Tables 1 and 2) were synthesized and purified by reverse-phase high-performance liquid chromatography by Genemed (San Francisco, CA). All peptides had >95% purity.
Table 1.
Peptide sequences and characterization of recurrent clones enriched from 17 phage-display peptide libraries by CL15*
| Panning, sequence | OD | Frequency | Clone designation |
|---|---|---|---|
| Pooled libraries | |||
| APVCV WERLS PYRER CV | 0.45 | 5/5 | CL151 |
| SCPAN TYACM | 0.47 | 5/5 | CL152 |
| VTVTY N | 0.45 | 3/4 | CL153 |
| EPTPS SFVHY RLNAA† | 0.49 | 1/4 | |
| None | 0.11 | NA | f88-4 vector |
| Individual library | |||
| HWDPF SLSAY FP | 0.51 | 5/8‡ | CL154 |
| None | 0.06 | NA | M13 |
Seventeen libraries were screened by CL15 in 6 pools of 2–3 libraries each. In addition, 2 of 17 libraries were screened individually. After 4 rounds of panning, 4–5 clones from each pool were isolated and sequenced. CL151 was from pool 2 (library X15CX), CL152 from pool 4 (library LX6), CL153 from pool 5 (library X6), and CL154 from the New England Biolabs linear 12-mer library. OD denotes the mean optical density of all clones with the indicated insert, as determined by the reversed phage enzyme-linked immunosorbent assay format. Frequency denotes the number of phage clones with identical inserts.
NA = not applicable.
This sequence occurred only one time, and thus was not studied and not assigned a name.
After 4 rounds of panning, 10 clones were isolated and sequenced.
Table 2.
CL15-reactive peptides and their modified ones*
| Sequence | Designation |
|---|---|
| APVCVWERLSPYRERCV | CL151A |
| APVCVWERLSPYRERCVPAEG | CL151E |
| HWDPFSLSAYFP | CL154A |
| HWDPFSLSAYFPGGGS | CL154E |
| CHWDPFSLSAYFPGGGSC | CL154C |
The carboxyl-termini of peptides CL151 and CL154 were amidated, and the modified peptides were designated CL151A and CL154A, respectively. CL151A and CL154A were modified by adding 4 extra residues to their carboxyl-termini (the added flanking residues are underlined), and the modified peptides were designated CL151E and CL154E (E denotes “extended”), respectively. To improve the stability of peptide CL154E in liquid phase, cysteine (Cys) was added to both ends of CL154E, and this further modification was designated CL154C (C denotes “Cys” and the added Cys are underlined). This was not done for CL151E, since it already had 2 Cys near its N- and C-terminals.
Analyses of peptide reactivity with CL15 by ELISA and competitive inhibition studies
High-binding Costar 96-well ELISA plates were coated with synthetic peptides at 10 μg/ml in phosphate buffered saline (PBS). After blocking with 1% skim milk in PBS, CL15 or normal human IgG (10 μg/ml) in 0.1% skim milk/PBS was distributed to wells in duplicate and bound IgG was quantitated with HRP-labeled antibodies against human IgG. For competitive inhibition studies, CL15 at 1.25 μg/ml was diluted in 0.1% skim milk/PBS and premixed with peptides serially diluted (from 400 μg/ml to 25.6 μg/ml) in PBS. After blocking, the peptide–antibody mix was then distributed to wells and bound IgG was quantitated as described above.
Competitive inhibition of CL15 binding to cardiolipin by peptides
To identify peptide mimetic for CL15, peptides were analyzed for their ability to inhibit the binding of CL15 to cardiolipin in the presence of bovine serum. Briefly, polysorp 96-well ELISA plates were coated with cardiolipin at 20 μg/ml in ethanol and blocked with 0.25% gelatin in PBS. CL15 at 1.25 μg/ml was diluted in PBS/5% fetal calf serum and premixed with peptides serially diluted in PBS. Then, the peptide–antibody mix was distributed to wells in duplicate, and bound IgG was quantitated. Inhibition of binding was calculated as follows: % inhibition of binding = [(OD from CL15 alone) − (OD from CL15 plus peptide)/(OD from CL15 alone)] × 100.
Patients and healthy controls
The present study was approved by the University of California at Los Angeles (UCLA) Institutional Review Board in the Office for the Protection of Research Subjects. Patients in this study were from the UCLA Medical Subspecialty Clinic Practice. All APS patients in this study satisfied the Sapporo classification criteria for definite APS (4), and all SLE patients satisfied the updated American College of Rheumatology criteria for SLE (23,24). Medical charts and laboratory test reports for each patient entered in this study were reviewed by a rheumatologist (JG or BH). Patients were then classified as having APS if they had no associated autoimmune disease, or secondary APS if they also fulfilled criteria for another autoimmune disease.
Nine patients with primary APS and 12 with secondary APS (all with SLE) were studied. The sex distribution was 14 women and 7 men. Fifteen of the 21 APS patients were positive for both aCL and LAC, 2 patients had aCL alone, and 4 patients had LAC alone. LAC were determined by either the dilute Russell’s viper venom time test or the rabbit brain neutralization procedure test (25). All patients had thrombosis (4 had both arterial and venous, 5 had arterial alone, 11 had venous alone, and 1 had small vessel occlusion). One of the 14 women had more than 2 unexplained fetal losses. The average age at diagnosis was 33.8 years (range 12–63 years).
A separate cohort of 12 patients with SLE but without APS was also studied, all of whom were female. Five of these SLE patients had aPL: 1 patient was positive for both aCL and LAC, 3 for aCL alone, and 1 for LAC alone. Seven of the 12 SLE patients had repeatedly tested negative for aPL. The average age at the time of sample donation was 44 years (range 26–68 years).
Twenty-five healthy donors were included as normal controls. The sex distribution was 6 men and 19 women. Their average age at the time of sample donation was 36 years (range 22–65 years).
Plasma samples (7 ml each) from each individual were collected in heparin or acid citrate dextrose–containing tubes. Tubes were centrifuged at 3,000 revolutions per minute for 15 minutes. Plasma was collected and stored at −20°C in aliquots.
Analysis of plasma samples for peptide CL154C-reactive IgG
Plasma samples were analyzed for the presence of CL154C-reactive IgG. This was done by an ELISA similar to the aforementioned one, to test for the reactivity of peptide CL154C with CL15. Briefly, polysorp 96-well ELISA plates were coated with CL154C peptide at 10 μg/ml in PBS and blocked with 0.3% gelatin in TBS. Plasma samples were analyzed at 1:100 dilutions in 0.1% gelatin/TBS, and bound IgG was measured.
Affinity purification and characterization of CL154C-reactive IgG
IgG was first isolated from 1 chosen plasma sample using the Pierce Immunopure IgG purification kit according to the manufacturer’s instruction. Peptide CL154C (3 mg) was linked to a sulfolink column using a sulfolink kit (Pierce, Rockford, IL) according to the manufacturer’s instruction. Briefly, the gel was equilibrated with coupling buffer (50 mM Tris, 5 mM EDTA-Na, pH 8.5), the peptide was incubated with gel for 45 minutes, and subsequently the nonspecific binding sites on the gel were blocked using L-cysteine HCl. For affinity purification, gel was equilibrated with PBS and 8 mg of purified IgG added to the column and incubated for 1 hour at RT. Bound IgG was eluted using 0.1M glycine, pH 2.5, neutralized with 1M Tris, pH 9.0, and dialyzed against PBS. The quantity of purified CL154C-reactive IgG was 40 μg (0.5%) as determined by ELISA. The unfractionated IgG, the affinity-purified CL154C-reactive IgG, and the pass IgG fraction were analyzed for reactivity with peptide CL154C and cardiolipin as described above.
Statistical analysis
The mean OD plus 3 SD of the 25 normal controls was used as the cutoff. Plasma samples with OD values that were consistently higher than the cutoff in 2 separate experiments were considered positive. A P value of less than 0.05 was considered significant. The Fisher’s exact test was used to determine the significance of anti-CL154C antibodies in APS patients versus those in SLE patients without APS (26).
Computer sequence analysis
To search for the homologous amino acid sequences of CL154, we used Wordsearch in the GCG software package (27) to compare the CL154 amino acid sequence with the 283,138 sequences in the Protein Information Resource databases (accessed on May 7, 2002).
RESULTS
Isolation and characterization of CL15-reactive phage clones from the pooled libraries
Initially, 17 peptide libraries combined into 6 pools were panned with CL15. After the third panning, the yields for pools 2, 4, and 5 were 3–4 logs higher than that of the f88-4 control (data not shown). The yields for these 3 pools further increased during the subsequent fourth round of panning, suggesting that phage displaying the CL15-binding peptides were enriched (data not shown). Thereafter, 5 clones from each of these 3 pools after the fourth panning, and 5 clones from pool 1 after the fourth panning (to serve as a control for CL15-nonreactive clones) were analyzed for their binding to CL15 by the reversed phage ELISA. In addition, 10 clones from pool 2 after the third panning that had the highest yield and reactivity with CL15 were also analyzed. The results of panning of the pooled libraries are summarized in Table 2.
A single CL15-reactive clone was isolated repeatedly from pool 2 and pool 4 and the inserted sequences were designated CL151 and CL152, respectively. Interestingly, 2 CL15-reactive clones were isolated from pool 5, one from the X6 library (designated CL153) and the other from the X15 library. The latter peptide sequence was found only once and thus was not studied further. In contrast, no CL15-reactive clones were isolated from pool 1, and all 5 clones had OD readings that were lower than those of the CL15-reactive clones. Moreover, 4 of these 5 clones did not have a peptide insertion, but rather had mutations within the pVIII signal peptide region (data not shown). It should be noted that a majority of the CL15-reactive clones had ODs ~4-fold higher than that of the f88-4 vector (Table 1). The specificity of these clones was confirmed by the minimal reactivity of the CL15-reactive clones with human IgG (data not shown).
In addition to screening libraries in pools, the NEB linear 12-mer (designated X12) and cyclic 7-mer libraries were also screened individually with CL15. After the fourth panning, CL15-reactive clones were isolated from both libraries. Of the 8 CL15-reactive clones originating from the X12 library, 5 clones shared an identical amino acid sequence. This sequence, HWDPF SLSAY FP, was designated CL154 (Table 1). Again, the specificity of these CL15-reactive clones was confirmed by the minimal reactivity of clones with human IgG (data not shown). In contrast, none of the CL15-reactive clones from the cyclic 7-mer library contained inserts.
Reactivity of synthetic peptides with CL15 and competitive inhibition studies
Peptides corresponding to all 4 recurrent CL15-reactive clones were synthesized and tested for reactivity with CL15 and a human monoclonal IgG3 (isotype control). CL15 bound to wells coated with peptides CL151 and CL154 at 10 μg/ml (Figure 1). However, CL15 did not bind to wells coated with either CL152 or CL153 at 10 μg/ml or 100 μg/ml (data not shown). In contrast, the isotype control IgG3 did not bind to any of the tested peptides (data not shown). Thereafter, we focused on peptides CL151 and CL154.
Figure 1.
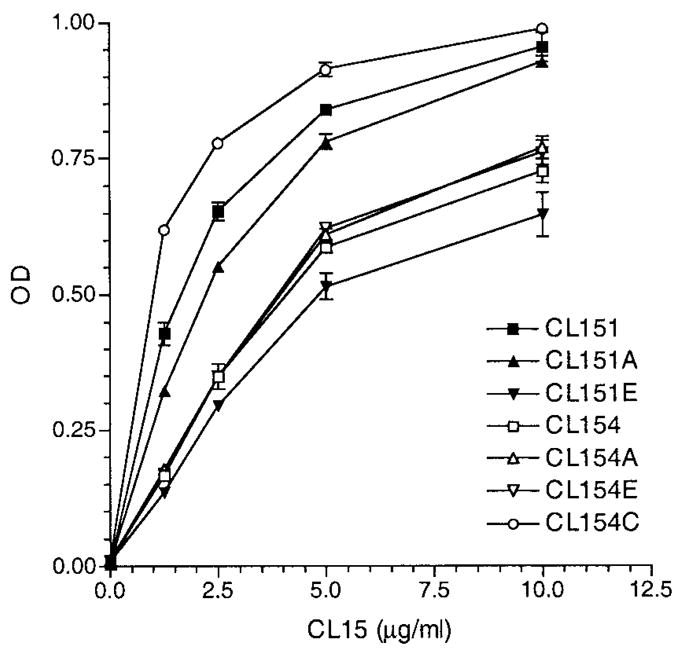
Reactivities of synthetic peptides with CL15. Enzyme-linked immunosorbent assay plates were coated with the indicated peptides at 10 μg/ml and CL15 was analyzed at 1.25–10 μg/ml (2-fold dilutions). Bound IgG of duplicate samples was measured and expressed in optical density (OD). Results are representative of 1 of 2 experiments.
Since phage-display peptides are fused to the major coat protein and thus are not charged at the peptidyl carboxyl-terminus, we amidated both the CL151 and the CL154 peptides (designated CL151A and CL154A, respectively; “A” denotes amidated). In addition, it was reported recently that incorporation of flanking amino acid residues from the host coat protein (i.e., coat protein III for the NEB library and coat protein VIII for the f88-4–based libraries) into the synthetic peptides derived from phage-display libraries significantly enhanced the binding affinity of peptides to the corresponding panning antibodies (28). Accordingly, 4 extra residues from the corresponding host coat proteins were added to the carboxyl-termini of CL151A and CL154A, resulting in CL151E and CL154E, respectively (Table 2 and Materials and Methods). Moreover, to improve the stability of peptides in liquid phase, Cys was added to both ends of CL154E, resulting in CL154C (Table 2 and Materials and Methods).
When these new peptides were tested for reactivity with CL15 and the control human IgG3, CL154C displayed the strongest reactivity with CL15 (Figure 1). In contrast, none of the new peptides reacted with the control IgG3 (data not shown). To determine the specificity of CL15 binding to solid-phase peptides, competitive inhibition was performed. All 5 peptides in fluid phase were able to inhibit binding of CL15 to corresponding peptides (data not shown).
Inhibition of CL15 reactivity with cardiolipin by synthetic peptides
To identify peptide mimetics for CL15, all 7 CL15-reactive peptides in fluid phase were tested for the ability to inhibit binding of CL15 to cardiolipin using the aCL ELISA. Only peptide CL154C was able to inhibit CL15 binding to cardiolipin (Figure 2) (the negative inhibition data are represented by the results using CL154A and CL154E). At 100 μM (193 μg/ml), CL154C inhibited ~47% of the CL15 binding to cardiolipin.
Figure 2.
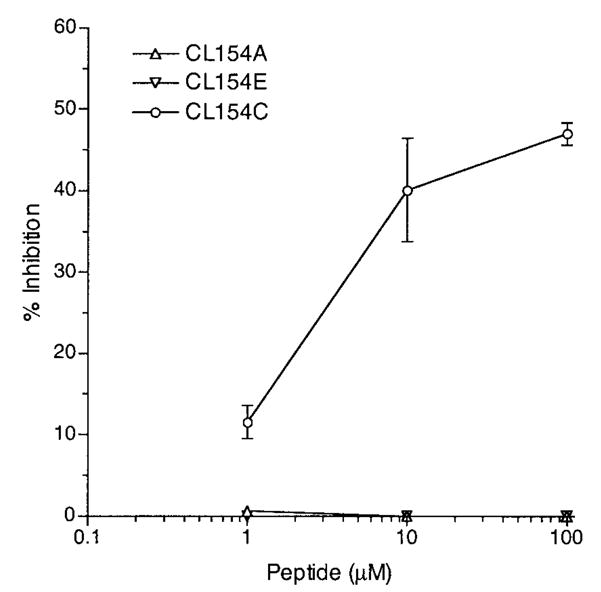
Inhibition of CL15 binding to cardiolipin by peptide CL154C, but not by peptides CL154A and CL154E. CL15 (1.25 μg/ml) was preincubated with the indicated peptides at the indicated concentrations (1–100 μM) and the peptide–antibody mix was distributed to cardiolipin-coated wells in duplicate. Bound IgG was measured, and the percentage of inhibition is reported. Results are representative of 1 of 2 experiments.
Presence of CL154C-reactive IgG in patient plasma
Thereafter, CL154C was examined for its potential in detecting CL15-like prothrombotic aCL in APS patients. Plasma samples from 21 APS patients (including those with SLE), 25 normal healthy donors, and 12 SLE patients without APS were analyzed for the presence of CL154C-reactive IgG. A cutoff of 3 SD above the mean OD of the 25 normal control subjects (0.31) was used to distinguish between patient plasma samples that were reactive with the peptide and those that were not. Fifteen (71%) of the 21 APS patients tested were positive for the presence of CL154C-reactive IgG, and of these 15, 14 were LAC positive. The frequency of CL154C-reactive IgG was significantly higher in APS patients than in SLE patients without APS (P = 0.0004, using the Fisher’s exact test). Of the APS patients showing positivity, 8 patients had high titers of CL154C-reactive IgG (OD >0.4) (Figure 3). Only 1 of the 12 SLE patients without APS had a higher titer of CL154C-reactive IgG.
Figure 3.
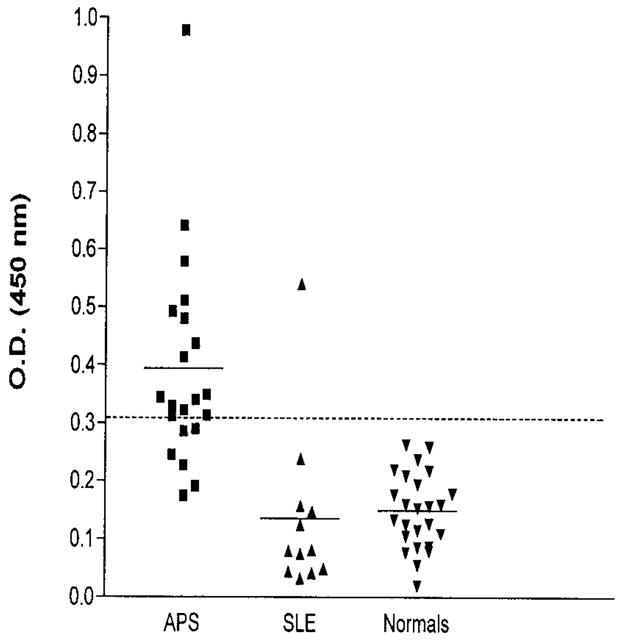
Reactivities of CL154C with plasma samples from patients with antiphospholipid syndrome (APS) (n = 21), normal healthy donors (n = 25), and systemic lupus erythematosus (SLE) patients without APS (n = 12). Enzyme-linked immunosorbent assay plates were coated with CL154C peptide (10 μg/ml) and plasma samples were analyzed at 1:100 dilution in Tris buffered saline/0.1% gelatin. Bound IgG was measured and reported. Each data point represents a test sample; the horizontal bars denote the mean of each group, and the dashed line represents the cutoff for positivity, that is, the mean optical density (O.D.) plus 3 SD of normal controls.
Isolation and characterization of CL154C-reactive IgG from a chosen APS patient plasma sample
To characterize the nature of CL154C-reactive IgG in a patient plasma sample, CL154C-reactive IgG from the reactive plasma sample of a chosen APS patient was affinity-purified. The unfractionated, the eluate (CL154C-reactive), and the pass (depleted of CL154C-reactive) IgG at a range of concentrations (1.25–5 μg/ml) were analyzed for binding to peptide CL154C and to cardiolipin. The eluate IgG bound strongly to peptide CL154C, whereas the pass IgG had minimal reactivity with the peptide (Figure 4). Similarly, while the eluate IgG bound strongly to cardiolipin, the pass IgG had minimal reactivity with cardiolipin (Figure 5). These data combined suggest that CL154C-reactive IgG aCL are immunologically homologous to the CL15 IgG aCL (i.e., CL15-like aCL).
Figure 4.
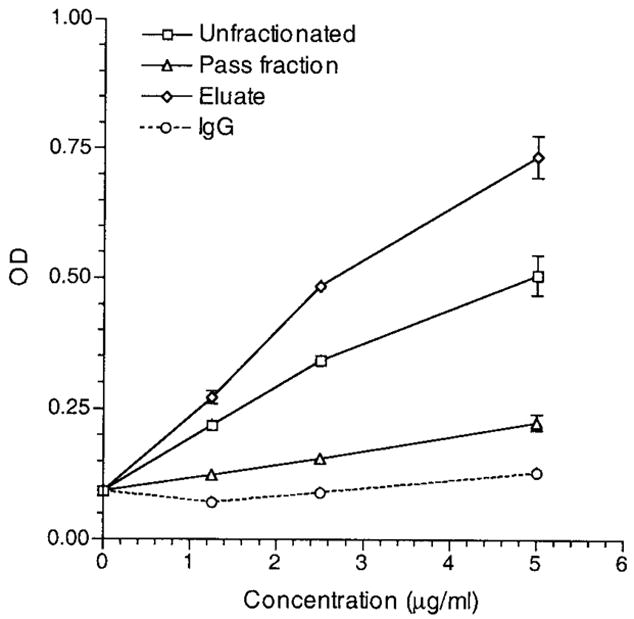
Reactivity of an affinity-purified CL154C-reactive IgG (from a patient with antiphospholipid syndrome) with CL154C. Enzyme-linked immunosorbent assay plates were coated with CL154C at 10 μg/ml. The unfractionated IgG, eluate (CL154C-reactive), and the pass IgG fractions, as well as control polyclonal human IgG were analyzed at the indicated concentrations (1.25–5 μg/ml) in duplicate. Bound IgG was measured and expressed in optical density (OD). Results are representative of 1 of 2 experiments.
Figure 5.
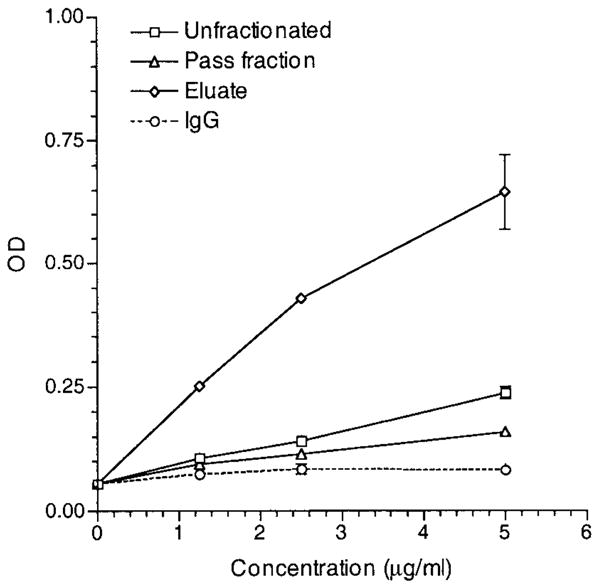
Reactivity of an affinity-purified CL154C-reactive IgG with cardiolipin. The unfractionated IgG, eluate (CL154C-reactive), and the pass IgG fractions, as well as control polyclonal human IgG were analyzed at the indicated concentrations (1.25–5 μg/ml) in duplicate. Bound IgG was measured and expressed in optical density (OD). Results are representative of 1 of 2 experiments.
DISCUSSION
To develop a specific ELISA for a single species of disease-associated aPL in APS patients, we used the CL15 prothrombotic monoclonal IgG aCL to screen 17 peptide libraries. Results of the peptide library screen with CL15 identified one diagnostic peptide mimetic, CL154C. This peptide inhibited ~47% of the binding of antibody CL15 to cardiolipin in the presence of bovine serum. Using the mean plus 3 SD of the 25 normal controls (OD 0.31) as the cutoff, 15 of 21 APS patients (71%) and 1 of 12 SLE patients without APS were positive for the presence of CL154C-reactive IgG. These preliminary data suggest that the anti-CL154C IgG ELISA can identify, with good sensitivity and specificity, patients with APS. The 1 SLE patient positive for CL154C-reactive IgG is a young woman (27 years old) who was diagnosed at age 23 with SLE and has had high titers of IgG aCL documented repeatedly over many years. Of note, patients with high titers of aCL but without thrombosis and/or recurrent fetal loss represent a confusing medical problem, since there is no consensus on how to treat such individuals. When CL154C-reactive IgG was affinity purified from the plasma of an APS patient, the IgG displayed aCL activity. In contrast, binding to cardiolipin by the pass IgG fraction (depleted of CL154C reactivity) was substantially reduced. Taken together, our data suggest that an ELISA for CL154C-reactive IgG may serve as a specific assay for a subset of CL15-like aCL that may be associated with disease in a majority of APS patients.
Of note, we recently found that CL15 binds to prothrombin and thrombin (29). Importantly, thrombin (at 16 μM, or 587 μg/ml) inhibited 90% of CL15 binding to thrombin, while prothrombin (at 16 μM, or 1.2 mg/ml) could only inhibit 36% of CL15 binding to prothrombin and 10% of CL15 binding to thrombin. Considering that CL15 does not bind to β2GPI alone, but binds strongly to cardiolipin in the presence of bovine serum (20), these data raise the possibility that the real autoantigen for CL15 may be thrombin or another, yet-unidentified autoantigen. In this context, peptide CL154C at 64–400 μg/ml could inhibit 76% of CL15 binding to thrombin (data not shown). The significance of this finding is not clear at this time, since CL15 could inhibit thrombin per se, as well as antithrombin inactivation of thrombin (29).
Previously, Blank et al used 3 pathogenic monoclonal IgM anti-β2GPI antibodies to screen a linear 6-mer library, and identified 3 recurring peptides: LK. TPRV for mAb ILA-1, KDKATF for mAb ILA-3, and TLRVYK for mAb H-3 (30). Each sequence closely resembled, or was identical to, segments of β2GPI. Sequence LK. TPRV (the dot denotes a gap that is introduced to maximize homology) resembled LKCTPRV of β2GPI at amino acid positions 58–63 (in the first domain), and KDKATF and TLRVYK were identical to β2GPI at positions 208–213 (in the fourth domain) and 133–138 (in the third domain), respectively (30). In contrast to these findings, peptides CL154, CL154E, and CL154C from our study were not homologous to any areas of β2GPI. This is consistent with the binding properties of CL15, which does not bind to β2GPI alone, but binds strongly to human β2GPI–cardiolipin complexes (20). Combined, our data suggest that peptide CL154C mimics an antigenic configuration of the β2GPI–cardiolipin complex that is recognized by CL15.
To test this hypothesis, we studied the ability of CL154C to inhibit CL15 binding to cardiolipin in the presence of human β2GPI, instead of bovine serum. The results showed that CL154C at 400 μg/ml inhibited ~53% of CL15 binding to β2GPI–cardiolipin (data not shown). In addition, the above contention is consistent with the identification of peptide mimetics for antibodies against polysaccharides and nucleic acids (16,17). For example, Gaynor et al used phage-display peptide libraries to study 3 structurally similar antibodies: the R4A anti-DNA antibodies that cause glomerulonephritis with a predominantly glomerular deposition, and R4A-derived mutants (by site-directed mutagenesis), designated 52b3 (17). Library screening with R4A and 52b3 defined 2 specific motifs: DWEYS for antibody R4A, and RHEDGDWPRV for antibody 52b3. Peptide DWEYS could inhibit only R4A (but not 52b3) from binding to DNA, while peptide RHEDGDWPRV could inhibit only 52b3 (but not R4A), thus clearly defining 2 specific mimetics (17). Since the native antigens for the above-studied antibodies were nucleic acids, not polypeptides, the peptide mimetics apparently mimic the conformational epitopes, but not the primary structures, of DNA molecules. Of note, a peptide mimetic for an antibody is defined as a peptide that can inhibit the antibody binding to its antigen, while a peptide mimotope is a peptide that can induce the corresponding antibody response (31).
Since the 3 peptide mimetics defined by monoclonal IgM anti-β2GPI antibodies bound to their corresponding mAb with low affinity, they all were lengthened by 4–6 amino acid residues (30). Among the elongated ones, 3 were found suitable; these were NTLKTPRVGGC, KDKATFGCHDGC, and CATLRVYKGG. When sera from 43 APS patients were analyzed for binding to the 3 elongated peptides, peptide-reactive antibodies were found in 10 of the 43 APS patients (30). However, the study did not include any serum samples from healthy controls.
In addition to the studies mentioned above, some investigators used the cardiolipin liposomes to affinity-purify polyclonal aCL from an APS patient; these antibodies were used to screen linear 8–15-mer and cyclic 6–13-mer libraries (32,33). The studies identified an 11-mer peptide GPCIL LAPDR C (LJP 685). The peptide LJP 685 at a concentration of 25 μg/well (~250 μg/ml) inhibited 50% of the binding of the parental patient serum aCL (at the 1:400 dilution) to β2GPI–cardiolipin (32). However, that study did not report the presence of the LJP 685–reactive antibodies in the sera of more than the 1 APS patient.
It is conceivable that immunologically distinct aPL could promote thrombosis via different mechanisms. For example, antibodies against β2GPI and/or its complexes with cardiolipin may interact with endothelial cells and monocytes and induce a tissue factor–dependent procoagulant state (34–38). On the other hand, antibodies against protein C and protein S may inhibit activation of protein C and/or function of the activated protein C, thus allowing an unchecked coagulation process once initiated; this would result in a hypercoagulable state (39–43). Thus, in the future, it will be important to identify peptide mimetics for each immunologically distinct prothrombotic aPL, and to determine their effectiveness for identifying corresponding prothrombotic aPL in APS patients. Hopefully, a complete panel of diagnostic peptide mimetics for most or all species of immunologically distinct, prothrombotic aPL should allow for specific identification of all prothrombotic aPL.
Acknowledgments
We thank Dr. M. Rosove, Dr. E. Brahn, Dr. A. Gorn, and Dr. K. Kalunian for recruiting patients and obtaining blood samples for the present study. We thank Mr. Oscar Pan and Mrs. Paifei Chen for technical assistance.
Supported by research grants from the Southern California Chapter of the Arthritis Foundation, by grants AI-32243, AR-42506, AR-36834, and R37 AI/AR-46776 from the NIH, and by funds from the Paxson family, the Dorough Foundation, and the Mitchell family. Dr. Visvanathan’s work is supported by an NIH training grant in clinical and fundamental immunology (T32 AI-07126).
References
- 1.Hahn BH. Pathogenesis of systemic lupus erythematosus. In: Ruddy S, Harris ED, Sledge CB, editors. Kelley’s textbook of rheumatology. Philadelphia: W. B. Saunders; 2001. pp. 1089–101. [Google Scholar]
- 2.Greaves M. Antiphospholipid antibodies and thrombosis. Lancet. 1999;353:1348–53. doi: 10.1016/S0140-6736(98)10362-8. [DOI] [PubMed] [Google Scholar]
- 3.Lockshin MD. Pregnancy loss in the antiphospholipid syndrome. Thromb Haemost. 1999;82:641–8. [PubMed] [Google Scholar]
- 4.Wilson WA, Gharavi AE, Koike T, Lockshin MD, Branch DW, Piette JC, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome. Arthritis Rheum. 1999;42:1309–11. doi: 10.1002/1529-0131(199907)42:7<1309::AID-ANR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 5.Pierangeli SS, Stewart M, Silva LK, Harris EN. An antiphospholipid wet workshop: 7th International Symposium on Antiphospholipid Antibodies. J Rheumatol. 1998;25:156–60. [PubMed] [Google Scholar]
- 6.McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Antiphospholipid antibodies are directed against a complex antigen that includes a lipid binding inhibitor of coagulation: β2-glycoprotein I (apolipoprotein H) Proc Natl Acad Sci U S A. 1990;87:4120–4. doi: 10.1073/pnas.87.11.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galli M, Comfurius P, Maassen C, Hemker HC, de Baets MH, van Breda-Vriesman PJC, et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. 1990;335:1544–7. doi: 10.1016/0140-6736(90)91374-j. [DOI] [PubMed] [Google Scholar]
- 8.Fleck RA, Rapaport SI, Rao LV. Antiprothrombin antibodies and the lupus anticoagulant. Blood. 1988;72:512–9. [PubMed] [Google Scholar]
- 9.Bevers EM, Galli M, Barui T, Comfurius P, Zwaal RFA. Lupus anticoagulant IgG’s (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb Haemost. 1991;66:629–32. [PubMed] [Google Scholar]
- 10.Oosting JD, Derksen RHWM, Bobbink IWG, Hackeng TM, Bouma BN, de Groot PG. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: an explanation for their pathogenic mechanism? Blood. 1993;81:2618–25. [PubMed] [Google Scholar]
- 11.Arvieux J, Pernod G, Regnault V, Darnige L, Garin J. Some anticardiolipin antibodies recognize a combination of phospholipids with thrombin-modified antithrombin, complement C4b-binding protein, and lipopolysaccharide binding protein. Blood. 1999;93:4248–55. [PubMed] [Google Scholar]
- 12.Cabiedes J, Cabral AR, Alarcón-Segovia D. Clinical manifestations of the antiphospholipid syndrome in patients with systemic lupus erythematosus associate more strongly with anti-β2-glycoprotein-I than with antiphospholipid antibodies. J Rheumatol. 1995;22:1899–906. [PubMed] [Google Scholar]
- 13.Roubey RAS, Maldonado MA, Byrd SN. Comparison of an enzyme-linked immunosorbent assay for antibodies to β2-glyco-protein I and a conventional anticardiolipin immunoassay. Arthritis Rheum. 1996;39:1606–7. doi: 10.1002/art.1780390922. [DOI] [PubMed] [Google Scholar]
- 14.Tsutsumi A, Matsuura E, Ichikawa K, Fujisaku A, Mukai M, Kobayashi S, et al. Antibodies to β2-glycoprotein I and clinical manifestations in patients with systemic lupus erythematosus. Arthritis Rheum. 1996;39:1466–74. doi: 10.1002/art.1780390905. [DOI] [PubMed] [Google Scholar]
- 15.Guerin J, Feighery C, Sim RB, Jackson J. Antibodies to β2-glycoprotein I: a specific marker for the antiphospholipid syndrome. Clin Exp Immunol. 1997;109:304–9. doi: 10.1046/j.1365-2249.1997.4601357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valadon P, Nussbaum G, Boyd LF, Margulies DH, Scharff MD. Peptide libraries define the fine specificity of antipolysaccharide antibodies to cryptococcus neoformans. J Mol Biol. 1996;261:11–22. doi: 10.1006/jmbi.1996.0438. [DOI] [PubMed] [Google Scholar]
- 17.Gaynor B, Putterman C, Valadon P, Spatz L, Scharff MD, Diamond B. Peptide inhibition of glomerular deposition of an anti-DNA antibody. Proc Natl Acad Sci U S A. 1997;94:1955–60. doi: 10.1073/pnas.94.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierangeli SS, Liu XW, Espinola R, Olee T, Zhu M, Harris NE, et al. Functional analyses of patient-derived IgG monoclonal anticardiolipin antibodies using in vivo thrombosis and in vivo microcirculation models. Thromb Haemost. 2000;84:388–95. [PubMed] [Google Scholar]
- 19.Rivadeneira AC, Cakir B, Hayes PM, Roubey RAS. An in vivo model of venous thrombosis induced by IgG from patients with the antiphospholipid syndrome [abstract] Arthritis Rheum. 1998;41(Suppl 9):S135. [Google Scholar]
- 20.Zhu M, Olee T, Le DT, Roubey RAS, Hahn BH, Woods VL, Jr, et al. Characterization of IgG monoclonal anticardiolipin/anti β2GPI antibodies from two patients with the antiphospholipid syndrome reveals three species of antibodies. Br J Haematol. 1999;105:102–9. [PubMed] [Google Scholar]
- 21.Zhong GM, Smith GP, Berry J, Brunham RC. Conformational mimicry of a chlamydial neutralization epitope on filamentous phage. J Biol Chem. 1994;269:24183–8. [PubMed] [Google Scholar]
- 22.Bonnycastle LLC, Mehroke JS, Rashed M, Gong X, Scott JK. Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. J Mol Biol. 1996;258:747–62. doi: 10.1006/jmbi.1996.0284. [DOI] [PubMed] [Google Scholar]
- 23.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 24.Hochberg MC for the Diagnostic and Therapeutic Criteria Committee of the American College of Rheumatology. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter] Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 25.Rosove MH, Ismail M, Koziol BJ, Runge A, Kasper CK. Lupus anticoagulants: improved diagnosis with a kaolin clotting time using rabbit brain phospholipid in standard and high concentrations. Blood. 1986;68:472–8. [PubMed] [Google Scholar]
- 26.Motulsky H. Intuitive biostatistics. New York: Oxford University Press; 1995. [Google Scholar]
- 27.Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–95. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venkatesh N, Im SH, Balass M, Fuchs S, Katchalski-Katzir E. Prevention of passively transferred experimental autoimmune myasthenia gravis by a phage library–derived cyclic peptide. Proc Natl Acad Sci U S A. 2000;97:761–6. doi: 10.1073/pnas.97.2.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang K, Grossman J, Visvanathan S, Chukwuocha R, Woods V, Le D, et al. Identification of antithrombin antibodies in the antiphospholipid syndrome that interfere with the inactivation of thrombin by antithrombin. J Immunol. 2001;167:7192–8. doi: 10.4049/jimmunol.167.12.7192. [DOI] [PubMed] [Google Scholar]
- 30.Blank M, Shoenfeld Y, Cabilly S, Heldman Y, Fridkin M, Katchalski-Katzir E. Prevention of experimental antiphospholipid syndrome and endothelial cell activation by synthetic peptides. Proc Natl Acad Sci U S A. 1999;96:5164–8. doi: 10.1073/pnas.96.9.5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valadon P, Nussbaum G, Oh J, Scharff MD. Aspects of antigen mimicry revealed by immunization with a peptide mimetic of cryptococcus neoformans polysaccharide. J Immunol. 1998;161:1829–36. [PubMed] [Google Scholar]
- 32.Iverson GM, Jones DS, Marquis D, Linnik MD, Victoria EJ. A chemically defined, toleragen-based approach for targeting anti-β2-glycoprotein I antibodies. Lupus. 1998;7:S166–9. doi: 10.1177/096120339800700236. [DOI] [PubMed] [Google Scholar]
- 33.Sem DS, Baker BL, Victoria EJ, Jones DS, Marquis D, Yu L, et al. Structural characterization and optimization of antibody-selected phage library mimotopes of an antigen associated with auto-immune recurrent thrombosis. Biochemistry. 1998;37:16069–81. doi: 10.1021/bi9807207. [DOI] [PubMed] [Google Scholar]
- 34.Simantov R, LaSala JM, Lo SK, Gharavi AE, Sammaritano LR, Salmon JE, et al. Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest. 1995;96:2211–9. doi: 10.1172/JCI118276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornberg A, Blank M, Kaufman S, Shoenfeld Y. Induction of tissue factor–like activity in monocytes by anticardiolipin antibodies. J Immunol. 1994;153:1328–32. [PubMed] [Google Scholar]
- 36.Del Papa N, Guidali L, Sala A, Buccellati C, Khamashta MA, Ichikawa K, et al. Endothelial cells as target for antiphospholipid antibodies: human polyclonal and monoclonal anti-β2-glycoprotein I antibodies react in vitro with endothelial cells through adherent β2-glycoprotein I and induce endothelial activation. Arthritis Rheum. 1997;40:551–61. doi: 10.1002/art.1780400322. [DOI] [PubMed] [Google Scholar]
- 37.Cuadrado MJ, López-Pedrera C, Khamashta MA, Camps MT, Tinahones F, Torres A, et al. Thrombosis in primary antiphospholipid syndrome: a pivotal role for monocyte tissue factor expression. Arthritis Rheum. 1997;40:834–41. doi: 10.1002/art.1780400509. [DOI] [PubMed] [Google Scholar]
- 38.Amengual O, Atsumi T, Khamashta MA, Hughes GRV. The role of the tissue factor pathway in the hypercoagulable state in patients with the antiphospholipid syndrome. Thromb Haemost. 1998;79:276–81. [PubMed] [Google Scholar]
- 39.Cariou R, Tobelem G, Bellucci S, Soria J, Soria C, Maclouf J, et al. Effect of lupus anticoagulant on antithrombogenic properties of endothelial cells: inhibition of thrombomodulin-dependent protein C activation. Thromb Haemost. 1988;60:54–8. [PubMed] [Google Scholar]
- 40.Marciniak E, Romond EH. Impaired catalytic function of activated protein C: a new in vitro manifestation of lupus anticoagulant. Blood. 1989;74:2426–32. [PubMed] [Google Scholar]
- 41.Malia RG, Kitchen S, Greaves M, Preston FE. Inhibition of activated protein C and its cofactor protein S by antiphospholipid antibodies. Br J Haematol. 1990;76:101–7. doi: 10.1111/j.1365-2141.1990.tb07843.x. [DOI] [PubMed] [Google Scholar]
- 42.Borrell M, Sala N, de Castellanau C, Lopez S, Gari M, Foncuberta J. Immunoglobulin fractions isolated from patients with antiphospholipid antibodies prevent the inactivation of factor Va by activated protein C on human endothelial cells. Thromb Haemost. 1992;68:268–72. [PubMed] [Google Scholar]
- 43.Smirnov MD, Triplett DT, Comp PC, Esmon NL, Esmon CT. On the role of phosphatidylethanolamine in the inhibition of activated protein C activity by antiphospholipid antibodies. J Clin Invest. 1995;95:309–16. doi: 10.1172/JCI117657. [DOI] [PMC free article] [PubMed] [Google Scholar]