

Abstract
The first step in T cell receptor for antigen (TCR) signaling is the activation of the receptor-bound Src kinases, Lck and Fyn. The exact mechanism of this process is unknown. Here, we report that the novel Src homology (SH) 3/SH2 ligand–Uncoordinated 119 (Unc119) associates with CD3 and CD4, and activates Lck and Fyn. Unc119 overexpression increases Lck/Fyn activity in T cells. In Unc119-deficient T cells, Lck/Fyn activity is dramatically reduced with concomitant decrease in interleukin 2 production and cellular proliferation. Reconstitution of cells with Unc119 reverses the signaling and functional outcome. Thus, Unc119 is a receptor-associated activator of Src-type kinases. It provides a novel mechanism of signal generation in the TCR complex.
Keywords: signal transduction, Src kinases, SH3 domain, T cell antigen receptor, IL-2
Introduction
The binding of an antigen to the TCR triggers a cascade of signaling pathways that leads to activation of T cells. The molecules that play a crucial role in initiating the signaling cascade are the Src family tyrosine kinases, Lck and Fyn (1–4). The fundamental role of Lck and Fyn for development and function of T cells was established in knockout models (1, 2, 3). Despite the growing knowledge about the importance of these kinases for T cell signaling, what triggers their activation is not fully known.
Src kinases are composed of five structural components as follows: a unique region, Src homology (SH) 2 and SH3 domains, a linker region, and the kinase domain. Two intramolecular interactions keep the enzyme in a closed, “off” conformation: (a) binding of the SH3 domain to the linker region and (b) binding of the SH2 domain to the phosphorylated COOH-terminal tyrosine residue (Y505 for Lck; references 5, 6). Dephosphorylation by CD45 phosphatase releases the COOH-terminal tyrosine from SH2, which is an important step for kinase activation (9). However, this dephosphorylation alone is not sufficient to activate Lck and Fyn (8, 9). The crystal structure predicts that disruption of the intramolecular SH3–linker interaction is essential for activation and autophosphorylation of the kinase. This disruption is likely to occur through binding to a higher affinity SH3 ligand (10).
Recently, we cloned and characterized a new SH3 ligand Uncoordinated 119 (Unc119; reference 11). Unc119 was identified as an IL-5 receptor α subunit interactive protein in the yeast two-hybrid screening of a hematopoietic cell cDNA library. Unc119 is expressed in myeloid and lymphoid cells. Unc119 was independently cloned by two other groups and was also given the name human retinal gene 4 (HRG4; references 12, 13). The exact function of Unc119 is unknown. In Caenorhabditis elegans, Unc119 was found to be involved in feeding, chemosensation, and locomotion of the worm (13). In transgenic mouse, expression of the mutated HRG4 under rhodopsin promoter caused retinal degeneration (14). The analysis of the Unc119 sequence revealed the presence of SH3- and SH2-binding sites. Unc119 bound and, more importantly, activated Lyn and Hck kinases through its SH3- and SH2-binding motif (11). Here, we demonstrate that Unc119 is crucial for signal generation by the TCR and is essential for T cell function.
Materials and Methods
Antibodies.
The mouse monoclonal anti-CD2, CD3, CD4, CD28, isotype controls, fluorescently labeled secondary antibodies, and anti–human IL-2 ELISA kit were obtained from BD Biosciences; mouse monoclonal antiphosphotyrosine (4G10) and anti-CD45 were obtained from Upstate Biotechnology; goat polyclonal anti–glutathione S-transferase (GST) was obtained from Amersham Biosciences; and mouse monoclonal anti-FLAG M2 was obtained from Sigma-Aldrich. The generation of rabbit polyclonal anti-Unc119 was described previously (11). All other antibodies were purchased from Santa Cruz Biotechnology, Inc.
Cells and Stimulation.
Mononuclear cells were isolated from peripheral blood of healthy donors by Ficoll-Hypaque (Sigma-Aldrich) separation. For T cell isolation, adherent cells and B cells were removed by successive incubations of mononuclear cells on uncoated and anti-CD19–coated plastic plates, respectively. If needed, T cells were stimulated with soluble anti-CD3 at 1 μg/ml for 3 min and lysed.
Immunoprecipitation, GST Pulldown, and Western Blotting.
These experiments were performed as described previously (11). For coprecipitation with CD3, we substituted 1% NP-40 with 1% Brij 35 in the lysis buffer, which improved the results.
Immune Complex Kinase Assay.
The immunoprecipitated kinases were assayed in the kinase buffer (100 mM Hepes, pH 7.4, 50 mM NaCl, 10 mM MgCl2, 100 μM Na3VO4, 500 μM DTT, 25 mM β-glycophosphate) containing additionally 5 μCi [32P]γ-ATP (Amersham Biosciences) and 2.5 μM ATP at 30°C for 5–10 min. The reactions were done in the presence or absence of the Src substrate enolase (Sigma-Aldrich) at 5 μM, recombinant Unc119, an SH3 peptide from CD2 (15), SH3 peptide (11), and SH3 mutant peptide (11) from Unc119. Samples were run on polyacrylamide gels, transferred, and autoradiographed.
Plasmids and Cloning.
For production of Jurkat transfectants, we used retroviral vectors pMSCVneo, pLEGFP-N1 (both obtained from CLONTECH Laboratories, Inc.), and green fluorescent protein (GFP)–RV (a gift from K. Murphy, Washington University, St. Louis, MO). The following inserts were ligated into pMSCVneo: complete Unc119 cDNA, recovered from pB42AD-Unc119, as described previously (11) and PCR-amplified 268-1108 bp fragment of Unc119/HRG4 cDNA (GenBank/EMBL/DDBJ accession no. U40998) inserted in the reversed orientation (antisense). Inverted Unc119 DNA was also inserted into bicistronic retroviral vector GFP-RV for the production of Unc119-deficient T cells. The open reading frame analysis showed that inverted fragments do not encode any protein or peptide. To create GFP-Unc119 fusion protein construct, 52-776 bp (U40998) Unc119 PCR fragment was ligated into pLEGFP-N1. GFP was fused to the COOH terminus of Unc119. The orientation of inserts was verified by PCR, and constructs were sequenced. Generation of the GST-Unc119 construct and production of recombinant GST-Unc119 and Unc119 were described previously (11).
Generation of Unc119 SH3 Motif Mutant.
As a template for mutagenesis (QuikChange; Stratagene) we used the Unc119-GFP-RV construct containing the complete coding sequence of Unc119 (U40998, 50-1036 bp). The first round of mutagenesis involved the inframe addition of FLAG (5′-gattacaaggatgacgacgataag-3′) to the 3′ terminus of the Unc119 sequence. SH3 motif modification was performed on Unc119-FLAG-GFP-RV plasmid. The NH2-terminal SH3 motif PQPP (residues 29–32, 5′-ccacagccgcct-3′) was mutated to AQPA (5′-gcacagccggca-3′). The wild-type Unc119-FLAG construct and the SH3 motif mutant plasmid were sequenced and used for the production of the retrovirus.
Generation of the Retrovirus and T Cell Infection.
We used previously described protocol with some modifications (16). For Jurkat cell infection, the 293 cells were transiently transfected with retroviral constructs as aforementioned, VSV-G (envelope) vector, and gag-pol (gifts from R. Davey, University of Texas Medical Branch, Galveston, TX). The supernatants were collected 48 h after transfection (viral titer = 108–109 CFU/ml) and sterilized with 0.45-μm filters. Cells were suspended in viral supernatants mixed 1:1 with growth media (RPMI 1640 + 10% FCS) at the final concentration of 8 μg/ml polybrene and centrifuged in plates at 1200 g for 90 min at 32°C (CLONTECH Laboratories, Inc. protocol). After 2 mg/ml G418 selection (4 wk), individual clones were isolated by the limiting dilution. Before infection, human primary T cells were stimulated with anti-CD3 and anti-CD28 antibodies (plate-bound) and 100 U/ml IL-2 (PeproTech) for 48 h. The infection was performed as aforementioned, but Phoenix Ampho packaging cell line (American Type Culture Collection on behalf of G. Nolan, Stanford University, Palo Alto, CA) was used, and viral media were supplemented with 100 U/ml of IL-2. GFP+ cells were sorted by flow cytometry 8 d after infection and analyzed 2–5 d after sorting.
Protein Transduction by Chariot.
Cells were incubated with Unc119 or human serum albumin (Sigma-Aldrich) in the presence of the protein-transducing reagent Chariot (Active Motif) as described previously (11).
Fluorescent Microscopy.
In translocation experiments, GFP or GFP-Unc119-expressing Jurkat cells, prepared as aforementioned, were mixed with anti-CD3, anti-CD28, or isotype-control antibody-coated protein A/G agarose beads and incubated at 37°C for 1–5 min. The bead–cell conjugates were fixed in 1% paraformaldehyde and examined under the microscope. Capping experiments were performed as described previously (17). For each antibody (bead or capping test), three independent experiments were performed, and 50–70 cells (or bead–cell conjugates) were examined per experiment. More than 85% of cells demonstrated interaction with bead or capping and >75% of bead-bound or capped cells showed GFP/GFP-IRIP localization similar to that shown in Fig. 2 (B and C), respectively.
Figure 2.
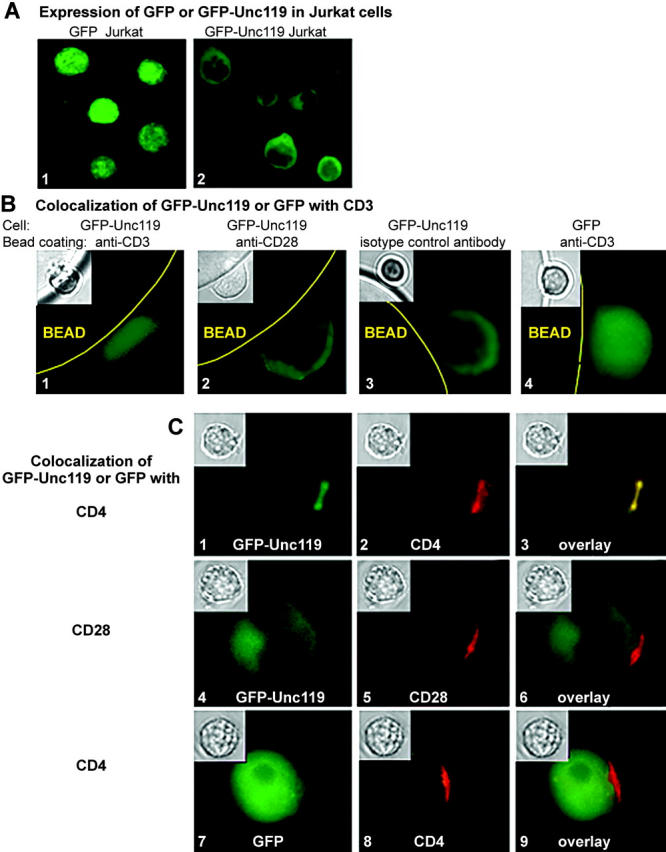
Unc119 translocates to the TCR complex upon anti-CD3 stimulation. (A) Expression of GFP-Unc119 fusion protein. Jurkat cells were stably transfected with pLEGFP-N1 containing cDNA for GFP alone (1) or GFP-Unc119 fusion protein (2) and examined by fluorescence microscopy. (B) Unc119 translocates to CD3 upon stimulation. GFP-Unc119 (1, 2, and 3) or GFP (4) Jurkat cells were incubated with anti-CD3 (1 and 4) or anti-CD28 (2) or isotype-control antibody (3)–coated protein A/G agarose beads, fixed, and analyzed by fluorescence and light (inset) microscopy. (C) Unc119 translocates to CD4 upon stimulation. Receptors of GFP-IRIP (1–6) or GFP (7–9) Jurkat cells were capped with anti-CD4 (1–3, 7–9), anti-CD28 (4–6), or isotype controls (not depicted) and examined by fluorescence or light (inset) microscopy. (left) GFP/GFP-IRIP fluorescence. (middle) Rhodamine (CD4 or CD28) fluorescence. (right) Color overlay.
Proliferation Assay.
T cells were cultured for 72 h on anti-CD3–coated 96-well plates or left unstimulated. [3H]Thymidine (Amersham Biosciences) was added at 1 μCi/well 12 h before the conclusion of the culture. Cells were harvested and 3H radiation was measured by liquid scintillation.
Cytokine ELISA.
T cells were cultured for 36 h on anti-CD3–coated plates or left unstimulated. IL-2 was measured in supernatants by ELISA.
Apoptosis Assay.
T cells were cultured for 72 h on anti-CD3–coated plates or left unstimulated. Annexin V/propidium iodide staining of the cells was performed according to the manufacturer's protocol (BD Biosciences) and analyzed by flow cytometry.
Surface Antigen Staining.
T cells were blocked with 30% human serum (AB+, Rh+) in RPMI 1640 for 30 min, followed by 1 h incubation with anti-CD3 or isotype control at 1 μg/106 cells on ice, and next with FITC-conjugated secondary antibody. Stained cells were analyzed by flow cytometry.
Data Presentation and Analyses.
Each experiment has been repeated 3–10 times, and a representative blot, image, or graph is shown in the figures.
Online Supplemental Material.
In Fig. S1, we present data regarding Unc119 expression in multiple antisense Unc119-overexpressing clones, Lck activation in Unc119-deficient Jurkat clones, and downstream signaling (LAT and ERK activation) in Unc119-deficient T cells. The description of generation of clones overexpressing sense and antisense cDNA for Unc119 has been aforementioned. The activation of Lck, LAT, and ERK in these clones was studied according to the aforementioned method. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20030589/DC1.
Results
Unc119 Is Present in T Cells.
We have shown previously that Unc119 activates Lyn kinase in eosinophils (11). To elucidate its function in other hematopoietic cells, we investigated the presence of Unc119 in human blood T cells and Jurkat T cells through Western blotting (Fig. 1 A). Sequence analysis predicts that Unc119 has a mass of ∼37–39 kD. We observed the appearance of three bands (one prominent and two faint, in this molecular weight range). The prominent middle band migrated in the gel in the same position as the recombinant Unc119. Furthermore, the anti-Unc119–depleted serum failed to detect the middle band and rUnc119 on the blot, whereas other bands were still present, suggesting that the middle band was indeed Unc119. Our results suggest that Unc119 is present in T cells.
Figure 1.
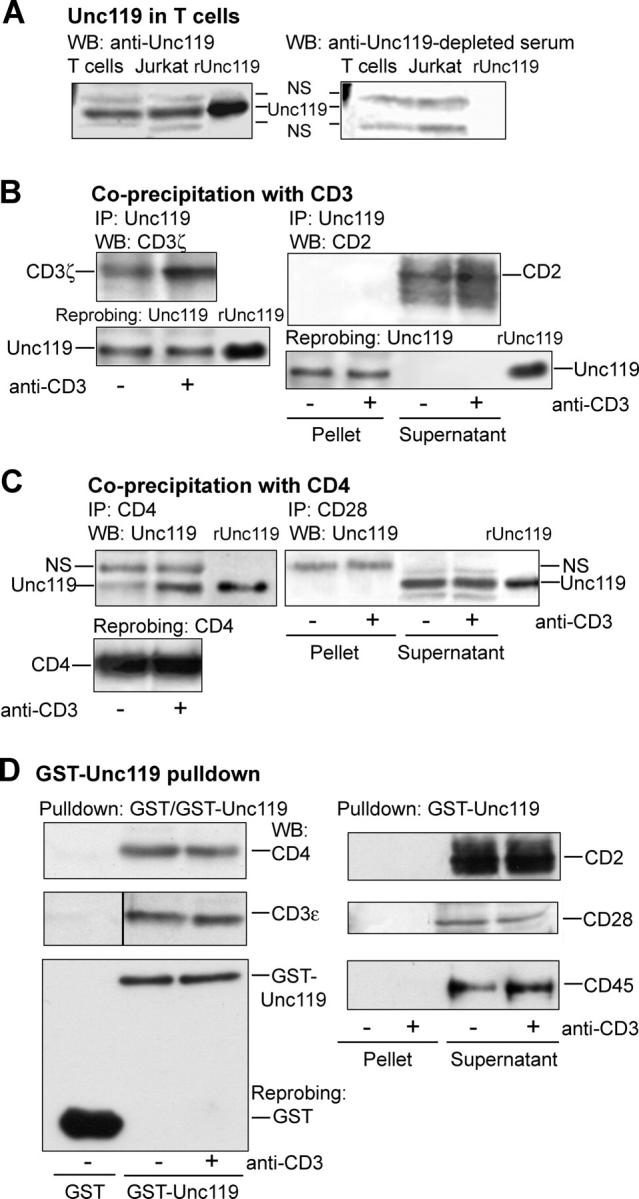
Unc119 interacts with the TCR complex. (A) Unc119 is expressed in human primary T cells and the Jurkat T cell line. Cell lysates were Western blotted with anti-Unc119 (left) or anti-Unc119 antibody–depleted (right) serum. Recombinant Unc119 (rUnc119) was electrophoresed to mark its position. NS, nonspecific band. (B) Unc119 specifically coprecipitates with CD3 in T cells. Human primary T cells were incubated with (+) or without (−) anti-CD3 and lysed. As a control, the anti-CD3 antibody was added to unstimulated sample after lysis. Lysates were immunoprecipitated (IP) with anti-Unc119 and Western blotted with antibodies against indicated molecules. The membranes were reprobed with anti-Unc119 to demonstrate equal protein loading (bottom). (Pellet) Immunoprecipitated proteins. (Supernatant) Cell lysate after removal of immunoprecipitated proteins. (C) Unc119 coprecipitates with CD4 in T cells. T cells, prepared as in B, were immunoprecipitated (IP) with indicated antibodies. Pellets and/or supernatants (B) were Western blotted with anti-Unc119. The membrane (left) was reprobed with anti-CD4. (D) GST-Unc119 interacts with CD3 and CD4. T cells, prepared as in B, were subjected to GST or GST-Unc119 pulldown. Pellets and/or supernatants (B) were Western blotted with antibodies against indicated molecules. Membranes from all experiments were reprobed with an anti-GST antibody. A representative blot is shown (bottom left).
Unc119 Interacts with TCR.
We investigated the association of TCR with Unc119 in primary T cells by coprecipitation and GST-Unc119 pulldown experiments. Unc119 interacted with CD3ζ and CD4 molecules under basal conditions and was additionally recruited to the receptors after anti-CD3 stimulation (Fig. 1, B and C, left). In parallel experiments, Unc119 failed to coprecipitate with CD2 (Fig. 1 B, right, pellet) or CD28 (Fig. 1 C, right, pellet). However, the antibodies detected target molecules in the supernatant fraction. We studied the association of Unc119 with another CD3 subunit. In pulldown experiments, GST-Unc119 interacted with CD3ɛ and CD4 (Fig. 1 D, left). Binding of Unc119 to CD3ɛ and CD4 was equally high in stimulated and nonstimulated cells, probably because of the use of excess GST-Unc119. Parallel pulldown experiments demonstrated lack of association of Unc119 with CD2, CD28, and CD45 (Fig. 1 D, right).
Unc119 Localizes to the CD3/CD4 Complex.
To examine its cellular distribution, we expressed Unc119 as the GFP fusion protein in Jurkat cells. GFP alone was distributed homogenously in all cellular compartments, including the nucleus (Fig. 2 A, image 1). In contrast, GFP-Unc119 was primarily cytoplasmic and was excluded from the nucleus (Fig. 2 A, image 2). To examine the localization of Unc119 upon TCR stimulation, we performed two types of experiment: (a) stimulation with anti-CD3–coated beads and (b) capping with a soluble anti-CD4 antibody. Upon TCR engagement, GFP-Unc119 translocated to the contact zone with anti-CD3–coated beads (Fig. 2 B, image 1). It also cocapped with anti-CD4 (Fig. 2 C, images 1–3). GFP-Unc119 localization did not change upon anti-CD28 stimulation or interaction of the cell with an isotype-matched control antibody (Fig. 2, B, images 2 and 3, and C, images 4–6). Similarly, GFP alone did not change upon anti-CD3 stimulation or CD4 ligation (Fig. 2, B, image 4, and C, images 7–9). The colocalization experiments provided the morphological confirmation of the results from coprecipitation and GST pulldown experiments.
Unc119 Specifically Interacts with Lck and Fyn.
CD3 and CD4 are known to interact with Fyn and Lck tyrosine kinases, respectively. We examined the association of Unc119 with Lck and Fyn. Coprecipitation (Fig. 3, A and B, left) and GST-Unc119 pulldown (Fig. 3 C, left) experiments with primary T cells showed the recruitment of Unc119 to Lck and Fyn upon anti-CD3 stimulation. We observed a low level of interaction between GST-Unc119 and the kinases in pulldown experiments with the unstimulated cell lysate. We believe that it is due to the excess of the recombinant protein used. Like other members of the Src-type kinases, Lck and Fyn have SH2 and SH3 domains in their sequence. We have shown previously that Unc119 interacts with the SH2 and SH3 domains of Lyn and Fyn (11). To determine specificity of this interaction, we examined the association of Unc119 with other SH2- and/or SH3-containing kinases as follows: p85 of phosphatidyl inositol-3 kinase (Fig. 3 A, right) and ZAP70 and Itk (Fig. 3 B, right) in coprecipitation experiments. ZAP70 as well as Abl, FAK, p85 subunit of phosphatidyl inositol-3 kinase, and the serine threonine kinases Raf-1, ERK2, and p38 MAPK were studied in GST-Unc119 pulldown experiments (Fig. 3 C, right). Unc119 and GST-Unc119 did not precipitate the foregoing kinases (pellet). However, our antibodies detected the kinases in the supernatant fraction. Previously, we reported that Unc119 did not interact with the SH2/SH3 adaptor proteins Grb2 and Shc (11). The results suggest that the interaction of Unc119 is relatively specific for the SH3/SH2 domains of Src family kinases.
Figure 3.
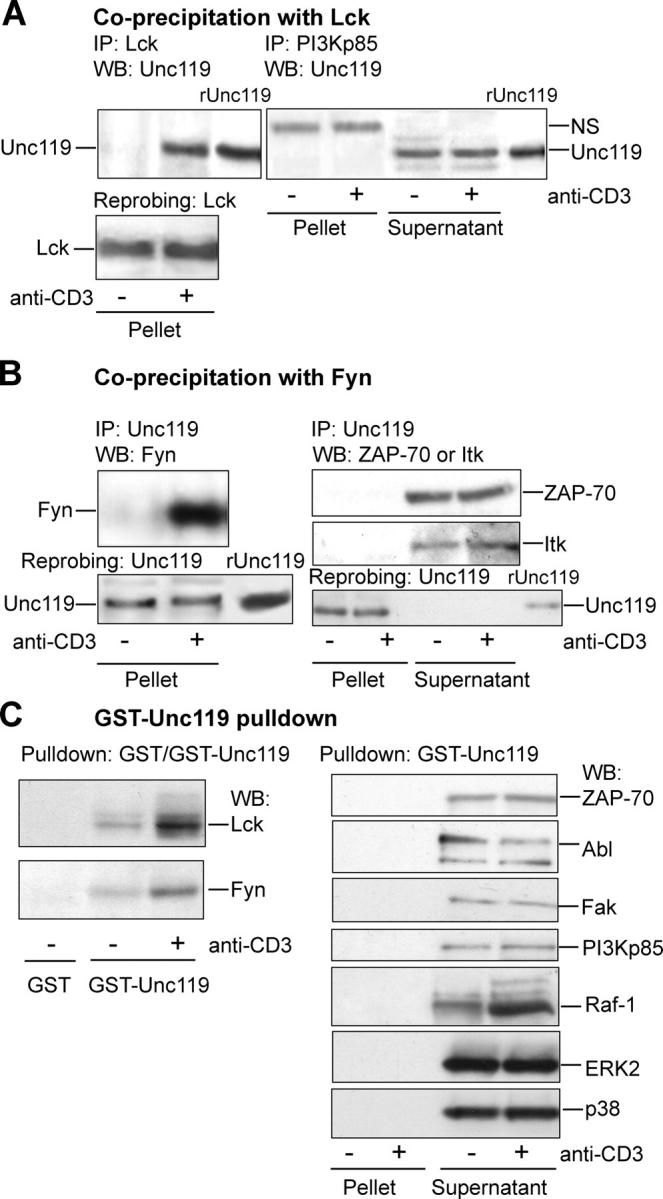
Unc119 specifically interacts with Lck and Fyn. (A) Unc119 specifically interacts with Lck in T cells. T cells were prepared as in Fig. 1 B, immunoprecipitated (IP) with indicated antibodies. Pellets and/or supernatants (see Fig. 1 B) were Western blotted with anti-Unc119 antibody and reprobed with anti-Lck. NS, nonspecific band. (B) Unc119 interacts with Fyn in T cells. T cells were prepared as in Fig. 1 B and immunoprecipitated (IP) with anti-Unc119. Pellets and/or supernatants (Fig. 1 B) were Western blotted with antibodies against indicated proteins and reprobed with anti-Unc119 (bottom). (C) GST-Unc119 interacts with Lck and Fyn. Cells lysates, prepared as aforementioned, were precipitated with GST or GST-Unc119. Pellets and/or supernatants were Western blotted with antibodies against indicated proteins.
Recruitment of Unc119 to Lck Is Concomitant with Elevated Kinase Activity.
Next, we asked if the association of Unc119 with Lck is correlated with an increase in the kinase activity and induction of downstream signaling events. Toward this goal, we stimulated peripheral blood T cells for increasing periods of time with anti-CD3 antibody. The lysates of these cells were used for Lck/Unc119 coprecipitation experiment, Lck kinase assay, and for the measurement of CD3ζ phosphorylation. Fig. 4 A shows that Unc119 associated with Lck from 30 s to 5 min of stimulation with the highest binding at the 2-min time point (Fig. 4 A, I). Interestingly, Lck activity slightly increased at 30 s as compared with nonstimulated kinase, reached maximum at 2 min, dramatically decreased at 10 min, and went back to basal level after the latter time point (Fig. 4 A, III). Therefore, the binding of Unc119 to Lck correlated well with the augmented kinase activity. Conversely, the dissociation of Unc119 from Lck correlated with the decrease in the kinase activity. We also examined the kinetics of CD3ζ phosphorylation in these cells. CD3ζ was phosphorylated to some extent at 30 s and maximally at 2 min, and became undetectable at 60 min (Fig. 4 A, V). Overall, the association of Unc119 with Lck was always concomitant with enhanced signaling through TCR. Both the Lck activity and CD3ζ phosphorylation reached the peak at the time when the Unc119–Lck interaction was maximum.
Figure 4.
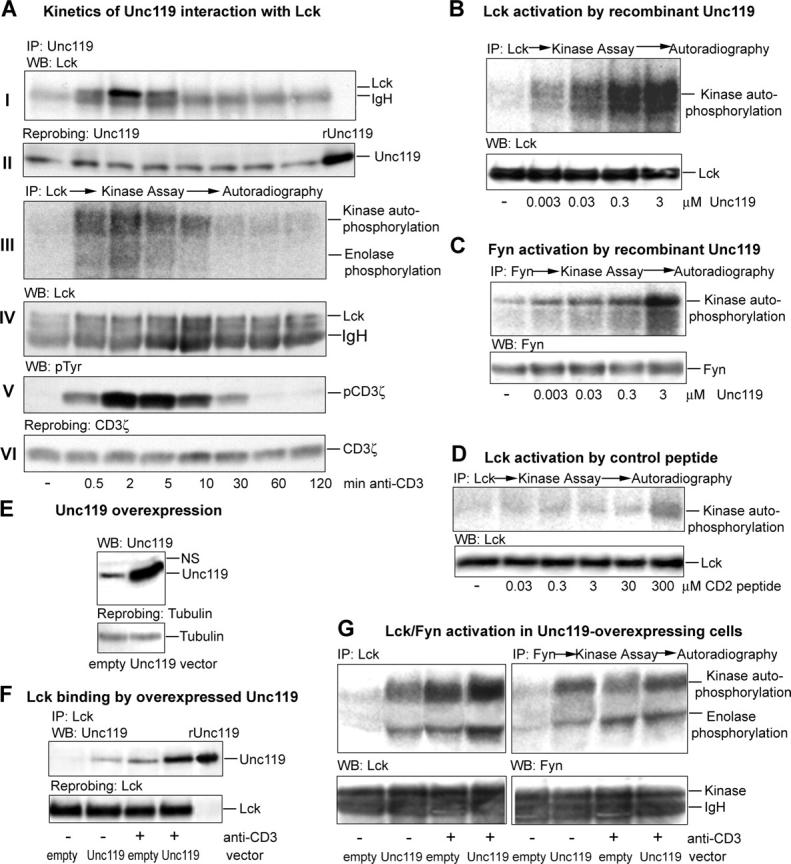
Unc119 activates Lck and Fyn. (A) Unc119-Lck association correlates with kinase activity. T cells were stimulated for indicated time and lysed. Lysates were immunoprecipitated with indicated antibodies. The anti-Unc119 and anti-CD3ζ precipitates were Western blotted with anti-Lck (I) and antiphosphotyrosine (V, pTyr), respectively, and reprobed with anti-Unc119 (II) and anti-CD3ζ (VI). Anti-Lck precipitates were used in the kinase assay with enolase as a substrate in the presence of [32P]γ-ATP and autoradiographed (III). The kinase assay membrane was blotted with monoclonal anti-Lck (IV). (B–D) Recombinant Unc119 activates Lck and Fyn. Lck (B and D) or Fyn (C) were immunoprecipitated (IP) with rabbit antikinase antibodies, subjected to the kinase assay in the presence of recombinant purified Unc119 (B and C) or a CD2-derived peptide (D), and autoradiographed. Membranes were Western blotted (bottom) with monoclonal anti-Lck (B and D) or anti-Fyn (C). (E) Generation of Unc119-overexpressing Jurkat clones. Jurkat cells were transfected with pMSCVneo (empty vector) or pMSCVneo-Unc119 (Unc119 vector). Lysates of select clones were Western blotted with anti-Unc119 and reprobed with antitubulin. (F) Recruitment of Unc119 to Lck is enhanced in overexpressing cells. Unc119-overexpressing (Unc119) and empty vector (empty) clones were incubated with (+) or without (−) anti-CD3 and lysed. Lysates were immunoprecipitated with anti-Lck, Western blotted with anti-Unc119 (top), and reprobed with anti-Lck (bottom). (G) Overexpressed Unc119 activates Lck and Fyn. Lck and Fyn was immunoprecipitated (IP) from Unc119-overexpressing and empty vector Jurkat lysates (F), assayed for kinase activity as in A, and autoradiographed. Membranes were Western blotted with rabbit anti-Lck or anti-Fyn antibodies (bottom). IgH, immunoglobulin heavy chain.
Exogenous Unc119 Activates Lck and Fyn.
The results of the aforementioned experiment point to the possible involvement of Unc119 in T cell Src kinase activation. Indeed, in an immune complex kinase assay, purified recombinant Unc119 stimulated Fyn and Lck autophosphorylation (Fig. 4, B and C). As a positive control, we used a previously described CD2-derived SH3 motif peptide that activated Fyn in vitro (15). In accordance with the foregoing publication, this peptide activated Lck (Fig. 4 D) and Fyn (not depicted), but required a relatively high concentration of 300 μM. In contrast, Unc119 activated the kinases at a concentration as low as 3 nM. We reported previously that Unc119 did not activate the nonSrc tyrosine kinase Itk and the serine-threonine kinase p38 MAP kinase (11). The results suggest that a physical interaction is essential for Unc119 activation of kinases.
Lck/Fyn Activation in Unc119-overexpressing Cells.
To test its effect on Lck and Fyn function in vivo, we stably overexpressed Unc119 in Jurkat cells using the retroviral vector pMSCV (Fig. 4 E). We have generated six stable Unc119-overexpressing Jurkat clones. These clones were examined for Unc119–Lck interaction and the activation of Lck and Fyn. A representative coprecipitation experiment and kinase assay with one select clone are shown in Fig. 4 F and G, respectively. As expected, Unc119 did not interact with Lck in unstimulated empty vector clone, and there was very little Lck activation. Stimulation with anti-CD3 resulted in the recruitment of Unc119 to Lck and significant increase in both kinase autophosphorylation and substrate (enolase) phosphorylation. In contrast, Unc119-overexpressing Jurkat clone demonstrated the binding of Unc119 to Lck already in the resting state, which was associated with increased kinase activity (Fig. 4 G). The coprecipitation experiment is in the agreement with GST-Unc119 pulldown data, showing that if Unc119 is present in excess, it associates with the kinase even in the resting cells. Stimulation of Unc119-overexpressing clone with anti-CD3 antibody caused additional recruitment of Unc119 to Lck. The recruitment was higher than in the empty vector cells. The increased binding of Unc119 to Lck in stimulated Unc119-overexpressing clone resulted in enhanced kinase activity. The activation of Fyn kinase in these cells followed Lck activity pattern (Fig. 4 G). Although overexpression is a nonphysiologic experimental system, the results of these experiments provide a proof of concept for Unc119 activation of Lck and Fyn in vivo.
Development of Unc119-deficient T Cells.
To develop Unc119-deficient T cells, we stably transfected human primary T cells and Jurkat cells with the bicistronic GFP-RV and pMSCV retroviral constructs, respectively, containing the Unc119 antisense cDNA. The antisense construct was created by ligating the Unc119 cDNA into vectors in reverse orientation. After transfection, GFP+ primary T cells were sorted by flow cytometry. Transfected Jurkat cells were selected on G418 and cloned. We have isolated five Unc119-deficient clones. The expression of Unc119 in sorted T cells (Fig. 5 A, left) and Jurkat clones (Fig. 5 A, right, and Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20030589/DC1) was substantially reduced. Reprobing the membrane with antitubulin ruled out the possibility of unequal protein loading (Fig. 5 A and Fig. S1 A, bottom). Unc119-deficient primary T cells and Jurkat clones demonstrated a dramatic reduction in anti-CD3–stimulated Fyn and/or Lck activation (Fig. 5 B, top, and Fig. S1 B, top, respectively). Because the introduction of the antisense vector did not alter total or surface (only slight elevation) CD3 (Fig. 5 A, bottom left and flow cytometry graph), CD4 (not depicted), and Lck and Fyn expression (Fig. 5 B, bottom), the inability of the anti-CD3 antibody to stimulate these kinases is likely due to the decrease in Unc119 concentration. The results strongly suggest that Unc119 is physiologically relevant for Lck and Fyn activation in T cells.
Figure 5.

Lck/Fyn activity in Unc119-deficient cells. (A) Unc119 level in antisense Unc119 cDNA-expressing T cells. Cells were transfected retrovirally with empty vectors (GFP-RV for primary T cells and pMSCV for Jurkat cells) or same vectors containing Unc119 antisense cDNA (antisense). GFP+ primary T cells were sorted by flow cytometry and infected Jurkat cells were selected and cloned on G418. The presence of Unc119 (Unc), CD3ζ, and tubulin was analyzed by Western blotting (left and middle). NS, nonspecific band. CD3 surface expression on Unc119-deficient cells was measured by flow cytometry (right graph, dashed line, isotype control; thin line, empty vector; and thick line, antisense vector). (B) Lck and Fyn activity in Unc119-deficient cells. GFP+ Unc119-deficient primary T cells were immunoprecipitated with rabbit anti-Lck and anti-Fyn, analyzed for kinase activity as in Fig. 4 A, and autoradiographed. Membranes were Western blotted with monoclonal anti-Lck or anti-Fyn antibodies (bottom).
Unc119 Deficiency Blocks TCR Signaling Pathways.
Next, we studied the effect of Unc119 deficiency on downstream signaling pathways. Jurkat cells (Fig. 6) and primary T cells (Fig. S1) demonstrated similar results. Unc119-deficient cells exhibited a substantial reduction in overall protein tyrosine phosphorylation (Fig. 6 A). The reduction correlated with the pronounced decline in the activation of key Lck/Fyn substrates as follows: tyrosine kinases ZAP-70 (Fig. 6 B, left) and Itk (Fig. 6 B, right). LAT is one of the most important signaling proteins phosphorylated by ZAP-70 upon TCR stimulation. Unc119-deficient cells showed a marked reduction in LAT phosphorylation (Fig. 6 C and Fig. S1 C). The foregoing signaling pathways contribute to MAP kinase activation. The activation of ERK kinases in antisense cells was also strikingly diminished (Fig. 6 D and Fig. S1 D). The results suggest that in the absence of Unc119, the TCR is unable to stimulate the most immediate (Lck/Fyn) as well as downstream signaling pathways. Because we have shown that Unc119 does not activate and/or interact with ZAP-70, Itk, nor MAP kinases (Fig. 3, B and C, and reference 11), we speculate that the observed global decrease in signaling activation is likely secondary to the lack of activation of Lck and Fyn.
Figure 6.

Downstream signaling and phenotype of Unc119-deficient cells. (A) Protein tyrosine phosphorylation in Unc119-deficient cells. Antisense Jurkat cells were incubated with (+) or without (−) anti-CD3 antibody, lysed, Western blotted with antiphosphotyrosine antibody, and reprobed with the antitubulin. (B) ZAP70 and Itk activity in Unc119-deficient cells. Antisense Jurkat cell lysates, prepared as in A, were immunoprecipitated with anti-ZAP70 or anti-Itk antibodies, analyzed for kinase activity as in Fig. 4 A, and autoradiographed. Membranes were Western blotted with anti-ZAP70 and anti-Itk antibodies (bottom). (C) LAT phosphorylation in Unc119-deficient cells. Antisense Jurkat cell lysates, prepared as in A, were immunoprecipitated with anti-LAT, Western blotted with antiphosphotyrosine, and reprobed with the anti-LAT antibody. (D) ERK phosphorylation in Unc119-deficient cells. Antisense Jurkat cell lysates, prepared as in A, were Western blotted with anti-phosphoERK and reprobed with anti-ERK2. (E) IL-2 production by Unc119-deficient cells. GFP+ primary T cells or Jurkat clones were cultured without (−) or with anti-CD3 (CD3) or Concanavalin A (left, ConA) or Phorbol myristate acetate + Ionomycin (right, PMA + Io) for 36 h. The IL-2 concentration (ELISA) in the supernatant was adjusted to the number of live cells (Trypan blue) at the conclusion of the culture. E, empty vector. A, antisense vector. (F) Proliferation of Unc119-deficient cells. GFP+ primary T cells were cultured for 72 h on anti-CD3 coated plates (+) or left unstimulated (−). [3H]Thymidine incorporation was measured. E, empty vector. A, anti-sense vector.
Unc119 Is Essential for T Cell Function.
Next, we examined the phenotypic outcome of Unc119 deficiency in T cells. Lck and Fyn regulate IL-2 production, T cell proliferation, and survival (1–3, 18). For this reason, we examined the effect of Unc119 deficiency on the foregoing T cell functions. Unc119-deficient primary T cells and Jurkat clones produced negligible amounts of IL-2 after anti-CD3 or Concanavalin A stimulation (Fig. 6 E, left). This blockade of IL-2 production was significantly reversed upon stimulation with ionomycin and phorbol ester, which activate signaling molecules downstream of the Src kinases (Fig. 6 E, right). The decrease in IL-2 production and its incomplete reversal after ionomycin/PMA treatment can be partially attributed to an increase in basal apoptosis of Jurkat cells (see Fig. 7 C). In contrast to Jurkat cells, Unc119-deficient primary T cells did not show elevated apoptosis (unpublished data). We observed a fourfold reduction in [3H]thymidine incorporation in antisense primary T cells, as compared with empty vector cells (Fig. 6 F). Overall, the phenotype of Unc119-deficient cells largely resembled the phenotype of T cell clones transfected with Lck antisense cDNA (18).
Figure 7.
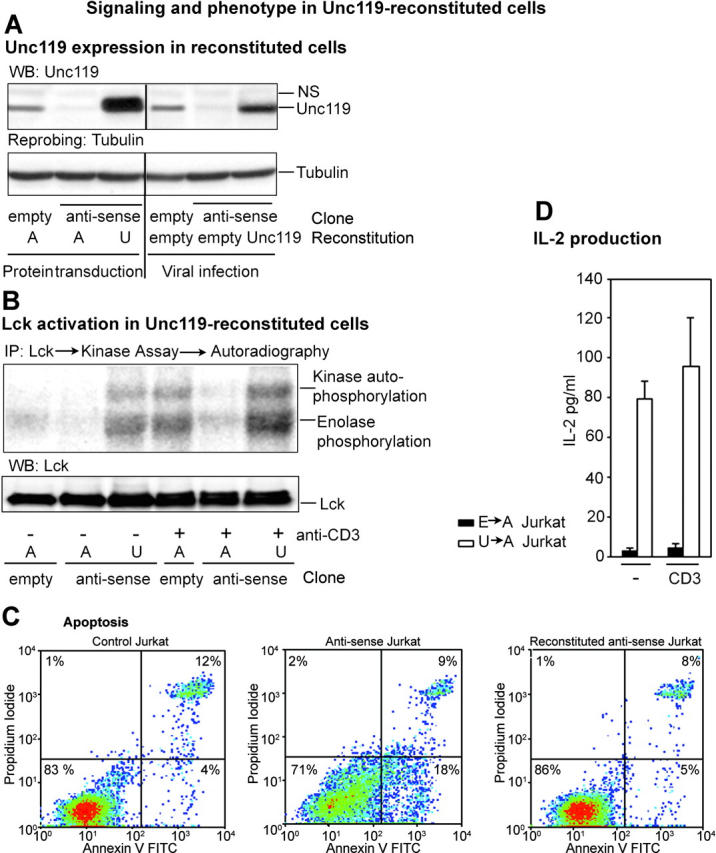
Reconstitution of Unc119-deficient Jurkat cells with Unc119. (A) Unc119 level in reconstituted cells. Unc119-deficient Jurkat clones were incubated with purified recombinant Unc119 (U) or human serum albumin (A) in the presence of the protein-transducing reagent Chariot (left area of top and bottom). The right area of the panels shows reconstitution by retroviral infection. Antisense clone was infected with the empty virus (pMSCV) or Unc119 virus (pMSCV-Unc119). An empty vector Jurkat clone was infected with empty virus. Reconstituted cell lysates were Western blotted with anti-Unc119 antibody (top) and reprobed with antitubulin (bottom). (B) Lck activity in Unc119 reconstituted cells. Unc119-deficient cells were reconstituted with Unc119 (U) or albumin (A) in the Chariot experiment and incubated with (+) or without (−) anti-CD3. Samples were analyzed for kinase activity as in Fig. 4 A, autoradiographed, and Western blotted with a monoclonal anti-Lck antibody. (C) Apoptosis in Unc119-deficient cells and the effect of Unc119 reconstitution. Empty vector and antisense Jurkat cells were reconstituted with albumin (left and middle) or Unc119 (right) as in A and B, cultured for 72 h, stained with annexin V–FITC/propidium iodide, and analyzed by flow cytometry. Anti-CD3 stimulation did not influence apoptosis in Unc119-deficient cells (not depicted). (D) Effect of Unc119 reconstitution on IL-2 production of antisense Jurkat cells. Antisense cells were infected with the empty virus (E→A Jurkat) or Unc119 virus (U→A Jurkat) with 30% infection efficiency as in A. The empty vector Jurkat clone was infected with empty virus (E→E Jurkat). Transiently infected cells were incubated on anti-CD3–coated plates (CD3) or left unstimulated (−). IL-2 production was measured as in Fig. 6 E. E→E Jurkat cells produced 30 ± 10 and 240 ± 20 pg/ml, without and with anti-CD3 stimulation, respectively (similar to noninfected cells).
Unc119 Reconstitution Rescues the Phenotype of Deficient Cells.
To determine whether the observed biochemical and phenotypic changes in the antisense clones are solely due to the lack of Unc119, we reconstituted Jurkat cells with Unc119 using the protein-transducing compound Chariot. We have reported previously successful internalization of proteins using Chariot (11). Fig. 7 A shows the level of Unc119 in the reconstituted cell population. We examined Src kinase activation in these cells. Internalization of the control protein (human serum albumin) did not influence Lck activity. In contrast, Unc119-transduced cells were fully able to activate Lck upon TCR stimulation (Fig. 7 B, top). Furthermore, the basal Lck activity in Unc119-reconstituted cells was comparable to that in stimulated control cells. These results are in agreement with Unc119 overexpression experiments, in which elevation of the basal kinase activity was also detected (see Fig. 4 G). We observed that antisense Jurkat cells had increased basal apoptosis (Fig. 7 C, middle vs. left panel). This increased apoptosis was completely reversed after Unc119 reconstitution (Fig. 7 C, right). To further study the phenotype of Unc119-reconstituted cells, we infected antisense cells with the Unc119 cDNA-containing vector pMSCV. The expression level of Unc119 in infected cells is shown in Fig. 7 A. Unc119-reconstituted cells demonstrated significant elevation in IL-2 production (22-fold; Fig. 7 D) in comparison to empty vector–transduced antisense cells. Unc119 reconstitution alone was sufficient to restore the foregoing deficiency. Stimulation of the reconstituted cells with the anti-CD3 antibody did not have any further effect. The reversal of function reached only 40% of IL-2 production of the control cell line (empty vector clone superinfected with the empty vector in the reconstitution phase). The reconstitution experiment for phenotype studies was performed with a mixed cell population. The efficiency of infection in these experiments was ∼30% (as measured by parallel infections with viruses containing cDNA for GFP), which is likely due to the slow cell proliferation rate of Unc119-deficient cells. It is known that the infection efficiency of retroviral viral vectors is highly dependent upon the rate of cell proliferation. Given this low infection efficiency, we believe that Unc119 reconstitution substantially reversed the phenotypic changes of antisense cells.
SH3 Motif Peptide from Unc119 Activates Lck In Vitro.
Previously, we reported activation of Lyn kinase by a SH3 motif peptide and to a lesser extent SH2 and phospho-SH2 peptides from Unc119 (11). We believe that Unc119 activation of Src kinases is mainly mediated through its interaction with the SH3 domain. In this paper, we examined the activation of Lck by the SH3 motif peptide from Unc119 (Fig. 8 A, left). To study the importance of motif-related conserved residues, we designed another peptide in which critical proline and arginine residues of the motif peptide were substituted with alanine (Fig. 8 A, right). The SH3 peptide, but not mutated peptide, activated Lck at a concentration as low as 0.01 μM. We obtained similar results for Fyn kinase (unpublished data).
Figure 8.

Unc119 binds and activates Lck through the SH3 motif. (A) An SH3 motif peptide from Unc119 activates Lck. Lck immunoprecipitates (IP) were assayed for kinase activity as in Fig. 4 A in the presence of the SH3 motif (left) or mutated peptide (right) from Unc119 (11). Membranes were Western blotted (bottom) with a monoclonal anti-Lck antibody. (B) Expression of a SH3 motif mutant in Jurkat cells. Jurkat cells (left, single transfection experiment) or the Unc119-deficient Jurkat clone (right, reconstitution experiment) were infected with the GFP-RV empty vector (empty) or the SH3 motif mutant of Unc119-FLAG-GFP-RV vector (mut) or the Unc119-FLAG-GFP-RV vector (wt). As an additional control, the empty vector clone was infected with GFP-RV. GFP+ cells (left and right) were sorted, Western blotted (top) with anti-FLAG antibody, and reprobed with antitubulin (bottom). (C) The SH3 motif mutant of Unc119 does not coprecipitate with Lck. GFP+ cells (B, single transfection) were incubated with (+) or without (−) anti-CD3, lysed, and immunoprecipitated with anti-Lck. Pellets and supernatants (Fig. 1 B) were Western blotted with anti-FLAG (top) and reprobed with anti-Lck (bottom). (D) The SH3 motif mutant inhibits Lck activation. GFP+ cells (B, single transfection) were treated as in C, analyzed for kinase activity as in Fig. 4 A, autoradiographed (top), and Western blotted with anti-Lck antibody (bottom). (E) The SH3 motif mutant is unable to rescue Lck activity in Unc119-deficient cells. GFP+ cells (B, reconstitution) were incubated with (+) or without (−) anti-CD3 antibody, analyzed for kinase activity as in Fig. 4 A, autoradiographed (top), and Western blotted with anti-Lck antibody (bottom).
Mutations in the SH3 Motif Sequence Abrogate Physical Interaction with and Activation of Lck.
To determine the importance of the SH3 motif sequence, we have created a construct wherein the proline residues in the canonical PXXP motif was substituted with alanine. We also added a FLAG tag at the COOH terminus to be able to distinguish from the native Unc119. The construct was ligated into the GFP-RV vector. The mutant and wild-type Unc119-containing virus was used to infect Jurkat T cells. Cells expressing GFP were sorted and used for subsequent studies. Fig. 8 B shows expression of the FLAG-tagged mutant and wild-type Unc119 in Jurkat cells. Coprecipitation studies demonstrated that the mutant Unc119 was unable to interact with Lck either under basal or stimulated (anti-CD3) conditions (Fig. 8 C). In contrast, FLAG-tagged wild-type Unc119 bound Lck modestly under basal conditions and more profoundly after anti-CD3 stimulation. Finally, we examined the effect of mutation of the SH3 motif on Lck activation (Fig. 8 D). Immunoprecipitated Lck from mutant Unc119-overexpressing cells showed lack of activation (autophosphorylation and substrate phosphorylation) regardless of cell stimulation status. Lck from cells overexpressing the empty construct showed activation mainly after anti-CD3 stimulation. In contrast, Lck from cells overexpressing the wild-type Unc119 demonstrated activation under basal conditions, which was further increased upon stimulation. We speculate that the lack of Lck activity in mutant-overexpressing T cells is due to the displacement of native, cell-derived Unc119 from its cellular partners by the mutated protein. Our results suggest that the interaction through the SH3 domain plays a crucial role in activation of Lck by Unc119.
The Unc119 SH3 Mutant Is Unable to Rescue Lck Activity in Unc119-deficient Cells.
To further investigate the role of SH3 motif mutation, we reconstituted Unc119-deficient Jurkat cells with the mutant virus. As a control, the Unc119-deficient clone was infected with the empty vector (GFP-RV) or Unc119-FLAG-GFP-RV vector. Simultaneously, the previously generated empty vector clone was retransfected with the empty vector virus. Infected cells were sorted and analyzed for FLAG fusion protein expression and Lck activity. Reconstituted cells demonstrated expression of FLAG fusion proteins (Fig. 8 B, right). The kinase assay showed lack of Lck activation in antisense cells transfected with the empty vector (Fig. 8 E). The introduction of the SH3 mutant did not cause any elevation of kinase activity in resting as well as stimulated cells. In contrast, introduction of the Unc119-FLAG-GFP-RV vector protein was concomitant with the increased kinase activation in resting cells, which was further augmented after CD3 stimulation. These results further document the importance of the Unc119 SH3 motif in Lck kinase activation.
Discussion
This manuscript addresses a fundamental problem of T cell biology: the signal generation in the TCR complex. How ligand binding to the TCR complex leads to the activation of Lck and Fyn is not fully known. Crystallographic analyses predict that an SH3 ligand–motif interaction is critical for Src kinase activation (5, 6). This prediction was confirmed elegantly by Moerafi et al. (10), who showed that the HIV protein Nef and its SH3 motif peptide directly activated Hck, a Src family kinase. In this work, we demonstrate that the novel TCR-associated SH3 ligand Unc119 activates Lck and Fyn. Unc119 is essential for initiation of TCR signaling and T cell function.
We provided biochemical (coprecipitation, GST pulldown, and mutational studies) and morphological evidence for the association of Unc119 with the CD3 complex. The essential role of Unc119 for activation of Lck and Fyn was demonstrated in vitro in a reductionist model and in vivo in overexpression, depletion, and reconstitution models. In Unc119-deficient cells, activation of ZAP70, Itk, LAT, and MAP kinases was greatly reduced. Biochemical changes in Unc119-deficient cells resemble the signaling pattern in T cells, which do not express Lck (4).
We have preliminary biochemical understanding of the mechanism of activation Src kinases by Unc119. Previously, we reported the interaction between the SH2 and SH3 domains of Src kinases (Lyn and Fyn) and Unc119 (11). Recombinant Unc119 interacted in a reductionist model with recombinant SH2 and SH3 domains of the Src kinases, but not with that from Grb2 and Shc, indicating a direct and specific physical interaction. Conversely, the SH3, SH2, and phosphorylated SH2 motif peptides from Unc119 pulled down Lyn from the cell lysate. The SH3 peptide and to a lower extent the SH2 peptides activated Lyn (11). The foregoing experiments established the biochemical basis for SH3-mediated activation of Src kinases by Unc119. In the present paper, we confirmed Lck activation by the motif peptide but not the mutated peptide. More importantly, we showed that overexpression of an Unc119 construct with substitution of the conserved proline residues completely abrogated its interaction with and activation of Lck. Unc119 has two SH3 motifs and the NH2-terminal motif was mutated in our studies. We do not know the importance of the other motif for Lck activation in vivo. Nonetheless, our results suggest that the NH2-terminal SH3 motif is essential for Lck activation.
Unc119 is essential for T cell function. Proliferation and IL-2 production was decreased in Unc119-deficient T cells. Additionally, Unc119-deficient Jurkat cells showed increased apoptosis. Jurkat is a tumor cell line, so it is not surprising that it behaves differently than nontransformed primary cells in some aspects. The phenotype of Unc119-deficient cells largely mimics the phenotype of Lck-deficient T cells. Lck antisense cDNA-transfected T cells demonstrate cell cycle arrest and hypersensitivity to apoptosis (18). The absence of Lck is associated with the profound reduction of T cell number in mice (1). Unc119 seems to regulate survival of nonhematopoietic cells as well. Organ-specific overexpression of a mutant Unc119 causes retinal degeneration (14). Interestingly, Lck is also expressed in retina and Lck-deficient mice show retinal dysplasia (19).
Recently, CD2 and CD28 were shown to activate Lck through their SH3-binding motifs (15). Unc119 is more potent than the CD2 peptide and activates the kinase at 4–5 orders of magnitude lower concentrations. More importantly, CD2 and CD28 are not directly involved in signal generation by TCR. CD4 was found to be essential for recruitment of Lck to the immunological synapse and its subsequent activation (17). However, the molecular mechanism by which CD4 activates Lck remains unclear. Unlike CD28 and CD2, CD4 does not have any classic consensus SH3-binding motifs (20). We believe that the recruitment of CD4 allows a direct interaction of Lck with Unc119, which leads to its activation.
Unc119 is the first receptor-associated activator of Lck and Fyn. It is essential for initiation of TCR signaling and T cell function. An understanding of Unc119 regulation of TCR signaling may help develop novel immunomodulators. Because Src-type kinases are associated with many cellular functions, including carcinogenesis, Unc119 may have a broader biological relevance.
Acknowledgments
The work was supported by the McLaughlin Fellowship Fund (to M.M. Gorska and O. Cen) and National Institute of Health grants AI46004 (to R. Alam, S.J. Stafford, and S. Sur), AI50179 (to R. Alam), and the John Sealy Memorial Endowment Fund (to R. Alam).
The online version of this article includes supplemental material.
Abbreviations used in this paper: GFP, green fluorescent protein; GST, glutathione S-transferase; HRG4, human retinal gene 4; SH, Src homology; Unc119, Uncoordinated 119.
References
- 1.Molina, T.J., K. Kishihara, D.P. Siderovski, W. van Ewijk, A. Narendran, E. Timms, A. Wakeham, C.J. Paige, K.U. Hartmann, A. Veillette, et al. 1992. Profound block in thymocyte development in mice lacking p56lck. Nature. 357:161–164. [DOI] [PubMed] [Google Scholar]
- 2.Appleby, M.W., J.A. Gross, M.P. Cooke, S.D. Levin, X. Qian, and R.M. Perlmutter. 1992. Defective T cell receptor signaling in mice lacking the thymic isoform of p59fyn. Cell. 70:751–763. [DOI] [PubMed] [Google Scholar]
- 3.Stein, P.L., H.M. Lee, S. Rich, and P. Soriano. 1992. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell. 70:741–750. [DOI] [PubMed] [Google Scholar]
- 4.Straus, D.B., and A. Weiss. 1992. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 70:585–593. [DOI] [PubMed] [Google Scholar]
- 5.Sicheri, F., I. Moarefi, and J. Kuriyan. 1997. Crystal structure of the Src family tyrosine kinase Hck. Nature. 385:602–609. [DOI] [PubMed] [Google Scholar]
- 6.Xu, W., S.C. Harrison, and M.J. Eck. 1997. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 385:595–602. [DOI] [PubMed] [Google Scholar]
- 7.Cahir McFarland, E.D., T.R. Hurley, J.T. Pingel, B.M. Sefton, A. Shaw, and M.L. Thomas. 1993. Correlation between Src family member regulation by the protein-tyrosine-phosphatase CD45 and transmembrane signaling through the T-cell receptor. Proc. Natl. Acad. Sci. USA. 90:1402–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Oro, U., and J.D. Ashwell. 1999. The CD45 tyrosine phosphatase is an inhibitor of Lck activity in thymocytes. J. Immunol. 162:1879–1883. [PubMed] [Google Scholar]
- 9.Rodgers, W., and J.K. Rose. 1996. Exclusion of CD45 inhibits activity of p56lck associated with glycolipid-enriched membrane domains. J. Cell Biol. 135:1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moarefi, I., M. LaFevre-Bernt, F. Sicheri, M. Huse, C.H. Lee, J. Kuriyan, and W.T. Miller. 1997. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 385:650–653. [DOI] [PubMed] [Google Scholar]
- 11.Cen, O., M.M. Gorska, S.J. Stafford, S. Sur, and R. Alam. 2003. Identification of Unc119 as a novel activator of Src-type tyrosine kinases. J. Biol. Chem. 278:8837–8845. [DOI] [PubMed] [Google Scholar]
- 12.Higashide, T., and G. Inana. 1999. Characterization of the gene for HRG4 (UNC119), a novel photoreceptor synaptic protein homologous to unc-119. Genomics. 57:446–450. [DOI] [PubMed] [Google Scholar]
- 13.Maduro, M., and D. Pilgrim. 1995. Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics. 141:977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi, A., T. Higashide, D. Hamasaki, S. Kubota, H. Sakuma, W. An, T. Fujimaki, M.J. McLaren, R.G. Weleber, and G. Inana. 2000. HRG4 (UNC119) mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Invest. Ophthalmol. Vis. Sci. 41:3268–3277. [PubMed] [Google Scholar]
- 15.Holdorf, A.D., J.M. Green, S.D. Levin, M.F. Denny, D.B. Straus, V. Link, P.S. Changelian, P.M. Allen, and A.S. Shaw. 1999. Proline residues in CD28 and the Src homology (SH)3 domain of Lck are required for T cell costimulation. J. Exp. Med. 190:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ouyang, W., S.H. Ranganath, K. Weindel, D. Bhattacharya, T.L. Murphy, W.C. Sha, and K.M. Murphy. 1998. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 9:745–755. [DOI] [PubMed] [Google Scholar]
- 17.Holdorf, A.D., K.H. Lee, W.R. Burack, P.M. Allen, and A.S. Shaw. 2002. Regulation of Lck activity by CD4 and CD28 in the immunological synapse. Nat. Immunol. 3:259–264. [DOI] [PubMed] [Google Scholar]
- 18.al-Ramadi, B.K., H. Zhang, and A.L. Bothwell. 1998. Cell-cycle arrest and apoptosis hypersusceptibility as a consequence of Lck deficiency in nontransformed T lymphocytes. Proc. Natl. Acad. Sci. USA. 95:12498–12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Omri, B., C. Blancher, B. Neron, M.C. Marty, J. Rutin, T.J. Molina, B. Pessac, and P. Crisanti. 1998. Retinal dysplasia in mice lacking p56lck. Oncogene. 16:2351–2356. [DOI] [PubMed] [Google Scholar]
- 20.Alexandropoulos, K., G. Cheng, and D. Baltimore. 1995. Proline-rich sequences that bind to Src homology 3 domains with individual specificities. Proc. Natl. Acad. Sci. USA. 92:3110–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]