

Abstract
A goal in cancer therapeutics is to develop targeted modalities that distinguish malignant from normal cells. T cells can discriminate diseased cells based on subtle alterations in peptides displayed in association with MHC molecules at the cell surface. Recent success using the adoptive transfer of tumor-specific T cells has fueled optimism that this approach may find a place as a targeted therapy for some human cancers.
The ability to adoptively transfer T cells to treat cancer and infections has been demonstrated in rodent models, but the task of translating the principles established in animal studies into the clinic has not been easy. A notable achievement in adoptive T cell therapy in humans was its use to control infections caused by cytomegalovirus (CMV) and Epstein Barr virus (EBV) in immunocompromised allogeneic bone marrow transplant recipients (1–3). This was a logical beginning for clinical application of T cell therapy because the pretransplant conditioning and immunosuppressive drugs administered to bone marrow transplant recipients cause a severe T cell deficiency that correlates with progressive infection. Moreover, virus-specific CD8+ T cells, which are the critical effector cells for controlling these viruses, could be readily isolated and expanded from the immunocompetent bone marrow donor for adoptive transfer. In the case of EBV infection in bone marrow transplant patients, a lymphoproliferative disease (EBV-LPD) of EBV-infected B cells was the target of therapy. The malignant B cells in EBV-LPD express a subset of EBV genes referred to as the latency III program, which includes the highly immunogenic EBNA-3A, 3B, and 3C proteins (4, 5). The adoptive transfer of EBV-specific T cells isolated from the transplant donor and consisting primarily of CD8+ T cells specific for EBNA 3 proteins promoted regression of established EBV-LPD in transplant recipients and prevented the development of tumors when given prophylactically (3, 6).
Extending the Role of EBV-specific T Cells for Therapy of EBV-associated Malignancies.
Several malignancies that occur in immunocompetent individuals also contain the EBV genome including a subset of Burkitt's lymphoma, Hodgkin's disease, and nasopharyngeal carcinoma. These malignancies express one or more of the EBNA-1, LMP-1, and LMP-2 viral proteins, which cooperate in establishing the malignancy (5). There has been pessimism regarding whether specific T cell therapy targeting either EBNA-1, LMP-1, or LMP-2 would be possible since these proteins are weakly immunogenic and the tumors evade immune recognition by a variety of mechanisms. In this issue, Bollard et al. show that the adoptive transfer of EBV-reactive T cells to patients with Hodgkin's disease can augment CD8+ T cell immunity to the weakly immunogenic LMP-2 protein, and that the infused T cells migrate to tumor sites and promote tumor regression (7). The authors appropriately urge caution in interpreting their study, which analyzed only a small number of patients who also had recent chemotherapy or radiation. Nevertheless, coupled with the recent success using adoptive T cell therapy to treat melanoma and leukemia (8–10) these results justify current efforts to discover antigens expressed by human tumors and improve the methodology for isolating tumor-reactive T cells that can survive and function in vivo after adoptive transfer.
Obstacles to Identifying Tumor-associated Antigens for T Cell Therapy.
A key step in the development of T cell therapy is the discovery of antigens expressed by malignant cells. Much of the focus has been on identifying antigens recognized by cytolytic CD8+ T cells because this subset is critical in murine models of T cell immunotherapy for cancer. Oncogenic viral proteins, such as EBNA-1, LMP-1, and LMP-2, that are expressed in the malignant Reed Sternberg cells in ∼40% of Hodgkin's disease patients represent logical target antigens because they are absent on normal cells and cooperate in cell transformation. However, targeting these antigens with adoptive T cell therapy has not been straightforward. The CD8+ T cell response to EBV is strongly focused on lytic cycle EBV proteins and the EBNA 3 antigens, with only weak responses detected to LMP-1 and LMP-2 (4). EBNA-1 was long considered a poor target for therapy because it contains a Gly-Ala repeat sequence that both inhibits proteosomal degradation needed to generate most immunogenic peptides and reduces EBNA-1 mRNA translation (11, 12). However, recent studies show that EBNA-1–specific CD8+ T cells can be detected in vivo and inhibit the outgrowth of EBV-transformed cells in vitro, resurrecting the notion that EBNA-1 might also be a useful target antigen (13, 14). Thus, although LMP-1 and LMP-2 are considered the most likely targets for effective immunotherapy of Hodgkin's disease, careful analysis of patients that receive adoptive T cell therapy will be required to determine the specificity of T cells that promote tumor regression, particularly if polyclonal populations of EBV-reactive T cells are administered.
Antigens expressed by human cancers that do not develop as a consequence of oncogenic viruses have also been identified as potential targets for T cell therapy, but many have conceptual or practical limitations. In allogeneic stem cell transplantation, T cells specific for minor histocompatibility antigens, which are peptides that differ between donor and recipient due to polymorphism in the genome, provide a potent graft versus leukemia effect (15). However, T cells specific for some minor histocompatibility antigens are also responsible for graft versus host disease, and a means of reliably segregating the beneficial graft versus leukemia effect from graft versus host disease has not yet been established.
Autologous T cells could potentially recognize tumor antigens that arise from mutated proteins or nonmutated self-proteins in human malignancies. Mutated proteins are conceptually attractive targets because they are often involved in cell transformation and could provide antigens to which the host is not tolerant. However, many mutations are specific for a single patient's tumor or involve point mutations or fusion proteins with limited immunogenicity, and identifying T cells reactive with these determinants has proven difficult in most patients.
Nonmutated self-proteins represent the largest group of tumor-associated antigens and include proteins that are expressed in a tissue-specific fashion such as the melanosome proteins in malignant melanoma, proteins that are overexpressed as a consequence of cellular transformation such as Her-2/neu in breast cancer, and proteins that are atypically expressed, such as the cancer-testes antigens, in many tumors (16). It was perceived that tolerance mechanisms would limit the repertoire or responsiveness of T cells specific for self-proteins. However, such T cells are increasingly being found in the blood or locally infiltrating tumors in cancer patients, providing evidence that the immune system detects the tumor even if the response is quantitatively or qualitatively insufficient to promote tumor regression. A critical issue is to identify which nonmutated self-antigens are best suited for targeted T cell therapy. Several factors related to antigen expression should be considered including expression on normal cells, homogeneity of expression on tumor cells and tumor stem cells, and the propensity for tumors to escape by loss of antigen expression.
The Isolation and Expansion of T Cells for Immunotherapy.
The in vitro expansion of T cells reactive with antigens expressed on tumor cells could overcome quantitative and qualitative defects but can represent a daunting obstacle because of the poor stimulatory capacity or availability of tumor cells. In the case of EBV-associated malignancies such as Hodgkin's disease, autologous B cells transformed in vitro with EBV (EBV-LCL) were used as APCs. EBV-LCL express all of the EBV-latent proteins in Hodgkin's disease and high levels of MHC molecules and ligands for T cell costimulatory receptors. However, EBV-LCL predominantly expand CD8+ T cells specific for the immunodominant EBNA-3 proteins that are not expressed in Hodgkin's disease. Indeed, in the study by Bollard et al. LMP-2–reactive T cells represented only a small minority of the infused T cell population (7), and one might speculate that greater antitumor activity may be achieved if methods to selectively enrich LMP-1–specific T cells were employed and greater numbers of tumor-reactive T cells were infused.
The molecular characterization of tumor-associated antigens has aided the development of methods for the selective isolation and expansion of tumor-reactive T cells in vitro. Stimulation of T cells with autologous or artificial APCs pulsed with antigenic peptides or transfected with genes that encode tumor or viral antigens has been used to enrich polyclonal T cells for a desired specificity (17, 18). Peptide–MHC tetramers and bispecific antibodies have also been developed for isolating specific T cells by flow cytometry or immunomagnetic selection (19, 20). It is possible to confer specificity for tumors by introducing the genes that encode the α and β chains of a T cell receptor of known specificity and MHC restriction into T cells (21), or by introducing engineered chimeric immunoreceptors that link a tumor-targeting antibody domain to the TCR ζ chain (22). These latter approaches diminish the arduous task of isolating tumor-reactive T cells from patients but have yet to be proven clinically effective and may impose additional risk due to gene insertion. Once tumor-reactive T cells are isolated, techniques have been established to efficiently propagate these T cells in vitro using IL-2 and antibodies that activate signaling through CD3 and costimulatory molecules. Thus, the ability to generate T cells for immunotherapy is progressively improving and will facilitate additional trials of targeted immunotherapy for human malignancy with autologous tumor-reactive T cells or T cell clones (Fig. 1).
Selecting the “Right” T Cells for Adoptive Therapy.
The adoptive transfer of T cells that recognize antigens expressed by malignant cells should in principle allow for precise control of the function and magnitude of T cell immunity and should correct quantitative or qualitative deficiencies in the host response. However, in many clinical trials cultured CD8+ T cells have engrafted poorly perhaps due to a lack of CD4+ T cells needed to provide helper functions or to a long duration of culture in excessive concentrations of IL-2, which drives T cells to terminal differentiation and conditions them for death with subsequent activation or cytokine withdrawal. CD4+ T cells are important for optimal CD8+ T cell immunity, and adoptive transfer studies in humans in which well-characterized populations of CD4+ T cells, alone or in combination with CD8+ T cells, are administered are necessary to define their potential role in improving the efficacy of adoptive therapy.
Antigen-specific T cells represent heterogeneous populations of differentiated cells that include effector, effector–memory, and central–memory subsets that can be distinguished by functional properties and the differential expression of homing receptors and costimulatory molecules (23). The transition between these subsets is not completely understood and has not been easy to orchestrate in vitro. The procedures for isolating and expanding T cells in culture generate effector cells, which might have optimal antitumor activity but exhibit a short survival time in vivo analogous to effector cells generated de novo in a primary immune response. In murine models, the transfer of CD8+ effector cells that express the IL-7Rα chain, which could provide both survival and proliferative signals, led to the establishment of persistent T cell memory (24), suggesting that subsets of effector cells might be selected with an improved ability to persist in vivo.
Careful immunologic monitoring of patients treated with adoptive immunotherapy is proving essential to provide insight into the qualities of T cells that predict long-term persistence in vivo after adoptive transfer. Flow cytometry with MHC tetramers or molecular monitoring to detect T cell receptor genes or an introduced marker gene has been used to track the fate of transferred T cells in vivo. These monitoring techniques are easily applied for tracking transferred T cells in the blood and can also be employed to examine migration to lymph nodes or tumor sites. The use of MHC tetramers for detecting transferred cells also allows the isolation of viable cells from the blood or tumor sites for functional analysis.
Monitoring of patients that receive adoptive immunotherapy has revealed the importance of costimulatory molecule expression in determining cell survival in vivo. Adoptively transferred melanoma-reactive T cells that persisted long term exhibited a CD28+ CD27+ phenotype, suggesting these costimulatory molecules were reacquired on surviving cells or that a subset of CD28+ CD27+ cells in the infused population selectively survived and expanded in vivo (25). In HIV-infected patients, CD27− HIV-specific T cells rapidly disappeared after adoptive transfer, whereas CD27+ cells derived from the same clone persisted at high frequency (26). Future studies are likely to use more sophisticated methods for monitoring, including gene expression arrays, and may define properties of antigen-specific T cells that correlate with improved persistence and antitumor efficacy.
Modifying the Host Environment to Promote the Efficacy of T Cell Therapy.
The environment into which T cells are adoptively transferred can also have a significant effect on cell survival and therapeutic efficacy. Transferred CD8+ CMV-specific T cell clones survived in vivo as functional memory cells only in patients with antigen-specific CD4+ T cell responses, suggesting an important role for CD4+ T cells in improving cell persistence (1). The administration of low dose IL-2, which is produced by CD4+ T cells and promotes T cell proliferation and survival in vitro, improved the survival of transferred melanoma-reactive T cells in vivo (9). Other cytokines that share the common γ chain receptor and regulate lymphocyte growth and survival, such as IL-7, IL-15, and IL-21, are also being investigated for improving the efficacy of adoptively transferred T cells.
Studies in murine models suggest that lymphodepletion induced by total body irradiation or other modalities might be exploited to improve the antitumor efficacy of adoptively transferred T cells (27). The lymphopenic environment may be favorable for several reasons including the availability of space for transferred T cells in the lymphoid compartment, less competition for homeostatic cytokines, such as IL-7 and IL-15, and the elimination of CD4+ CD25+ regulatory T cells. This approach has been examined recently in human trials of adoptive T cell therapy for melanoma. Patients were first treated with lymphodepleting chemotherapy followed by the administration of tumor-reactive T cells expanded in vitro from tumor infiltrates and systemic high-dose IL-2. Transferred T cells underwent dramatic in vivo expansion in a subset of patients, infiltrated into tumor sites, and promoted tumor regression (8). In some patients, transferred T cells comprised a large fraction (up to 70%) of the total peripheral blood lymphocytes for several months after T cell infusion. These results although encouraging were not without complications as many responding patients exhibited autoimmune destruction of normal melanocytes (8). Additional studies are needed to determine if lower doses of IL-2 or other cytokines would be effective in promoting T cell persistence after transfer into a lymphopenic environment and to determine if autoimmunity can be controlled without compromising antitumor activity.
Overcoming Tumor Immune Evasion.
Although progress has been made in tumor-specific T cell therapy, malignancies are formidable adversaries, and the majority of treated patients still fail to completely respond to therapy or progress after an initial response. This likely reflects strategies that tumors utilize to overcome immune recognition, which may include defects in the processing or presentation of tumor antigens, production of factors that disable tumor-reactive T cells, and encasement in a stromal environment that is inhospitable to immune attack (28, 29). This illustrates the importance of monitoring not only the transferred T cells but also the characteristics of tumors that respond or fail to respond to therapy to derive insights into the critical immune evasion mechanisms that must be overcome to improve efficacy. T cells can be readily modified by gene insertion to introduce molecules that promote T cell signaling or homing to tumor sites or interfere with signaling by inhibitory molecules (30). Thus, tools are available to enhance the efficacy of T cell therapy, which based on progress in immunology and tumor cell biology appears to be gradually assuming a place in the armamentarium of targeted cancer therapies.
Figure .
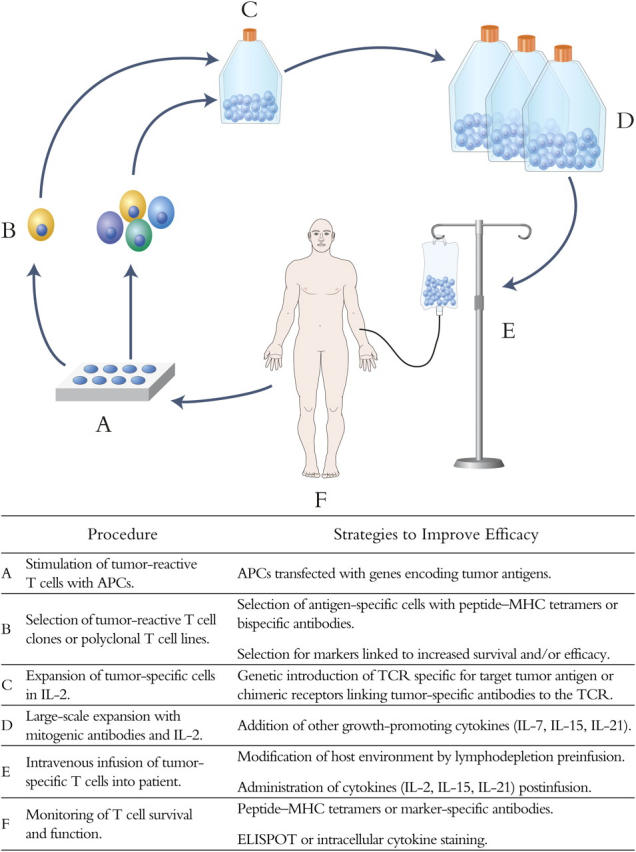
Strategies to Improve Efficacy
References
- 1.Walter, E.A., P.D. Greenberg, M.J. Gilbert, R.J. Finch, K.S. Watanabe, E.D. Thomas, and S.R. Riddell. 1995. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 333:1038–1044. [DOI] [PubMed] [Google Scholar]
- 2.Peggs, K.S., S. Verfuerth, A. Pizzey, N. Khan, M. Guiver, P.A. Moss, and S. Mackinnon. 2003. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. 362:1375–1377. [DOI] [PubMed] [Google Scholar]
- 3.Heslop, H.E., C.Y. Ng, C. Li, C.A. Smith, S.K. Loftin, R.A. Krance, M.K. Brenner, and C.M. Rooney. 1996. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat. Med. 2:551–555. [DOI] [PubMed] [Google Scholar]
- 4.Hislop, A.D., N.E. Annels, N.H. Gudgeon, A.M. Leese, and A.B. Rickinson. 2002. Epitope-specific evolution of human CD8(+) T cell responses from primary to persistent phases of Epstein-Barr virus infection. J. Exp. Med. 195:893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young, L.S., and A.B. Rickinson. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer. 4:757–768. [DOI] [PubMed] [Google Scholar]
- 6.Rooney, C.M., C.A. Smith, C.Y. Ng, S.K. Loftin, J.W. Sixbey, Y. Gan, D.K. Srivastava, L.C. Bowman, R.A. Krance, M.K. Brenner, et al. 1998. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 92:1549–1555. [PubMed] [Google Scholar]
- 7.Bollard, C.M., L. Aguilar, K.C. Straathof, B. Gahn, M.H. Huls, A. Rousseau, J. Sixbey, M.V. Gresik, G. Carrum, M. Hudson, et al. 2004. Cytotoxic T lymphocyte therapy for EBV-positive Hodgkin's Disease. J. Exp. Med. 200:1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudley, M.E., J.R. Wunderlich, P.F. Robbins, J.C. Yang, P. Hwu, D.J. Schwartzentruber, S.L. Topalian, R. Sherry, N.P. Restifo, A.M. Hubicki, et al. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 298:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yee, C., J.A. Thompson, D. Byrd, S.R. Riddell, P. Roche, E. Celis, and P.D. Greenberg. 2002. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci. USA. 99:16168–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marijt, W.A., M.H. Heemskerk, F.M. Kloosterboer, E. Goulmy, M.G. Kester, M.A. van der Hoorn, S.A. van Luxemburg-Heys, M. Hoogeboom, T. Mutis, J.W. Drijfhout, et al. 2003. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc. Natl. Acad. Sci. USA. 100:2742–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levitskaya, J., A. Sharipo, A. Leonchiks, A. Ciechanover, and M.G. Masucci. 1997. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA. 94:12616–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin, Y., B. Manoury, and R. Fahraeus. 2003. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science. 301:1371–1374. [DOI] [PubMed] [Google Scholar]
- 13.Lee, S.P., J.M. Brooks, H. Al-Jarrah, W.A. Thomas, T.A. Haigh, G.S. Taylor, S. Humme, A. Schepers, W. Hammerschmidt, J.L. Yates, et al. 2004. CD8 T cell recognition of endogenously expressed Epstein-Barr virus nuclear antigen 1. J. Exp. Med. 199:1409–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voo, K.S., T. Fu, H.Y. Wang, J. Tellam, H.E. Heslop, M.K. Brenner, C.M. Rooney, and R.F. Wang. 2004. Evidence for the presentation of major histocompatibility complex class I–restricted Epstein-Barr virus nuclear antigen 1 peptides to CD8+ T lymphocytes. J. Exp. Med. 199:459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bleakley, M., and S.R. Riddell. 2004. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat. Rev. Cancer. 4:371–380. [DOI] [PubMed] [Google Scholar]
- 16.Rosenberg, S.A. 1999. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 10:281–287. [DOI] [PubMed] [Google Scholar]
- 17.Oelke, M., M.V. Maus, D. Didiano, C.H. June, A. Mackensen, and J.P. Schneck. 2003. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat. Med. 9:619–624. [DOI] [PubMed] [Google Scholar]
- 18.Maus, M.V., A.K. Thomas, D.G. Leonard, D. Allman, K. Addya, K. Schlienger, J.L. Riley, and C.H. June. 2002. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat. Biotechnol. 20:143–148. [DOI] [PubMed] [Google Scholar]
- 19.Dunbar, P.R., J.L. Chen, D. Chao, N. Rust, H. Teisserenc, G.S. Ogg, P. Romero, P. Weynants, and V. Cerundolo. 1999. Cutting edge: rapid cloning of tumor-specific CTL suitable for adoptive immunotherapy of melanoma. J. Immunol. 162:6959–6962. [PubMed] [Google Scholar]
- 20.Rubio, V., T.B. Stuge, N. Singh, M.R. Betts, J.S. Weber, M. Roederer, and P.P. Lee. 2003. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat. Med. 9:1377–1382. [DOI] [PubMed] [Google Scholar]
- 21.Stanislawski, T., R.H. Voss, C. Lotz, E. Sadovnikova, R.A. Willemsen, J. Kuball, T. Ruppert, R.L. Bolhuis, C.J. Melief, C. Huber, et al. 2001. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat. Immunol. 2:962–970. [DOI] [PubMed] [Google Scholar]
- 22.Wang, J., O.W. Press, C.G. Lindgren, P. Greenberg, S. Riddell, X. Qian, C. Laugen, A. Raubitschek, S.J. Forman, and M.C. Jensen. 2004. Cellular immunotherapy for follicular lymphoma using genetically modified CD20-specific CD8+ cytotoxic T lymphocytes. Mol. Ther. 9:577–586. [DOI] [PubMed] [Google Scholar]
- 23.Lanzavecchia, A., and F. Sallusto. 2002. Progressive differentiation and selection of the fittest in the immune response. Nat. Rev. Immunol. 2:982–987. [DOI] [PubMed] [Google Scholar]
- 24.Kaech, S.M., J.T. Tan, E.J. Wherry, B.T. Konieczny, C.D. Surh, and R. Ahmed. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198. [DOI] [PubMed] [Google Scholar]
- 25.Powell, D.J., Jr., M.E. Dudley, P.F. Robbins, and S.A. Rosenberg. 2004. Transition of late stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochsenbein, A.F., S.R. Riddell, M. Brown, G.M. Baerlocher, P.M. Lansdor, and P.D. Greenberg. 2004. CD27 expression promotes long-term survival of functional effector/memory CD8+ CTL in HIV-infected patients. J. Exp. Med. 200:1395-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dudley, M.E., and S.A. Rosenberg. 2003. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer. 3:666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groh, V., J. Wu, C. Yee, and T. Spies. 2002. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 419:734–738. [DOI] [PubMed] [Google Scholar]
- 29.Khong, H.T., and N.P. Restifo. 2002. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 3:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blattman, J.N., and P.D. Greenberg. 2004. Cancer immunotherapy: a treatment for the masses. Science. 305:200–205. [DOI] [PubMed] [Google Scholar]