

Abstract
Natural killer (NK) cells are thought to provide the first line of defence against tumors, particularly major histocompatibility complex (MHC) class I− variants. We have confirmed in C57BL/6 (B6) mice lacking perforin that peritoneal growth of MHC class I− RMA-S tumor cells in unprimed mice is controlled by perforin-dependent cytotoxicity mediated by CD3− NK1.1+ cells. Furthermore, we demonstrate that B6 mice lacking tumor necrosis factor (TNF) are also significantly defective in their rejection of RMA-S, despite the fact that RMA-S is insensitive to TNF in vitro and that spleen NK cells from B6 and TNF-deficient mice are equally lytic towards RMA-S. NK cell recruitment into the peritoneum was abrogated in TNF-deficient mice challenged with RMA-S or RM-1, a B6 MHC class I− prostate carcinoma, compared with B6 or perforin-deficient mice. The reduced NK cell migration to the peritoneum of TNF-deficient mice correlated with the defective NK cell response to tumor in these mice. By contrast, a lack of TNF did not affect peptide-specific cytotoxic T lymphocyte–mediated rejection of tumor from the peritoneum of preimmunized mice. Overall, these data show that NK cells delivering perforin are the major effectors of class I− tumor rejection in the peritoneum, and that TNF is specifically critical for their recruitment to the peritoneum.
Keywords: tumor, natural killer cell, recruitment, tumor necrosis factor, perforin
atural killer (NK) cells are large granular lymphocytes, distinguishable from T or B lymphocytes by their surface phenotype, cytokine profile, and the ability to mediate spontaneous cytotoxicity against a broad range of targets, including MHC class I− tumor cells (1). NK cells mainly circulate in the blood, where they account for ∼5–15% of circulating lymphocytes (2); however, in certain pathologic conditions, including viral and bacterial infections, NK cells selectively accumulate into the infected tissue sites (3–5). Several studies have shown that NK cells express many known adhesion molecules that bind to ligands present on both resting and inflamed endothelium (6–8). Furthermore, NK cells express many chemokine receptors and are responsive to several chemokines, including macrophage inflammatory protein (MIP)-1α, IFN-inducible protein (IP)-10, and monocyte chemoattractant protein (MCP) family members (9). Several cytokines have been shown to modulate these adhesive as well as chemotactic functions, including IL-2, IL-4, IL-8, IFN-γ, IL-12, and TNF (10–15). Activated NK cells produce a spectrum of cytokines and can also respond rapidly to exogenous signals such as those delivered by tumor cells, by upregulation of cytokine production and by increased migration to tissue sites (16). This migration has been demonstrated to be dependent on the activation of NK cells; however, the mechanisms underlying selective recruitment of NK cells from the blood vessels to the tissues have not been elucidated.
The ability to spontaneously lyse tumor cells is the best known functional attribute of NK cells, and particular tumor escape variants that have lost MHC class I expression are efficiently controlled in vivo by NK cells (17). This model of NK cell function has been used in perforin-deficient (P0)1 mice to demonstrate that NK cell–mediated lysis of class I− RMA-S tumor cells in mice is perforin dependent (18). Indeed, perforin gene knockout mice have allowed assessment of the relative contribution of the granule exocytosis pathway in a variety of immune responses in vitro and in vivo (19–22). Activated NK cells also produce the potentially cytolytic molecules, TNF and Fas ligand (FasL [1, 23]). Herein, we use TNF-deficient (TNF0) and a variety of other gene-targeted or mutant mice to define a critical role for TNF in the specific migration of NK cells mediating MHC class I− tumor rejection.
Materials and Methods
Mice.
Inbred C57BL/6 mice were purchased from The Walter and Eliza Hall Institute of Medical Research (Melbourne, Australia). C57BL/6 perforin-deficient (B6.P0) mice (19; from Dr. Guna Karupiah, John Curtin School of Medical Research, Canberra, Australia) and C57BL/6 IL-12p40–deficient (B6.IL-12p400) mice (24; Hoffman-La Roche, Nutley, NJ) were bred at the Austin Research Institute Biological Research Laboratories (ARI-BRL). C57BL/6 gld (FasL mutant; breeding colonies obtained from The Jackson Laboratory, Bar Harbor, ME) and C57BL/6 TNF-deficient (B6.TNF0) mice (25, 26) were obtained from the Centenary Institute of Cancer Medicine and Cell Biology. Male mice of 5–6 wk of age were used in all experiments, which were performed according to animal experimental ethics committee guidelines.
Cell Culture and Reagents.
The mouse WEHI 164 fibrosarcoma (H-2d), RM-1 prostate carcinoma (H-2b; reference 27), YAC-1 (H-2a) lymphoma, RMA (H-2b) lymphoma, RMA-S (H-2b) mutant lymphoma (derived from the Raucher virus- induced murine cell line RBL-5 and defective for peptide loading of MHC class I molecules [28]), and an E7-transfected clone of RMA, RMA-E7 (29), were grown in RPMI medium supplemented with 10% (vol/vol) FCS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO BRL, Gaithersburg, MD). Soluble recombinant human FasL (srFasL; produced in COS cell supernatants and used neat), and mouse Fas–Fc and human TNF p80 receptor (TNFR)–Fc fusion proteins were a gift from Dr. David Lynch (Immunex Corp., Seattle, WA). Recombinant human IL-2 was a gift from Chiron Corp. (Emeryville, CA). Spleen cells were harvested from B6, B6.P0, and B6.TNF0 mice, and IL-2–activated adherent NK cells were cultured and prepared for adoptive transfer as described previously (30). Recombinant mouse TNF-α was a gift from the Biological Response Modifier's Program Repository (Frederick Cancer Research and Development Center, Frederick, MD). Polyinosinic-polycytidylic acid (poly-IC) was purchased from Sigma Chemical Co. (St. Louis, MO). Peptide of human papilloma virus protein 16 (HPV-16) protein E749–57 (RAHYNIVTF) was synthesized (>98% pure) on an automated peptide synthesizer (model 430A; Applied Biosystems, Inc., Foster City, CA). In some experiments, to deplete T lymphocytes and NK cells in vivo, mice were treated with mAb (200 μg) anti-CD3 (KT3, rat IgG2a), anti-CD4 (H129.19, rat IgG2a), anti-CD8 (1803, rat IgG2a), or anti-NK1.1 (PK136, mouse IgG2a; all from the American Type Culture Collection, Rockville, MD) on days −4, −2, 0 (day of tumor inoculation), and weekly thereafter. Depletions were monitored by the analysis of spleens of treated mice by immunofluorescence using FITC-labeled mAbs anti-CD3 (29B, rat IgG2b; Sigma Chemical Co.); anti-CD4 (H129.19; Sigma Chemical Co.); anti-CD8 (53-6.7, rat IgG2a; Sigma Chemical Co.); and anti-NK1.1 (PK136).
51Cr-release Assays.
The cytotoxicity of NK cells, responder CTL, perforin/granzyme B, soluble FasL, or mouse TNF were assessed by 51Cr-release assays against labeled targets (RMA-S, YAC-1, RMA, WEHI 164, or RMA-E7). Spontaneous release of 51Cr was determined by incubating the target cells with medium alone, and maximum release was determined by adding SDS to a final concentration of 5%. The percent specific lysis was calculated as follows: 100 × ([experimental release − spontaneous release]/[maximum release − spontaneous release]). Each experiment was performed twice using triplicate samples.
Peptide Immunization and Induction of B6, B6.TNF0, and B6.P0 Anti-E749–57 CTL.
100 μg (100 μl of 1 mg/ml peptide solution) peptide was extensively mixed with 100 μl IFA and 0.5% BSA. The 200 μl mixture was injected subcutaneously in B6, B6.TNF0, or B6.P0 mice and the procedure repeated after 2 wk. 1 wk after the second immunization, mice were either challenged with 105 RMA-E7 tumor cells intraperitoneally or killed. The spleen cells from killed mice were cultured overnight on plastic flasks to remove adherent cells. Responder cells were then treated with anti-CD4 (H129.19, rat IgG2a; Sigma Chemical Co.) and C′ (1:30 dilution of normal rabbit serum) as described previously (30). Stimulator RMA-S cells were cultured at 25°C for 24 h and incubated with 100 μM E749–57 peptide for 2–4 h at 33°C. Stimulator cells (106) were extensively washed, irradiated (20,000 rad), and then cultured (25-cm2 culture flasks) with responder cells (2 × 107) in RPMI supplemented with 10% FCS and 5 μM peptide. After 5 d, the CTL activity of responder cells was determined in a 4-h 51Cr-release assay using labeled RMA-E7 target cells.
Tumor Control In Vivo.
Groups of five untreated (B6, B6.gld, B6.TNF0, or B6.P0), mAb-depleted (B6), or peptide-immunized (B6, B6.TNF0, or B6.P0) mice were injected intraperitoneally with RMA-S, RMA, or RMA-E7 tumor cells in 0.2 ml PBS as indicated. Mice were observed daily for tumor growth for 70–100 d by monitoring body weight and development of ascites in mice. Mice were killed when first distressed by tumor growth.
Flow Cytometry.
For multiparameter analysis of lymphocytes, cells were stained with anti-CD3–biotin (clone 145-2C11; PharMingen, San Diego, CA), anti-NK1.1–PE, anti-CD4–FITC (clone RM4-5; PharMingen), or anti-CD8α APC (clone 53-6.7; PharMingen). Biotinylated mAbs were detected with streptavidin-conjugated Texas red (SA-TXR; Molecular Probes, Inc., Eugene, OR). Analysis was performed on a FACStar PLUS (Becton Dickinson, San Jose, CA).
NK Cell Migration to the Peritoneum.
The number of NK cells migrating to the peritoneum was evaluated in groups of five B6, B6.P0, or B6.TNF0 mice that had received PBS (0.2 ml) or RMA-S tumor cells (102–104)/0.2 ml i.p. After 24–144 h (72 h optimal for NK cell migration), mice were killed by CO2 asphyxiation, and their peritoneal cavity flushed with 0.5 ml RPMI 1640 and 10% FCS and aspirated with a syringe. Cytospins were performed to make sure no blood leakage into the peritoneal cavity had occurred; if so, these mice were discarded. Analysis of peritoneal NK1.1+ cell content was not practical by flow cytometry due to relatively low cell numbers; thus, single color immunofluorescence was performed upon cells harvested from the peritoneum by fluorescence microscopy. Peritoneal contents were analyzed for NK1.1+ cell numbers by detecting the percentage of NK cells with FITC-labeled anti-NK1.1 (PK136; relative to a negative control) and converting this value into an absolute number of NK1.1+ cells on the basis of a simultaneously obtained cell count. Results are expressed as the mean ± SE of five mice. The NK activity and number of cells collected in the peritoneum were measured against YAC-1 after overnight culture in IL-2 (1,000 U/ml) and the results recorded as LU/107 cells, where one LU is the number of effector cells required to lyse 25% of the target cells.
Results
Class I− Tumor Control in the Peritoneum Is Perforin and TNF Dependent.
To investigate the effector mechanisms used to reject class I− tumors in vivo, we injected RMA-S cells intraperitoneally into B6, B6.P0, B6.TNF0, and B6.gld mice (Fig. 1 A). B6 mice that received ≤105 cells died within 18–33 d, whereas 104 cells were controlled to some extent (death between 35 and 50 d after inoculation). An inoculum of ≤103 injected cells was controlled beyond 100 d in wild-type B6 mice. By contrast, intraperitoneal injection of 102 RMA-S tumor cells resulted in uncontrolled tumor growth in B6.P0 mice (all dead within 22–41 d), and even as few as 10 RMA-S tumor cells caused the death of 2/5 B6.P0 mice. B6.P0 mice that received greater numbers of RMA-S cells (>102) all died within 31 d. In accordance with van den Broek et al. (18), these data confirmed that MHC class I− RMA-S tumor growth is controlled by perforin-dependent cytotoxicity. NK cells have also been demonstrated to display cytotoxicity via two members of the TNF superfamily, FasL and TNF (1, 23), and thus the contribution of these molecules to class I− tumor rejection was examined. FasL mutant B6.gld mice rejected RMA-S cells in a similar fashion to B6 mice, and there was no apparent role for NK cell FasL in this process (Fig. 1 A). The data were in agreement with the finding that NK cell cytotoxicity against many tumor target cells appears to be predominantly perforin mediated (30), but contrast a possible role for CTL FasL in tumor control in vivo (32). Clearly however, B6.TNF0 mice were unable to control the peritoneal growth of RMA-S tumor cells, even at inoculations as low as 102 cells (death within 38–50 d). The defective control of RMA-S cells in B6.TNF0 mice was not as dramatic as in B6.P0 mice, but was nonetheless significantly compromised compared with rejection in B6 and B6.gld mice. It should be noted that all of these strains of B6 mice cleared class I+ RMA tumor cells to a similar degree (≤103 cells; data not shown).
Figure 1.


Elimination of intraperitoneally administered MHC class I− syngeneic tumors (RMA-S) in vivo: dependence on perforin and TNF. (A) B6, B6.P0, B6.TNF0, and B6.gld mice (n = 5/group) were injected intraperitoneally with live tumor cells (10–106) in 0.2 ml PBS as indicated. Mice were observed daily for tumor growth for up to 100 d by monitoring body weight and development of ascites in mice. Individual mice are represented by each symbol. (B) B6 mice were inoculated with RMA-S as above, except that B6 mice were treated on days −4, −2, the day of tumor inoculation, and weekly thereafter with anti-CD3, anti-CD4, anti-CD8, or anti-NK1.1 mAb. Complete depletion of lymphocyte subsets was observed throughout the course of the experiment, and anti-NK1.1–treated mice were shown to lack any NK cytotoxicity against YAC-1 target cells.
NK1.1+ NK Cells Control Class I− Tumor Growth In Vivo.
To exclude the possibility that growth of RMA-S was controlled by T cells expressing perforin and recognizing residual empty MHC class I in an allotype-like reaction, B6 mice were depleted of CD3+, CD4+, CD8+, or NK1.1+ cells before and after tumor inoculation (Fig. 1 B). Depletions were complete (≤1% remaining) as monitored by fluorescence microscopy and splenic NK activity (data not shown). Mice depleted of NK1.1+ cells were unable to control RMA-S tumor growth, with all mice receiving 102 cells surviving only 21–47 d. The remarkably similar profile of B6.P0 mice (Fig. 1 A) and B6 mice treated with anti-NK1.1 (Fig. 1 B) suggested that T cells expressing perforin were not responsible for RMA-S tumor rejection. This conclusion was supported by the lack of effect of depleting CD3+, CD4+, or CD8+ T cells on RMA-S tumor rejection (Fig. 1 B). It should also be noted that B6.P0 mice were just as ineffective in controlling RMA-S tumor growth as B6.P0 mice treated with anti-NK1.1 mAb (data not shown). It is unlikely that IL-12–responsive NK1.1+ T cells expressing perforin (33, 34) were responsible for the rejection of RMA-S cells, since RMA-S is CD1−, anti-CD3 mAb depletion of T cells was without effect, and B6.IL-12p400 mice rejected RMA-S in a similar manner to wild-type B6 mice (data not shown). The use of B6.CD1-deficient mice lacking NK1.1+ T cells (35) will provide confirmatory evidence of the role of this cytotoxic subset in RMA-S tumor rejection.
RMA-S Cells Are Not Sensitive to TNF.
A simple explanation for defective class I− tumor control in B6.P0 and B6.TNF0 mice might be that in wild-type mice, RMA-S tumor cells are directly lysed by perforin and/or TNF derived from NK cells. Indeed, RMA-S cells were sensitive to a sublytic concentration of perforin combined with increasing concentrations of granzyme B (Fig. 2 A), and NK cells from the spleens of B6.P0 mice were not cytolytic to RMA-S tumor cells (Fig. 2 D). Although soluble TNF is not a good effector of TNF cytotoxicity compared with membrane-bound TNF (36), RMA-S cells were not sensitive to soluble TNF compared with WEHI 164 cells (Fig. 2 B). Furthermore, NK cells from B6.TNF0 mice were as cytolytic towards RMA-S as NK cells from B6 mice, whereas NK cells from B6.P0 (that presumably still express membrane TNF) were not cytolytic in 4-h (Fig. 2 D) or longer assays (data not shown). B6 NK cell lysis of RMA-S was not inhibited by a TNFR–Fc fusion protein that significantly inhibited TNF-mediated lysis of WEHI 164 cells (data not shown). RMA-S tumor cells were sensitive to FasL (Fig. 2 C), but NK cells expressing nonfunctional mutant gld-FasL still effectively lysed RMA-S cells (Fig. 2 D). The data further supported a role for NK cell perforin in direct lysis of RMA-S in vivo, but suggested that direct lysis by NK cell TNF was not critical. Furthermore, the relative insensitivity of RMA-S to TNF suggested that direct lysis of RMA-S in vivo by other leukocyte subsets expressing TNF, including T cells and activated macrophages, was unlikely. Together, the in vitro and in vivo data suggested that the role of TNF in NK cell–mediated tumor rejection may not be related to direct cytotoxicity.
Figure 2.
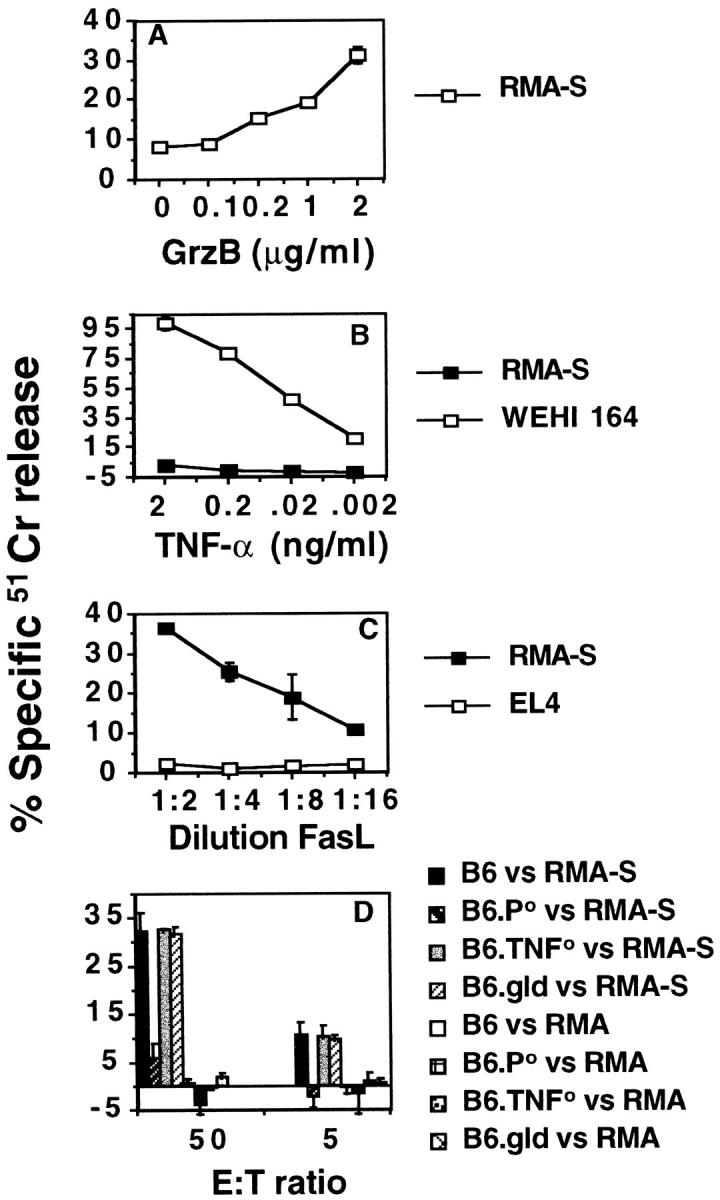
Sensitivity of RMA-S tumor cells to granule- and FasL- mediated cytotoxicity. The cytotoxicity of various cytotoxic molecules or NK cells was assessed by 51Cr-release assays against labeled targets as indicated. (A) Perforin (30 U) and increasing concentrations of granzyme B (GrzB, 0–2 μg/ml) versus RMA-S, (B) murine TNF (0.002–2 ng/ml) versus RMA-S or TNF-sensitive WEHI 164, (C) soluble FasL versus RMA-S or FasL-insensitive EL-4, and (D) NK cells from B6, B6.P0, B6.TNF0, or B6.gld mice versus MHC class I− RMA-S or MHC class I+ RMA. The spontaneous release of 51Cr was always <15%, and each experiment was performed twice using triplicate samples.
Defective Migration of NK Cells in Response to Tumor in the Peritoneum of B6.TNF0 Mice.
Given that TNF has previously been reported to be chemokinetic for NK cells in vitro (37), a possible mode of action of TNF could be to recruit NK cells to the peritoneum upon tumor inoculation. Since members of the TNF family of cytokines influence lymphoid architecture (26, 38, 39), it was important to initially evaluate B6 and B6.TNF0 mice for NK1.1+ cell number and distribution. Flow cytometric analysis of NK1.1+ NK cells and NK1.1+ T cells in the spleen, thymus, lymph nodes, blood, and bone marrow of unchallenged B6 versus B6.TNF0 mice revealed no statistical differences (data not shown). Migration of NK1.1+ cells to the peritoneum was observed in B6 mice that had received RMA-S tumor cells (Table 1). Inoculation with RMA-S cells increased NK1.1+ cell numbers by almost 10-fold after 72 h. By contrast, NK cell recruitment to the peritoneum was significantly reduced and delayed in B6.TNF0 mice receiving RMA-S tumor cells. Despite the reduction in NK1.1+ cell migration, RMA-S tumor inoculation stimulated similar total leukocyte migration in B6.TNF0 and B6 mice (Table 1). The NK activity recovered, as measured by LU25, correlated with the number of NK1.1+ cells in the peritoneum, thus indicating that the NK cells migrating to the peritoneum by 72 h displayed NK cell cytotoxicity (Table 1). Irrespective of the RMA-S cell number inoculated (102–104), migration of NK1.1+ cells to the peritoneum was observed in both B6 and B6.P0 mice, but was abrogated in B6.TNF0 mice (Table 2). NK cell cytotoxicity recovered from B6.TNF0 mice was minimal but measurable compared with undetectable levels of NK cytotoxicity in the peritoneum of B6.P0 mice challenged with RMA-S cells (Table 3). NK cell migration in response to RMA-S tumor inoculation appeared normal in B6.P0, B6.IL-12p400, and B6.gld mice (Table 3).
Table 1.
Kinetics of NK Cell Migration in Response to RMA-S Tumor Cells
| Strain | Treatment | Time | % NK1.1+ | No. NK1.1+≳ | Cell no.‡ | LU25 § | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| h | ||||||||||||
| B6 | ||||||||||||
| PBS | 24 | 1.1 ± 0.1 | 3.1 ± 0.3 | 2.8 ± 0.4 | 1.8 | |||||||
| RMA-S 103 | 24 | 3.8 ± 0.4 | 12.4 ± 1.3 | 3.3 ± 0.2 | 7.1 | |||||||
| PBS | 72 | 1.1 ± 0.2 | 3.5 ± 0.6 | 3.2 ± 0.3 | 1.9 | |||||||
| RMA-S 103 | 72 | 3.3 ± 0.3 | 25.4 ± 2.3 | 7.7 ± 0.7 | 22.3 | |||||||
| PBS | 144 | 1.2 ± 0.1 | 3.4 ± 0.3 | 2.9 ± 0.1 | 1.5 | |||||||
| RMA-S 103 | 144 | 2.2 ± 0.3 | 14.2 ± 1.9 | 6.4 ± 0.4 | 17.6 | |||||||
| B6.TNF0 | ||||||||||||
| RMA-S 103 | 24 | 0.9 ± 0.3 | 3.1 ± 1.0 | 3.4 ± 0.1 | 1.7 | |||||||
| PBS | 72 | 0.9 ± 0.1 | 3.2 ± 0.4 | 3.6 ± 0.2 | 1.6 | |||||||
| RMA-S 103 | 72 | 0.6 + 0.3 | 4.2 + 2.1 | 7.1 + 0.5 | 1.3 | |||||||
| RMA-S 103 | 144 | 1.3 ± 0.2 | 8.5 ± 1.3 | 6.5 ± 0.5 | 3.5 |
Number of NK1.1+ cells (× 10−4) in the peritoneum ± SE, calculated as described in Materials and Methods from five mice per group.
Total cell number (× 10−6) in the peritoneum.
NK activity in lytic units (25%) against YAC-1 tumor target cells. Peritoneal cells were cultured overnight in IL-2 (1,000 U/ml).
Table 2.
NK Cell Migration to Increasing Numbers of RMA-S Tumor Cells
| Strain | Treatment | % NK1.1+≳ | No. NK1.1+ | Cell no. | LU25 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| B6 | ||||||||||
| PBS | 1.1 ± 0.2 | 3.5 ± 0.6 | 3.2 ± 0.3 | 1.9 | ||||||
| RMA-S 102 | 2.9 ± 0.3 | 20.8 ± 2.2 | 7.2 ± 0.6 | 12.3 | ||||||
| RMA-S 103 | 3.3 ± 0.3 | 25.4 ± 2.3 | 7.7 ± 0.7 | 22.3 | ||||||
| RMA-S 104 | 3.5 ± 0.3 | 28.9 ± 2.5 | 8.2 ± 0.2 | 20.7 | ||||||
| B6.TNF0 | ||||||||||
| PBS | 0.9 ± 0.1 | 3.2 ± 0.4 | 3.6 ± 0.2 | 1.6 | ||||||
| RMA-S 102 | 0.6 ± 0.2 | 3.9 ± 1.3 | 6.4 ± 0.2 | 1.8 | ||||||
| RMA-S 103 | 0.6 ± 0.3 | 4.2 ± 2.1 | 7.1 ± 0.5 | 1.3 | ||||||
| RMA-S 104 | 0.8 ± 0.2 | 6.3 ± 1.6 | 7.9 ± 0.1 | 1.7 |
Number of NK1.1+ cells (× 10−4) in the peritoneum ± SE after 72 h, calculated as described in Materials and Methods from five mice per group.
Table 3.
NK Cell Migration in Various Gene-targeted/Mutant Mice
| Strain | Treatment | % NK1.1+≳ | No. NK1.1+ | Cell no. | LU25 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| B6 | ||||||||||
| PBS | 1.1 ± 0.2 | 3.5 ± 0.6 | 3.2 ± 0.3 | 1.9 | ||||||
| RMA-S 103 | 3.3 ± 0.3 | 25.4 ± 2.3 | 7.7 ± 0.7 | 22.3 | ||||||
| B6.TNF0 | ||||||||||
| PBS | 0.9 ± 0.1 | 3.2 ± 0.4 | 3.6 ± 0.2 | 1.6 | ||||||
| RMA-S 103 | 0.6 ± 0.3 | 4.2 ± 2.1 | 7.1 ± 0.5 | 1.3 | ||||||
| B6.P0 | ||||||||||
| PBS | 0.9 ± 0.1 | 3.1 ± 0.3 | 3.4 ± 0.3 | bd‡ | ||||||
| RMA-S 103 | 3.6 ± 0.3 | 26.6 ± 2.2 | 7.4 ± 0.3 | bd | ||||||
| B6.gld | ||||||||||
| PBS | 0.9 ± 0.2 | 2.7 ± 0.3 | 3.0 ± 0.3 | 1.4 | ||||||
| RMA-S 103 | 3.2 ± 0.4 | 24.5 ± 0.8 | 7.7 ± 0.2 | 16.5 | ||||||
| B6.IL-12p400 | ||||||||||
| PBS | 1.0 ± 0.1 | 3.2 ± 0.3 | 3.2 ± 0.3 | 1.7 | ||||||
| RMA-S 103 | 3.1 ± 0.2 | 22.5 ± 0.8 | 7.3 ± 0.3 | 24.2 |
Number of NK1.1+ cells (× 10−4) in the peritoneum ± SE after 72 h, calculated as described in Materials and Methods from five mice per group.
Below detection (bd), LU25 could not be calculated.
It was important to establish that the role of TNF in tumor rejection and NK cell recruitment was not specific for RMA-S tumor target cells. We have previously demonstrated that the B6 MHC class I− prostate carcinoma RM-1 was NK cell sensitive and when injected into the peritoneum was cleared by perforin expressing CD3−NK1.1+ cells in a TNF-dependent manner (data not shown). Migration of NK1.1+ cells to the peritoneum was also observed in B6 mice in response to class I− RM-1 prostate carcinoma cells (Table 4). Inoculation with RM-1 cells increased NK1.1+ cell numbers by approximately sixfold after 72 h. RM-1 tumor inoculation again stimulated similar total leukocyte migration in B6.TNF 0 and B6 mice (Table 4), however NK cell recruitment to the peritoneum was significantly reduced in B6.TNF0 mice receiving RM-1 tumor cells. The data indicated that endogenous TNF was a major contributor to NK cell accumulation in the peritoneum specifically in response to syngeneic class I− tumor cells.
Table 4.
NK Cell Migration in B6.TNF0 Mice to Other Stimuli
| Strain | Treatment | % NK1.1+≳ | No. NK1.1+ | Cell no. | LU25 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| B6 | ||||||||||
| PBS | 1.0 ± 0.1 | 2.6 ± 0.2 | 2.6 ± 0.2 | 1.8 | ||||||
| RM-1 103 | 2.6 ± 0.2 | 16.2 ± 0.8 | 6.2 ± 0.3 | 17.7 | ||||||
| Poly-IC | 3.8 ± 0.2 | 42.3 ± 1.3 | 11.1 ± 0.4 | 37.8 | ||||||
| B6.TNF0 | ||||||||||
| PBS | 1.0 ± 0.1 | 2.6 ± 0.1 | 2.6 ± 0.1 | 1.7 | ||||||
| RM-1 103 | 1.0 ± 0.3 | 5.7 ± 0.2 | 5.7 ± 0.2 | 3.5 | ||||||
| Poly-IC | 3.1 ± 0.3 | 29.2 ± 1.8 | 9.4 ± 0.6 | 17.8 |
Number of NK1.1+ cells (× 10−4) in the peritoneum ± SE after 72 h, calculated as described in Materials and Methods from five mice per group.
Consistent with earlier reports (37), cell recruitment, in particular NK1.1+ cell migration, was greatly enhanced in B6 mice inoculated with poly-IC, a powerful stimulator of NK cell migration and cytotoxicity (Table 4). Peritoneal inoculation with poly-IC increased total leukocyte migration in B6.TNF0 and B6 mice by 3–5-fold and NK1.1+ cell numbers in B6 mice by >15-fold (Table 4). In contrast to tumor inoculation, poly-IC did stimulate significant NK1.1+ cell numbers in the peritoneum of B6.TNF0 mice (∼11-fold above controls); however, numbers of NK1.1+ cells were still reduced compared with B6 mice inoculated with poly-IC. These data further served to illustrate that TNF is not critical for all NK cell recruitment, even into the peritoneum.
CTL-mediated Tumor Rejection in the Peritoneum Is Normal in B6.TNF0 Mice.
The striking need for TNF in effective NK cell–mediated rejection of class I− RMA-S cells prompted the question whether TNF was also necessary for CTL rejection of tumor localized in the peritoneum. To minimize tumor variation, RMA cells expressing the HPV-16 E7 protein were used as the tumor challenge to mice preimmunized with the E749–57 H-2Db–presented 9-mer peptide. Importantly, mouse CD8+ CTL reactive with E749–57 were generated from the spleen cells of preimmunized B6, B6.P0, and B6.TNF0 mice (Fig. 3 A). Thus, TNF was not absolutely necessary for the generation of peptide-specific CTL. CD8+ CTL generated from B6 and B6.TNF0 mice displayed peptide-specific perforin- and FasL-mediated lysis of RMA-E7 (as determined by inhibition with Mg-EGTA or Fas–Fc, respectively), whereas CD8+ CTL from B6.P0 mice lysed RMA-E7 via FasL exclusively (data not shown). Preimmunized B6 and B6.TNF0 mice effectively cleared intraperitoneal challenge with 105 RMA-E7 cells, whereas B6.P0 mice did not and died within 20–30 d (Fig. 3 B). Unprimed B6, B6.TNF0, and B6.P0 mice challenged with RMA-E7 all died within 17– 28 d. Thus, the data indicated that RMA-E7 tumor rejection in the peritoneum was mediated by CTL expressing perforin and showed that a lack of host TNF did not reduce CTL effector function at this site in vivo.
Figure 3.


CTL-mediated peritoneal tumor rejection is normal in B6.TNF0 mice. B6, B6.P0, and B6.TNF0 mice were immunized twice (2 wk apart) with E7 peptide and their spleen cells harvested 1 wk later and stimulated in vitro (as described in Materials and Methods). 5 d after stimulation, the CTL activity of responder cells was determined in a 4-h 51Cr-release assay (at E/T ratios indicated) using labeled RMA-E7 target cells, RMA-S pulsed with E749–57 peptide, or RMA-S pulsed with OVA257–264. The spontaneous release of 51Cr was <15%, and the experiment was performed using triplicate samples. (B) Untreated B6, B6.P0, and B6.TNF0 mice (n = 5/group) or those immunized as above were challenged intraperitoneally 1 wk after the second immunization with 105 RMA-E7 tumor cells in 0.2 ml PBS. Mice were observed daily for tumor growth for up to 70 d by monitoring body weight and development of ascites in mice. Individual mice are represented by each symbol.
Discussion
NK cells have been shown to accumulate locally and in various organs after animals are exposed to viruses (4, 40) or biological response modifiers (37, 41, 42). Where evaluated, it has become apparent that TNF may (37) or may not (40, 42) be critical for NK cell migration in response to these stimuli; therefore, NK cell emigration does not appear to be generally affected by TNF. However, the role of TNF in NK cell recruitment to and rejection of tumor has been comparatively poorly understood. We have used two different MHC class I− tumors and TNF0 mice to illustrate an essential role for TNF in NK cell antitumor activity in the peritoneum. Given a previously demonstrated role for TNF in leukocyte trafficking (25, 26, 38, 39, 43) and our observation that B6.TNF0 mice displayed reduced numbers of NK1.1+ cells in the peritoneum in response to tumor inoculation, we believe TNF is specifically vital for the migration of NK cells into the peritoneum.
Several other possibilities may explain a critical role for TNF in NK cell–mediated tumor rejection, including (a) a direct effect of NK cell TNF secretion on tumor growth; (b) a direct enhancement of the NK cell activity of resident NK cells by TNF; (c) local proliferation or enhanced survival of peritoneal NK cells in response to TNF; or (d) decreased adherence of NK cells to endothelial cells in the presence of TNF. However, most experimental evidence that we and others have generated would undermine these possibilities. A direct cytotoxic effect of NK cell, or host, TNF on RMA-S tumor growth is unlikely, given that this tumor appears insensitive to TNF. NK cells from TNF0 mice have normal levels of NK activity in the spleen and thus a general defect in the activation of NK cell cytotoxic potential in mice lacking TNF was not apparent. In addition, when spleen NK cells from B6.TNF0 mice were expanded by culture in IL-2, the proliferation of NK1.1+ cells and their lytic activity were comparable with the same cells from wild-type B6 mice (data not shown). Similarly, exogenous TNF did not support the survival of B6 or B6.TNF0 peritoneal cells taken 24–72 h after tumor inoculation (data not shown). Furthermore, when adoptively transferred intraperitoneally into B6.TNF0 mice, cultured B6.TNF0 and B6 NK cells, but not B6.P0 NK cells, were capable of mediating RMA-S tumor rejection (data not shown). Therefore, our data would support a previous study that demonstrated TNF did not enhance NK cell survival (44). Whether exogenous soluble TNF could mimic local signals that control cytokine networks, resulting in NK cell emigration, is debatable; however, B6 or B6.TNF0 mice that received TNF intraperitoneally did not display increased numbers of NK cells or enhanced lytic activity in response to RMA-S (data not shown). It appears doubtful that TNF reduced adhesion and increased the peritoneal lavage recovery of NK1.1+ cells considering that TNF has previously been observed to increase NK cell adherence (37).
There may be several processes whereby TNF regulates NK cell trafficking to tumor in the peritoneum. We have not attempted to determine the original source of NK1.1+ NK cells which accumulate in the peritoneal cavity after tumor inoculation; however, previous studies of NK cell migration in response to viral infection would favor the bone marrow as a source over peripheral lymphoid organs (45, 46). TNF induces expression of the vascular adhesion molecule (VCAM-1) on vascular endothelial cells, and VCAM-1/ very late antigen (VLA)-4 interactions are required for migration of NK cells from the blood into lung and liver after poly-ICLC treatment (41, 47). Nonetheless, binding to vascular endothelium is only the first step in NK cell infiltration, and subsequent events must result in transit across the basement membrane and chemotaxis to the tumor. Recent studies in autoimmune inflammation in the B6.TNF0 central nervous system (CNS) indicated not only that VCAM-1 is upregulated on CNS endothelium normally (25), but that leukocytes extravasate and accumulate in the CNS perivascular region yet fail to move out into the tissue in a normal fashion (25, 43). Specific leukocyte populations infiltrating lesions were not defined in these studies, but the contrasting roles of NK1.1+ cells reportedly controlling the severity of experimental allergic encephalomyelitis (EAE [48]) and TNF increasing the onset of EAE (43) suggest that TNF is not regulating EAE by affecting NK1.1+ cell emigration.
NK cell availability, activity, and chemotaxis have been demonstrated to be regulated by cytokines of the C-C family made in NK cells (49), and macrophages or dendritic cells (15, 51, 52). Thus, it is possible that TNF may serve to regulate macrophages or dendritic cell production of chemokines that in turn recruit NK cells. Although IL-12 does not appear necessary, tumor rejection experiments in various C-C chemokine–deficient mice may be very informative. Pathways for NK cell trafficking are likely to be complex, and the chemokine network elicited will very much depend on the form and site of the challenge. Our data indicated that NK1.1+ cell recruitment in response to tumor was virtually abolished in the absence of TNF, whereas NK cell migration to the peritoneum was still significant in response to poly-IC. Poly-IC elicits many cytokines that directly activate NK cells; thus, TNF may play a relatively minor role in the migration of NK cells into the peritoneum in response to poly-IC. Subcutaneous inoculation of various gene-targeted mice or B6 mice treated with anti-NK1.1 mAb with RMA-S tumor cells revealed that TNF was not necessary for NK1.1+ cells to reject RMA-S in a perforin-dependent manner (data not shown). Thus, in the absence of further data at other tumor sites, it appears that the absolute requirement for TNF in NK cell antitumor activity may be restricted to the peritoneum.
However, NK1.1+ cells with antitumor activity are heterogeneous (18, 33, 34), as are dendritic cells and macrophages in various tissues (52); therefore, future studies will be required to resolve the apparent anomaly that TNF is not universally critical for all sites of tumor rejection. Although all the mediators involved in NK cell responses to class I− tumor are unclear, our work has highlighted a critical role for TNF in NK cell rejection of class I–deficient tumors by virtue of its capacity to regulate cell recruitment. Importantly, the requirement for TNF appears specific for NK cells responding to tumor. This finding now will stimulate further examination of a possible role for TNF in controlling NK cell responses to tumor initiation, growth, and metastasis.
Acknowledgments
We thank Dr. Joe Trapani for purified perforin and granzyme B, Dr. David Lynch for recombinant FasL, Dr. Timothy Thompson for the RM-1 prostate carcinoma cells, and Drs. Ricky Johnstone and Thomas Sayers for helpful discussion. We also thank Dr. Jeanne Magram (Hoffman-La Roche, Nutley, NJ) for providing the IL-12 gene knockout mice.
Abbreviations used in this paper
- B6
C57BL/6
- CNS
central nervous system
- EAE
experimental allergic encephalomyelitis
- FasL
Fas ligand
- LU25
the number of effector cells required to lyse 25% of the target cells
- P0
perforin-deficient
- poly-IC
polyinosinic-polycytidylic acid
- TNF0
TNF-deficient
- VCAM-1
vascular adhesion molecule 1
Footnotes
M.J. Smyth is currently supported by Wellcome Trust Australasian Senior Research Fellowship and by Project Grants from the National Health and Medical Research Council of Australia. J.D. Sedgwick and H. Körner were supported by a Program Grant from the National Health and Medical Research Council of Australia and Project Grants from the Multiple Sclerosis Society of Australia.
Dr. Korner's current address is the Institut für Klinische Mikrobiologie, Immunologie und Hygiene, Wasserturmstrasse 3, D-91054 Erlangen, Germany. Dr. Sedgwick's current address is DNAX Research Institute of Molecular and Cellular Biology, Inc., Palo Alto, CA 94304-1104.
References
- 1.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whiteside TL, Herberman RB. Human natural killer cells in health and disease. Biology and therapeutic potential. Clin Immunother. 1994;1:56–66. doi: 10.1128/cdli.1.2.125-133.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmberg LA, Springer KA, Ault KA. Natural killer cell activity in the peritoneal exudate of mice infected with Listeria monocytogenes. . J Immunol. 1981;127:1792–1799. [PubMed] [Google Scholar]
- 4.McIntyre KW, Welsh RW. Accumulation of natural killer and cytotoxic T large granular lymphocytes in the liver during virus infection. J Exp Med. 1986;164:1667–1681. doi: 10.1084/jem.164.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nemlander A, Saksela E, Hayrj P. Are natural killer cells involved in allograft rejection? . Eur J Immunol. 1983;13:348–350. doi: 10.1002/eji.1830130415. [DOI] [PubMed] [Google Scholar]
- 6.Bender JR, Pardi R, Karasek MA, Engleman EG. Phenotypic and functional characterization of lymphocytes that bind human microvascular endothelial cells in vitro. J Clin Invest. 1987;79:1679–1688. doi: 10.1172/JCI113007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allavena P, Paganin C, Martin-Padura I, Peri G, Gaboli M, Dejana E, Marchisio PC, Mantovani A. Molecules and structures involved in the adhesion of natural killer cells to vascular endothelium. J Exp Med. 1991;173:439–448. doi: 10.1084/jem.173.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gismondi A, Morrone S, Humphries MJ, Piccoli M, Frati L, Santoni A. Human natural killer cells express VLA-4 and VLA-5, which mediate their adhesion to fibronectin. J Immunol. 1991;146:384–392. [PubMed] [Google Scholar]
- 9.Taub DD, Sayers TJ, Carter CRD, Ortaldo JR. α and β chemokines induce NK cell migration and enhance NK-mediated cytolysis. J Immunol. 1995;155:3877–3888. [PubMed] [Google Scholar]
- 10.Maghazachi AA. Tumor necrosis factor-α is chemokinetic for lymphokine-activated killer cells: regulation by cyclic adenosine monophosphate. J Leukocyte Biol. 1991;49:302–308. doi: 10.1002/jlb.49.3.302. [DOI] [PubMed] [Google Scholar]
- 11.Bianchi G, Sironi M, Ghibaudi E, Selvaggini C, Elices M, Allavena P, Mantovani A. Migration of NK cells across endothelial cell monolayers. J Immunol. 1993;151:5135–5144. [PubMed] [Google Scholar]
- 12.Bottazzi B, Introna M, Allavena P, Villa A, Mantovani A. In vitro migration of human large granular lymphocytes. J Immunol. 1985;134:2316–2321. [PubMed] [Google Scholar]
- 13.Pohajdak B, Gomez J, Orr FW, Khalil N, Talgoy M, Greenberg AH. Chemotaxis of large granular lymphocytes. J Immunol. 1986;136:278–284. [PubMed] [Google Scholar]
- 14.Sebok K, Woodside D, al-Aoukaty A, Ho AD, Gluck S, Maghazachi AA. IL-8 induces the locomotion of human IL-2-activated natural killer cells. Involvement of a guanine nucleotide binding (Go) protein. J Immunol. 1993;150:1524–1534. [PubMed] [Google Scholar]
- 15.Allavena P, Paganin C, Zhou D, Bianchi G, Sozzani S, Mantovani A. Interleukin-12 is chemotactic for natural killer cells and stimulates interaction with vascular endothelium. Blood. 1994;84:2261–2268. [PubMed] [Google Scholar]
- 16.Whiteside TL, Herberman RB. Extravasation of antitumor effector cells. Invasion Metastasis. 1992;12:128–146. [PubMed] [Google Scholar]
- 17.Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immunedefence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 18.van den Broek MF, Kagi D, Zinkernagel RM, Hengartner H. Perforin dependence of natural killer cell-mediated tumor control in vivo. . Eur J Immunol. 1995;25:3514–3516. doi: 10.1002/eji.1830251246. [DOI] [PubMed] [Google Scholar]
- 19.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 20.Kagi D, Odermatt B, Ohashi PS, Zinkernagel RM, Hengartner H. Development of insulitis without diabetes in transgenic mice lacking perforin-dependent cytotoxicity. J Exp Med. 1996;183:2143–2152. doi: 10.1084/jem.183.5.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kagi D, Ledermann B, Burki K, Hengartner H, Zinkernagel RM. CD8+ T cell-mediated protection against an intracellular bacterium by perforin. Eur J Immunol. 1994;24:3068–3072. doi: 10.1002/eji.1830241223. [DOI] [PubMed] [Google Scholar]
- 22.Schulz M, Schuurman HJ, Joergensen J, Steiner C, Meerloo T, Kagi D, Hengartner H, Zinkernagel RM, Schreier M, Burki K, Ledermann B. Acute rejection of vascular heart allografts by perforin-deficient mice. Eur J Immunol. 1995;25:474–480. doi: 10.1002/eji.1830250225. [DOI] [PubMed] [Google Scholar]
- 23.Oshimi Y, Oda S, Honda Y, Nagata S, Miyazaki S. Involvement of Fas ligand and Fas-mediated pathway in the cytotoxicity of human natural killer cells. J Immunol. 1996;157:2909–2915. [PubMed] [Google Scholar]
- 24.Magram J, Connaughton SE, Warrier WW, Carvajal DM, Wu CY, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFN-γ production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 25.Korner H, Riminton DS, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD. Critical points of tumor necrosis factor action in central nervous system autoimmune inflammation defined by gene targeting. J Exp Med. 1997;186:1585–1590. doi: 10.1084/jem.186.9.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korner H, Cook M, Riminton DS, Lemckert FA, Hoek B, Ledermann B, Kontgen F, Fazekas de St B, Groth, Sedgwick JD. Distinct roles for lymphotoxin-α and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur J Immunol. 1997;27:2600–2609. doi: 10.1002/eji.1830271020. [DOI] [PubMed] [Google Scholar]
- 27.Baley PA, Yoshida K, Qian W, Sehgal I, Thompson TC. Progression to androgen insensitivity in a novel in vitro mouse model for prostate cancer. J Steroid Biochem Mol Biol. 1995;52:403–413. doi: 10.1016/0960-0760(95)00001-g. [DOI] [PubMed] [Google Scholar]
- 28.Ljunggren HG, Karre K. Host resistance directed selectively against H-2–deficient lymphoma variants. Analysis of the mechanism. J Exp Med. 1985;162:1745–1759. doi: 10.1084/jem.162.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu X, Tommasino M, Vousden K, Sadovnikava E, Rappuoli R, Crawford L, Kast M, Melief CJ, Beverley PC, Stauss HJ. Both immunization with protein and recombinant vaccinia virus can stimulate CTL specific for the E7 protein of human papilloma virus 16 in H-2d mice. Scand J Immunol. 1995;42:557–563. doi: 10.1111/j.1365-3083.1995.tb03696.x. [DOI] [PubMed] [Google Scholar]
- 30.Smyth MJ, Thia KYT, Kershaw MH. Xenogeneic mouse anti-human NK cytotoxicity is mediated via perforin. Xenotransplantation. 1997;4:78–84. [Google Scholar]
- 31.Smyth MJ, Sutton VR, Kershaw MH, Trapani JA. Xenospecific cytotoxic T lymphocytes use perforin- and Fas-mediated lytic pathways. Transplantation (Baltimore) 1996;62:1529–1532. doi: 10.1097/00007890-199611270-00030. [DOI] [PubMed] [Google Scholar]
- 32.van den Broek MF, Kagi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, Melief CJM, Zinkernagel RM, Hengartner H. Decreased tumor surveillance in perforin-deficient mice. J Exp Med. 1996;184:1781–1790. doi: 10.1084/jem.184.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, Kaneko Y, Koseki H, Kanno M, Taniguchi M. Requirement for Vα14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–1626. doi: 10.1126/science.278.5343.1623. [DOI] [PubMed] [Google Scholar]
- 34.Kawamura T, Takeda K, Mendiratta SK, Kawamura H, Van Kaer L, Yagita H, Abo T, Okumura K. Critical role of NK1+T cells in IL-12-induced immune responses in vivo. J Immunol. 1998;160:16–19. [PubMed] [Google Scholar]
- 35.Chen Y-H, Chiu NM, Mandal M, Wang N, Wang C-R. Impaired NK1+T cell development and early IL-4 production in CD1-deficient mice. Immunity. 1997;6:459–467. doi: 10.1016/s1074-7613(00)80289-7. [DOI] [PubMed] [Google Scholar]
- 36.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 37.Pilaro AM, Taub DD, McCormick KL, Williams HM, Sayers TJ, Fogler WE, Wiltrout RH. TNF-α is a principal cytokine involved in recruitment of NK cells to liver parenchyma. J Immunol. 1994;153:333–342. [PubMed] [Google Scholar]
- 38.Pasparikis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF-α– deficient mice: a critical requirement for TNF-α in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumoto M, Fu YX, Molina H, Chaplin DD. Lymphotoxin-α-deficient and TNF receptor-I-deficient mice define developmental and functional characteristics of germinal centers. Immunol Rev. 1997;156:137–144. doi: 10.1111/j.1600-065x.1997.tb00965.x. [DOI] [PubMed] [Google Scholar]
- 40.Salazar-Mather TP, Orange JS, Biron CA. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1α (MIP-1α)- dependent pathways. J Exp Med. 1998;187:1–14. doi: 10.1084/jem.187.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiltrout RH, Mathieson BJ, Talmadge JE, Reynolds CW, Zhang SR, Herberman RB, Ortaldo JR. Augmentation of organ-associated natural killer activity by biological response modifiers. Isolation and characterization of large granular lymphocytes from the liver. J Exp Med. 1984;160:1431–1449. doi: 10.1084/jem.160.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sayers TJ, Mason LL, Wiltrout TA. Trafficking and activation of murine natural killer cells: differing roles for IFN-γ and IL-2. Cell Immunol. 1990;127:311–326. doi: 10.1016/0008-8749(90)90135-e. [DOI] [PubMed] [Google Scholar]
- 43.Riminton DS, Korner H, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD. Challenging cytokine redundancy: inflammatory cell movement and clinical course of experimental autoimmune encephalomyelitis are normal in lymphotoxin-deficient but not TNF-deficient mice. J Exp Med. 1998;187:1517–1528. doi: 10.1084/jem.187.9.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carson WE, Fehniger TA, Haldar S, Eckhert K, Lindemann MJ, Lai CF, Croce CM, Baumann H, Caligiuri MA. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J Clin Invest. 1997;99:937–943. doi: 10.1172/JCI119258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biron CA, Sonnenfeld G, Welsh RM. Interferon induces natural killer cell blastogenesis in vivo. J Leukoc Biol. 1984;35:31–37. doi: 10.1002/jlb.35.1.31. [DOI] [PubMed] [Google Scholar]
- 46.Hackett, J., Jr., M. Tutt, M. Lipscomb, M. Bennett, G. Koo, and V. Kumar. Origin and differentiation of natural killer cells. II. Functional and morphologic studies of purified NK-1.1+ cells. J. Immunol. 136:3124–3131. [PubMed]
- 47.Fogler WE, Volker K, McCormick KL, Watanabe M, Ortaldo JR, Wiltrout RH. NK cell infiltration into lung, liver, and subcutaneous B16 melanoma is mediated by VCAM-1/VLA-4 interaction. J Immunol. 1996;156:4707–4714. [PubMed] [Google Scholar]
- 48.Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med. 1997;186:1677–1687. doi: 10.1084/jem.186.10.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hedrick JA, Saylor V, Figueroa D, Mizoue L, Xu Y, Menon S, Abrams J, Handel T, Zlotnik A. Lymphotactin is produced by NK cells and attracts both NK cells and T cells in vivo. J Immunol. 1997;158:1533–1540. [PubMed] [Google Scholar]
- 50.Allavena P, Bianchi G, Zhou D, van Damme J, Jilek P, Sozzani S, Mantovani A. Induction of natural killer cell migration by monocyte chemotactic protein-1, -2 and -3. Eur J Immunol. 1994;24:3233–3236. doi: 10.1002/eji.1830241249. [DOI] [PubMed] [Google Scholar]
- 51.Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Activation of NK cells by CC chemokines. Chemotaxis, Ca2+mobilization, and enzyme release. J Immunol. 1996;156:322–327. [PubMed] [Google Scholar]
- 52.Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol. 1997;9:10–16. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]