

Abstract
During pathologic vessel remodeling, vascular smooth muscle cells (VSMCs) embedded within the collagen-rich matrix of the artery wall mobilize uncharacterized proteolytic systems to infiltrate the subendothelial space and generate neointimal lesions. Although the VSMC-derived serine proteinases, plasminogen activator and plasminogen, the cysteine proteinases, cathepsins L, S, and K, and the matrix metalloproteinases MMP-2 and MMP-9 have each been linked to pathologic matrix-remodeling states in vitro and in vivo, the role that these or other proteinases play in allowing VSMCs to negotiate the three-dimensional (3-D) cross-linked extracellular matrix of the arterial wall remains undefined. Herein, we demonstrate that VSMCs proteolytically remodel and invade collagenous barriers independently of plasmin, cathepsins L, S, or K, MMP-2, or MMP-9. Instead, we identify the membrane-anchored matrix metalloproteinase, MT1-MMP, as the key pericellular collagenolysin that controls the ability of VSMCs to degrade and infiltrate 3-D barriers of interstitial collagen, including the arterial wall. Furthermore, genetic deletion of the proteinase affords mice with a protected status against neointimal hyperplasia and lumen narrowing in vivo. These studies suggest that therapeutic interventions designed to target MT1-MMP could prove beneficial in a range of human vascular disease states associated with the destructive remodeling of the vessel wall extracellular matrix.
In disease states ranging from atherosclerosis to postangioplasty restenosis, vascular smooth muscle cells (VSMCs) embedded in a dense, three-dimensional (3-D) matrix of interstitial collagens activate a tissue-invasive program that supports migration from the vessel wall media into the subendothelial intimal space (1). Within this compartment, smooth muscle cells proliferate and deposit extracellular matrix molecules, ultimately leading to the formation of neointimal lesions that can occlude the arterial lumen directly or precipitate catastrophic occlusive events by triggering thrombosis (1, 2). Although proteolytic enzymes are assumed to play a critical role in conferring VSMCs with the ability to traverse the type I collagen–rich media of the arterial wall (3–14), the identity of the matrix-degrading proteinases that confer tissue-invasive activity have remained the subject of speculation.
To date, efforts to characterize the matrix remodeling activities of VSMCs have emphasized potential roles for the serine proteinases, plasminogen activator and plasminogen, the cysteine proteinases, cathepsins K, L, and S, or the matrix metalloproteinases (MMPs) MMP-2 and MMP-9 (3–14). However, which, if any, of these proteinases participate directly in the collagen-degradative events necessary to drive VSMC invasion through 3-D matrix barriers is unknown. Herein, we demonstrate that VSMCs mobilize a pericellular proteolytic activity that allows them to degrade and invade collagen-rich tissues in 3-D explants via a process that operates independently of the plasminogen activator–plasminogen axis, cysteine proteinases, MMP-2, or MMP-9. Instead, VSMCs rely on the pericellular collagenase, membrane type I (MT1)-MMP, to infiltrate 3-D barriers of type I collagen, composites of type I and III collagen, or the arterial wall itself. Further, using MT1-MMP+/− heterozygote mice, we demonstrate that even a partial reduction in MT1-MMP expression levels ameliorates the vessel wall damage and remodeling associated with pathologic VSMC invasion in vivo.
RESULTS
In pathologic states in vivo, medial VSMCs gain access to the subintimal space by expressing motile activity within the confines of the 3-D matrix largely comprised of type I collagen (1, 15). Although most in vitro analyses of VSMC function have been performed under two-dimensional (2-D) culture conditions wherein cells are plated atop a matrix substratum (3–14), recent studies indicate that mesenchymal cell phenotype is altered significantly when cells are embedded within, rather than cultured atop, a 3-D extracellular matrix (16, 17). Hence, to recapitulate the collagen-rich, 3-D interstitial matrix confronted by VSMCs exposed to invasion-promoting growth factors in situ, medial explants recovered from WT mice were embedded within a cross-linked matrix of type I collagen (Fig. 1 a). The transition of VSMCs from a quiescent contractile state to a proliferating invasive phenotype was triggered by the addition of autologous serum supplemented with platelet-derived growth factor (PDGF)-BB and fibroblast growth factor 2 (FGF-2; PDGF/FGF-2) (18). After a 3-d lag, VSMCs begin to emigrate from the tissue explant and to infiltrate the surrounding type I collagen matrix (Fig. 1 b). By 8 d, a dense cloud of VSMCs surround the explant, with the leading front of the advancing cells having traversed >1,000 μm of dense collagen (Fig. 1 c). The tissue-invasive VSMCs acquire a spindle cell–like morphology as they negotiate the surrounding 3-D matrix while leaving tunnels of immunodetectable collagen degradation products in their wake (Fig. 1, d and f).
Figure 1.
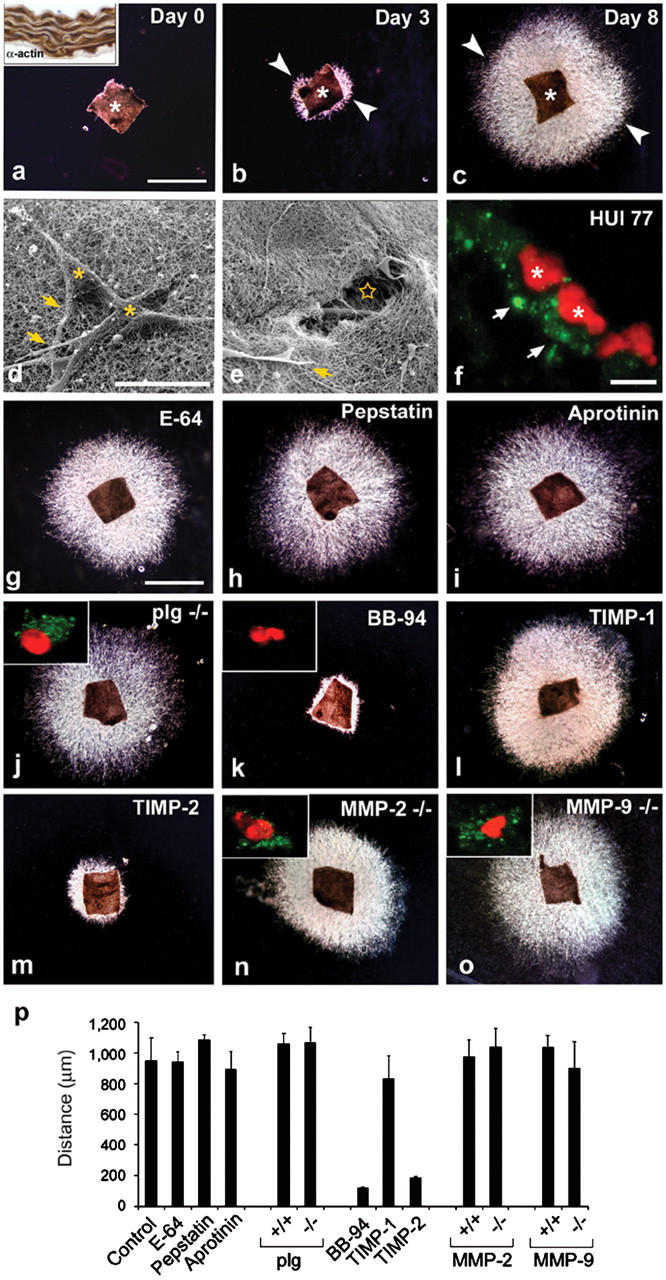
Ex vivo invasive activity of VSMCs in 3-D collagen. (a–c) Media explants of mouse aorta were embedded within a 3-D gel of type I collagen, VSMC egress was initiated by exogenous PDGF/FGF-2, and outgrowth into the translucent collagen matrix was visualized by darkfield microscopy at 0, 3, and 8 d. Asterisk indicates explant , and arrowheads point at the leading edge of the invading SMC. Bar, 1 mm. Inset in (a) shows anti-SM α-actin staining (brown) of the media explant. (d and e) Scanning electron micrograph of freeze-fractured explant cultures. Asterisks mark invading VSMCs that have infiltrated into the field of densely packed type I collagen fibrils. Arrows highlight VSMC cytoplasmic extensions. Tunnels (star) in the collagen matrix are seen in areas surrounding the embedded explant. Bar, 20 μm. (f) Degraded collagen (detected by mAb HUI77) appears as punctate green staining (arrows) surrounding propidium iodide–labeled cells (red, asterisks). Bar, 20 μm. (g–o) Media explants from WT, plasminogen (plg)-, MMP-9–, or MMP-2–null mice were suspended in collagen in autologous sera and cultured for 8 d in the absence or presence of E-64, pepstatin, aprotinin, BB-94, TIMP-1, or TIMP-2 as described in Materials and methods. Insets in j, k, n, and o show immunostains for degraded collagen (mAb HUI77) surrounding propidium iodide–labeled cells. Bar, 1 mm. (p) Quantitative analysis of VSMC invasive activity in aortic explant cultures after an 8-d culture period. Results are expressed as the mean ± SEM (n = 5).
VSMCs, like other mesenchymal cell populations, can mobilize multiple proteolytic systems to degrade and migrate through collagenous barriers (3–14). To identify the major proteinase classes involved in regulating 3-D invasion, explants were embedded in collagen gels in the presence of inhibitors directed against cysteine-, aspartyl-, serine-, or metalloproteinases (8, 19–21), and migratory responses were monitored over the course of an 8-d incubation period. At concentrations previously demonstrated to block cysteine proteinase or aspartyl proteinase activity effectively (8, 19–21), neither E-64 nor pepstatin A affect VSMC invasion (Fig. 1, g, h, and p). Further, VSMC emigration proceeds in an unabated fashion either in the presence of the plasmin inhibitor aprotinin (19, 20) or when explants are recovered from plasminogen-null mice and suspended in plasminogen-null serum (Fig. 1, i, j, and p). In contrast, both the tissue-invasive activity expressed by VSMC outgrowth and VSMC collagen-degradative activity are ablated by the MMP inhibitor BB-94 (Fig. 1, k and p).
MMPs currently are classified as a family of more than 20 proteinases whose members are expressed either as secreted or membrane-anchored enzymes (22). The endogenous tissue inhibitor of metalloproteinases (TIMP-1), preferentially targets secreted MMPs as well as the glycophosphatidylinositol-anchored MMPs (i.e., MT4-MMP and MT6-MMP) (22–24). A second member of the TIMP family, TIMP-2, more potently inhibits secreted MMPs, MMP-2, and MMP-9, as well as the type I membrane–anchored MMPs (i.e., MT1-, 2-, 3-, and 5-MMPs) (22–24). Although VSMC invasion is unaffected by TIMP-1 (Fig. 1, l and p), equimolar concentrations of active TIMP-2 exerted an inhibitory effect indistinguishable from that observed with BB-94 (Fig. 1, m and p).
Recently, a series of studies have concluded that MMP-2 and MMP-9 play key roles in regulating the 2-D migration of VSMCs (3–5, 9–14). However, although both MMP-2 and MMP-9 are preferentially inhibited by TIMP-2 (23), VSMC outgrowth from either collagen-embedded MMP-2−/− or MMP-9−/− explants cultured, respectively, in MMP-2−/− or MMP-9−/− autologous serum proceeds in a fashion indistinguishable from littermate controls (Fig. 1, n–p). Likewise, neither of the KO explants displayed outgrowth defects when embedded in a 3-D matrix of Matrigel (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050607/DC1). Hence, MMP-2 and MMP-9 are not necessary in regulating the tissue-invasive machinery mobilized by smooth muscle cells during 3-D invasion.
Although the ability of TIMP-2 to block VSMC invasion cannot be ascribed to inhibitory effects on either MMP-2 or MMP-9, at least two TIMP-2–sensitive members of the membrane-anchored MMP family, i.e., MT1-MMP and MT2-MMP, have been described recently as potent collagenolysins (19, 25–28). Because WT explants express only MT1-MMP and MT3-MMP (Fig. 2 a), and MT3-MMP does not express type I collagenolytic activity (25, 26, 28, 29), we next determined the collagen-invasive potential of explants recovered from MT1-MMP–null mice (30). Although MT1-MMP–null VSMCs express MMP-2, MMP-9, and MMP-13 as well as MT3-MMP mRNA at comparable levels, MT1-MMP−/− explants fail to display invasive activity either in 3-D matrices of type I collagen or in a composite gel of type I and III collagens (Fig. 2, a and b). Further, in contrast with the behavior of WT VSMCs, MT1-MMP−/− VSMCs are confined to the interface between the explant surface and the surrounding matrix; collagen-degradation products are not detected in association with the immobile cells (Fig. 2, c and d). In electron micrographs of the explant cultures, WT VSMCs are surrounded by collagen-free zones generated as a consequence of pericellular collagenolysis, but intact collagen fibrils are consistently found in juxtaposition to the surface of MT1-MMP−/− VSMC (Fig. 2, c and d). Importantly, the invasion-null phenotype is confined to interstitial collagen barriers because MT1-MMP−/− VSMCs readily traverse a 3-D gel of cross-linked fibrin, a physiologically relevant matrix whose proteolysis is supported equally well by either MT1-MMP or MT3-MMP (Fig. 2, a and b; [29]). Likewise, MT1-MMP–null VSMC traverse dense matrices of Matrigel, a basement membrane extract whose remodeling proceeds independently of MT1-MMP (Fig. 2, a and b), MMP-2, or MMP-9 (unpublished data; [19, 25, 29]).
Figure 2.

MT1-MMP–dependent control of VSMC invasion. (a and b) Media explants from MT1-MMP+/+ and MT1-MMP−/− mice were suspended within 3-D gels of type I collagen, a mixture of type I and III collagens, fibrin, or Matrigel. Asterisks indicate the position of the explants, and arrowheads mark the leading edge of egressing VSMC. Bars, 1 mm. Upper right panels show RT-PCR analysis of MMPs expressed by invading VSMCs. (c and d) Semi-thin sections (upper left panels) and transmission electron micrographs (right side-panels) of media explant (stars) cultured within 3-D collagen matrix for 8 d. (c) Zones of degraded collagen lie juxtaposed to the WT VSMCs as assessed by staining for denatured collagen (green staining, arrows) around propidium iodide–labeled cells (red, asterisk) or by areas of collagen clearing in transmission electron micrographs. (d) Intact collagen fibrils lie adjacent to MT1-MMP−/− VSMCs. Bars, 20 μm for two left panels and 2 μm for right panel.
To determine if defects in collagenolysis and invasive activity can be ascribed directly to isolated VSMCs, smooth muscle cells were recovered from MT1-MMP+/+ or MT1-MMP−/− explants and were cultured atop a subjacent bed of type I collagen fibrils. Under these conditions, WT but not MT1-MMP−/− VSMCs express collagenolytic activity and invade type I collagen gels (Fig. 3, a and b). The inability of MT1-MMP−/− VSMC to negotiate 3-D collagen gels is not limited to in vitro ECM constructs, because these findings could be extended to the vessel wall itself. Whereas WT fluorescent-tagged VSMCs invade explants of devitalized aorta, MT1-MMP−/− VSMCs remain confined to the surface of the vessel wall (Fig. 3 a, lower row). After reconstitution of MT1-MMP expression in MT1-MMP−/− VSMCs by retroviral transduction, the null cells fully recover the ability to degrade subjacent collagen and to invade either the reconstituted collagen matrices or the vessel wall explants (Fig. 3, a and b). Despite obvious defects in the ability of MT1-MMP−/− VSMCs to remodel collagen or to invade collagen-rich tissues, rates of proliferation, apoptosis, and 2-D migration across collagen-coated surfaces are indistinguishable from littermate controls (Fig. 3 c).
Figure 3.

Collagenolytic activity of isolated MT1-MMP−/− VSMCs. (a) Isolated WT cells, MT1-MMP-null VSMCs, or MT1-MMP–transduced null cells (rMT1-MMP) were cultured atop a film of type I collagen (upper row) or a 3-D gel of type I collagen (middle row) in the absence or presence of BB-94. Upper row: yellow arrowheads mark zones of collagen proteolysis. Insets are phase contrast micrographs of MT1-MMP+/+ and MT1-MMP−/− VSMCs that were cultured atop the collagen film substratum. Middle row: black arrows indicate the position of VSMCs that have invaded the collagen gels. Bottom row: fluorescently labeled VSMCs (green) were cultured atop devitalized aorta. White arrowheads mark the position of VSMCs that invaded the aortic tissue. Bars, 50 μm. (b) Quantitative analysis of the MT1-MMP–dependent collagenolytic and invasive activities displayed by smooth muscle cells cultured as described. Results are expressed as the mean ± SEM (n = 5). (c) VSMC proliferation (BrdU) and apoptosis (TUNEL) in WT and MT1-MMP−/− cultures established atop type I collagen gels. TUNEL-positive VSMCs (arrowheads) and BrdU-labeled VSMCs (arrows) are shown with propidium iodide counterstaining (red) used to visualize cells. Bar, 50 μm. Right panels: motility of MT1-MMP+/+ and MT1-MMP−/− VSMCs across a type I collagen–coated substratum. The dashed yellow line marks the position of cells at the start of the assay; the red arrow indicates the position of the leading front of cells after 72 h in culture. Bar, 0.5 mm.
In humans, the intima-media thickening that occurs as a consequence of VSMC migration and proliferation within the carotid artery is an important predictive phenotype for cardiovascular disease (1, 2, 31). Because elevated MT1-MMP expression has been localized at pathologic sites of vascular remodeling in vivo (32, 33 ), we next sought to determine whether an MT1-MMP–deficient status would affect VSMC behavior in vivo during neointima formation. Although the morbid status and decreased lifespan of MT1-MMP−/− mice complicate the use of the homozygote-null animals (30), pilot experiments were initiated in an attempt to gauge the response of the null animals to the surgical procedure. However, none of the MT1-MMP−/− mice recovered from even a brief period of anesthesia (n = 4). Hence, the phenotypically normal heterozygotes were selected for further study because the isolated cells displayed an ∼25% reduction in collagen-invasive activity in vitro (unpublished data). Hence, MT1-MMP+/+ and MT1-MMP+/− littermates underwent unilateral common carotid artery ligation to induce neointima formation via a hemodynamically initiated process that involves minimal endothelial cell damage or inflammation (34). The total number of cells that migrate into the subendothelial space during the first 5 d after ligation is strikingly reduced in heterozygous arteries (Fig. 4, a and d). Further, at day 14 after ligation, MT1-MMP haploinsufficient mice remain protected against occlusive vascular remodeling (Fig. 4, a and d). At 2 wk after surgery, the ligated arteries of MT1-MMP+/+ mice display marked neointimal hyperplasia and outward geometric remodeling accompanied by local increases in MMP-2 and MMP-9 expression (Fig. 4, a and b [12, 13]). By contrast, in the ligated carotids of MT1-MMP+/− mice, neointimal size and cell number, as well as the intima/media ratios, are reduced by >50% despite similar, if not enhanced, increases in MMP-2 and MMP-9 expression (Fig. 4, a and b). Intimal thickening is not detected in the contralateral carotids of either genotype (Fig. 4 a, insets).
Figure 4.
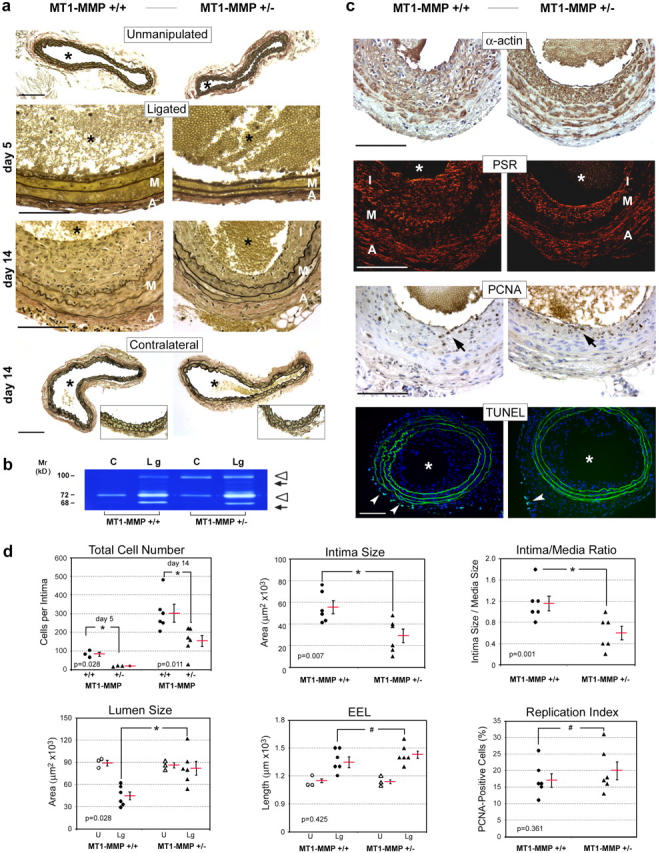
MT1-MMP deficiency reduces neointima formation in vivo. (a) Verhoeff Van Gieson's staining of unmanipulated controls as well as contralateral and ligated common carotid arteries from MT1-MMP+/+ and MT1-MMP+/− mice 5 d and 14 d after ligation. Elastic fibers are stained black; nuclei are stained brown. Bottom row insets demonstrate intact intima in contralateral carotids of either genotype. *, vessel lumen; A, adventitia; I, intima; M, media. (b) Gelatin zymography of carotid artery extracts 14 d after ligation. The pro- (open arrowheads) and processed (arrows) forms of MMP-9 (upper arrowhead and arrow) and MMP-2 (lower arrowhead and arrow) are indicated. C, contralateral artery; Lg, ligated artery. (c) 14 d after ligation, paraffin sections of ligated carotid arteries were probed with anti-smooth muscle α-actin polyclonal antibody (α-actin; brown stain) or stained with Picro-Sirius red (PSR; red stain). Cell proliferation and apoptosis were determined with anti-PCNA mAb (PCNA) and TUNEL, respectively. PCNA-positive nuclei (brown, arrows) and TUNEL-positive cells (arrowheads) are shown. (d) Quantitative assessment of vascular remodeling. Charts show individual numbers (six for each group) with the mean indicated by a red bar ± SEM. All data were obtained at day 14 after ligation except for total cell numbers, for which values are shown for both days 5 and 14. EEL, external elastic lumina. Bars, 100 μm.
Consistent with a specific role for MT1-MMP in regulating VSMC invasion alone, neither cell replication nor apoptotic rates were altered at day 5 (17 ± 1 versus 16 ± 2 proliferating cell nuclear antigen [PCNA]-positive cells/vessel cross section in MT1-MMP+/+ and MT1-MMP+/− mice, respectively, and 24 ± 1 versus 24 ± 3 TUNEL-positive cells/vessel cross section in MT1-MMP+/+ and MT1-MMP+/− mice, respectively) or at day 14 after ligation in MT1-MMP heterozygote mice (Fig. 4, c and d). Likewise, no differences in rates of macrophage influx were detected between MT1-MMP+/+ and MT1-MMP+/− mice (unpublished data). Interestingly, although intimal size is decreased significantly in the heterozygote mice, the ligated vessel undergoes compensatory outward remodeling to a degree similar to that exhibited by the WT animals (as assessed by length of the external elastic lamina) coincident with indistinguishable rates of collagen deposition in the remodeling vessels (Fig. 4, c and d). Consequently, and in marked contrast to the response of the ligated MT1-MMP+/+ carotids, a reduction in intimal expansion in MT1-MMP+/− mice is coupled with active outward geometric remodeling that results in an almost complete retention of the lumen diameter relative to unmanipulated controls (i.e., whereas lumen diameter decreases by ∼50% in MT1-MMP+/+ ligated carotids, lumen size remains intact in MT1-MMP+/− mice; Fig. 4 d). Hence, a partial reduction in MT1-MMP expression protects heterozygote mice against neointimal hyperplasia and arterial lumen narrowing.
DISCUSSION
A wide range of pathologic insults to the arterial wall induces VSMCs to infiltrate the intimal space and mount a hyperplastic response that narrows the artery lumen and alters vessel wall geometry (1–14). Multiple proteolytic systems have been posited to participate in VSMC migration or invasion (3–14), but prior studies have focused on assessing the behavior of passaged VSMCs cultured atop 2-D substrata in short-term assays that do not recapitulate the 3-D matrix environment in which the cells are embedded normally (1, 16, 17). In our studies designed specifically to recapitulate the in vivo environment, proteinases linked previously to VSMC migration, including plasmin, cysteine proteinases, MMP-2, and MMP-9, do not play a critical role in supporting the 3-D invasive phenotype. Instead, MT1-MMP confers VSMCs with the ability to degrade and invade either a composite extracellular matrix of purified type I and III collagens or the vessel wall itself. Although recent studies demonstrate that MT1-MMP−/− VSMCs exhibit a stimulus-specific defect in PDGF-BB–mediated signaling under serum-free conditions (35), the ability of the null cells to respond to a mixture of PDGF-BB and FGF-2 in a serum milieu is unperturbed with regard to 2-D motility and proliferation. Further, MT1-MMP−/− VSMCs do not display a global defect in their ability to mount invasive responses, because migration through 3-D barriers of fibrin or Matrigel were unaffected. Rather, MT1-MMP seems primarily to regulate invasion through type I/III collagen–rich barriers regardless of the initiating stimulus. We do note that mouse and human VSMCs can display distinct properties (1, 2), but preliminary studies indicate that MT1-MMP likewise regulates the collagen-invasive activity of human aortic smooth muscle cells (unpublished observation). Although recent studies have suggested that mesenchymal cells may mobilize nonproteolytic systems to infiltrate connective tissue barriers (36), no compensatory mechanisms were identified that proved able to rescue the null phenotype of the invasion-incompetent MT1-MMP−/− VSMCs.
MT1-MMP−/− mice are runted, infertile, and have a shortened lifespan, whereas the heterozygote mice exhibit a normal phenotype (30). Further, in contrast with MT1-MMP–null mice that harbor defects in endothelial cell–mural cell interactions as a consequence of a specific defect in PDGF-BB signaling, the vasculature of the heterozygotes is normal, and PDGF-BB (35) responses are indistinguishable from those in WT mice. Hence, the MT1-MMP+/− mice afforded the opportunity to assess the role of the proteinase in the in vivo setting. Consistent with our ex vivo findings that highlight the importance of MT1-MMP in conferring VSMCs with collagenolytic and invasive activities, an MT1-MMP+/− status confers mice with a resistant phenotype against ligation-induced neointimal hyperplasia. Although others have reported that MMP-2−/− or MMP-9−/− mice also exhibit a protected status in this model and have concluded that these enzymes control invasive activity (11–14), our findings suggest that these proteinases do not play direct or necessary roles in regulating VSMC migration. Instead, we posit that MMP-2 and MMP-9 more likely affect the activity or availability of cell- or matrix-bound growth factor/growth factor receptors, chemokines, or cytokines (37). Because both MMP-2– and MMP-9–null mice also suffer from a number of developmental defects that can affect events ranging from the mobilization of progenitor cells to immune function (38–40), the direct or indirect mechanisms by which these metalloenzymes affect neointimal hyperplasia deserve further study. These issues notwithstanding, MT1-MMP controls VSMC invasive activity in vitro and in vivo independently of MMP-2 or MMP-9 activity. Further, the ability of MT1-MMP, rather than MMP-2 or MMP-9, to regulate the collagen-degradative and invasive activities of VSMCs is consistent with more recent studies of angiogenesis and with fibroblast–extracellular matrix interactions (19, 26, 41–43).
Increasing evidence suggests that the intimal hyperplasia probably is not restricted to the participation of VSMCs alone and may involve adventitial fibroblasts, marrow-derived smooth muscle precursors, or macrophages (1, 2, 44). Because MT1-MMP may serve as the dominant determinant of cellular motility within collagen-rich environments, this proteinase may well play similar roles in regulating the invasive properties of multiple cell types within the context of the arterial matrix. Given that even a partial reduction in MT1-MMP expression affords the vessel wall lumen of heterozygote animals with a protected status in vivo, therapeutic interventions directed against this proteinase may prove beneficial in human vascular disease states.
MATERIALS AND METHODS
Mice.
All ex vivo and in vivo studies were performed with 4- to 6-wk-old male plasminogen−/− C57BL6 mice (7), MMP-2−/− C57BL6 mice (13), MMP-9−/− 129SvEv mice (12), or MT1-MMP−/− or MT1-MMP+/− Swiss Black mice (30). Age-matched C57BL6 animals were used as controls for plasminogen−/− and MMP-2−/− mice, and 129SvEv animals were used as controls for MMP-9−/− mice. For studies of MT1-MMP−/− or MT1-MMP+/− mice, paired analyses were performed with WT littermates.
3-D culture conditions and invasion assays.
For 3-D ex vivo invasion assay, type I collagen (acid extracted from rat tail tendons [25]) or type III collagen (Sigma-Aldrich) was dissolved in 0.2% acetic acid to a final concentration of 2.7 mg/ml (25). Before assay, fragments of mouse thoracic aorta were stripped of intima and adventitia, and the media of the vessel wall was dissected into 1 × 1–mm fragments. Media explants were then suspended within a solution of type I collagen alone, a composite of type I/type III collagens (3:1), 12 mg/ml Matrigel (Becton Dickinson), or 3 mg/ml cross-linked fibrin prepared as described (25, 29) and were cultured for 8 d in DMEM medium supplemented with 10% FBS, autologous plasminogen-, MMP-9–, or MMP-2–null mouse sera. A PDGF-BB/FGF-2 mixture (10 ng/ml each; R&D Systems) was added to explant cultures to initiate VSMC outgrowth and invasion. Invasive activity was quantified by measuring the distance migrated by the leading front of VSMCs from the explanted tissue.
To assess the invasive activity of isolated VSMCs, homogeneous cultures were established from collagenase type 2 (1.5 mg/ml; Worthington Biochemical Corporation) digests of vessel wall explants as described (16). VSMCs were seeded atop 3-D gels of type I collagen or fibrin in the upper well of 24-mm Transwell dishes (3-μm pore size; Corning, Inc.). After a 24-h incubation period, a PDGF/FGF-2 mixture was added to the lower compartment of the Transwell chambers. The number of invasive foci was determined in randomly selected fields by phase-contrast microscopy.
Where indicated, protease inhibitors were added to media explants or isolated VSMCs at the following final concentrations: 3 μM BB-94 (in 0.1% DMSO final; gift of British Biotechnology Ltd.), 5 μg/ml TIMP-2, 12.5 μg/ml TIMP-1 (equimolar as determined by active site titration [25]; endotoxin-free; Fuji Industries Co., Ltd.), 100 μM E-64 (in 0.05% ethanol final, Sigma-Aldrich), 100 μg/ml aprotinin (Roche), or 50 μM pepstatin (in 0.05% ethanol final; Roche).
VSMCs labeled with fluorescent microspheres (Fluoresbrite, Polysciences, Inc.; [19]) were seeded atop segments of dog aorta that had been devitalized after three rounds of freezing in liquid N2 and thawing. The cocultures were suspended in DMEM/10% FCS in the absence or presence of 3 μm BB-94 and placed into the upper compartment of 24-mm Transwell dishes. PDGF/FGF-2 (10 ng each) was added to the lower compartment of the dishes to initiate invasion.
Retroviral-gene transfer.
Hemagglutinin-tagged human MT1-MMP cDNA was subcloned into the pRET2 retroviral vector derived from the Moloney murine leukemia virus–based MFG backbone, and polyclonal ecotropic producer cell lines were established as described (26). Subconfluent monolayers of the isolated VSMCs were cultured in the retroviral supernatant for 12 h, and collagen invasion and degradation assays were performed 24 h later.
RT PCR analysis.
RNA was isolated from MT1-MMP WT or null explant cultures using TRIzol reagent (Life Technologies). RT and PCR amplification using specific oligonucleotide primers for MMP-9, MMP-2, MMP-8, MMP-13, mCol A, MT1-MMP, MT2-MMP, or MT3-MMP was performed as described (19, 29).
Transmission and scanning electron microscopy.
3-D cultures of primary aortic media explants were prepared for transmission and scanning electron microscopy as described previously (25, 29). For freeze-fracture scanning electron microscopy, gels were immersed in liquid N2 and fractured.
Immunofluorescence, proliferation, and apoptosis assays.
To detect proteolyzed collagen, immunofluorescence was performed on frozen sections that were fixed in 1% paraformaldehyde, incubated overnight at 4°C with mAb HUI77 (100 μg/ml; gift of Cell-Matrix, Inc., a subsidiary of CancerVax Corp.), and incubated with FITC-conjugated secondary antibody (1:400). Bromodeoxyuridine (BrdU) incorporation was determined after a 60-min pulse with 10 μM BrdU in isolated VSMC cultures. Apoptosis was assessed by TUNEL assay (Fluorescein Direct Apoptag; Intergen, Limited; [45]). Proliferative indices in the intima of remodeled carotids were quantified by staining sections with PCNA mAb (clone PC10, DakoCytomation; [45]). VSMC and macrophages were visualized, respectively, with anti-smooth muscle α-actin mAb (clone 1A4; DakoCytomation) and anti-Mac-2 mAb (CL8942AP; Cedarlane Laboratories Limited).
Subjacent collagenolysis and cell motility.
Isolated VSMCs (5 × 104) were stimulated with a PDGF/FGF-2 mixture atop a thin film of type I collagen (100 μg/2.2 cm2) in the absence or presence of BB-94 (3 μM). After 5 d in culture, cells were dislodged from the collagen substratum with 10 mM EDTA, and the integrity of the underlying matrix was assessed by Coomassie staining. Zones of cleared collagen were counted in 10 randomly selected fields. VSMC migration atop a collagen-coated surface was assayed as described (29). In brief, VSMC monolayers were established on a collagen substratum whose surface was decorated with small cloning chips. When the cultures were confluent, the cloning chips were removed, leaving a well-demarcated denuded zone wherein VSMC migration could be monitored (29).
Animal model, tissue processing and morphometric analysis.
All animal protocols were approved by the University of Michigan Committee on Use and Care of Animals. Mice were housed in the American Association for Accreditation of Laboratory Animal Care–approved facility of the University of Michigan. Left common carotid arteries of MT1-MMP+/+ and MT1-MMP+/− littermates (n = six each) were ligated for 2 wk as described (34). Mouse tissues were perfusion fixed with methanol-Carnoy's fixative (methanol/chloroform/glacial acetic acid in a 60:30:10 volume ratio). Unmanipulated control contralateral and ligated carotids were removed, paraffin embedded, and sectioned (5 μm thick). Vessel wall elastin and collagen were visualized with Verhoeff Van Gieson's (Accustain Elastic Stain, Sigma-Aldrich) or Picro-Sirius red stains, respectively (46). Groups of four consecutive carotid artery tissue sections spaced at equal intervals (150 μm) were analyzed using SPOT image software (SPOT 3.4, Diagnostic Instruments). The lumen circumference, the length of the internal elastic lamina, and external elastic lamina were determined as described (34) with lumen circumference used to calculate the lumen area. The intima was determined as the area defined by the luminal surface and internal elastic lamina with the medial area defined by the internal elastic lamina and external elastic lamina.
For gelatin zymography, ligated carotid arteries were collected separately, pulverized in liquid N2 and equal amounts of tissue extract protein (10 μg), and assayed as described (29).
Statistical analysis.
All data, expressed as mean ± SEM, in MT1-MMP+/+ and MT1-MMP+/− mice, were analyzed by the paired Student's t test. Data were considered statistically significant at P < 0.05.
Online supplemental material.
Fig. S1 shows ex vivo invasive activity of VSMCs in 3-D Matrigel. Media explants from WT, MMP-9–, or MMP-2–null mice were embedded within a 3-D gel of Matrigel. VSMC egress was initiated by exogenous (PDGF/FGF-2), and outgrowth into the translucent Matrigel matrix was visualized by phase contrast microscopy at 8 d. Asterisk indicates the explant tissue, and arrowheads point at the leading edge of the invading SMC. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050607/DC1.
Acknowledgments
We thank L. Peng and J. Sun for help with animal surgery and V. Krivtsov for advice on statistical analysis.
This study was supported in part by NIH grants CA088308 and CA71699 to S.J. Weiss and grants HL65224 and HL57346 to W.P. Fay.
The authors have no conflicting financial interests.
Abbreviations used: 2-D, two-dimensional; 3-D, three- dimensional; BrdU, bromodeoxyuridine; FGF, fibroblast growth factor; MMP, matrix metalloproteinase; MT1, membrane type I; PCNA, proliferating cell nuclear antigen; PDGF, platelet-derived growth factor; TIMP, tissue inhibitor of metalloproteinases; VSMC, vascular smooth muscle cell.
References
- 1.Ross, R. 1999. Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 340:115–126. [DOI] [PubMed] [Google Scholar]
- 2.Lusis, A.J. 2000. Atherosclerosis. Nature. 407:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zempo, N., N. Koyama, R.D. Kenagy, H.J. Lea, and A.W. Clowes. 1996. Regulation of vascular smooth muscle cell migration and proliferation in vitro and in injured rat arteries by a synthetic matrix metalloproteinase inhibitor. Arterioscler. Thromb. Vasc. Biol. 16:28–33. [DOI] [PubMed] [Google Scholar]
- 4.Kenagy, R.D., S. Vergel, E. Mattsson, M. Bendeck, M.A. Reidy, and A.W. Clowes. 1996. The role of plasminogen, plasminogen activators, and matrix metalloproteinases in primate arterial smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 16:1373–1382. [DOI] [PubMed] [Google Scholar]
- 5.Kenagy, R.D., C.E. Hart, W.G. Stetler-Stevenson, and A.W. Clowes. 1997. Primate smooth muscle cell migration from aortic explants is mediated by endogenous platelet-derived growth factor and basic fibroblast growth factor acting through matrix metalloproteinases 2 and 9. Circulation. 96:3555–3560. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet, P., L. Moons, V. Ploplis, E. Plow, and D. Collen. 1997. Impaired arterial neointima formation in mice with disruption of the plasminogen gene. J. Clin. Invest. 99:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lijnen, H.R., B. Van Hoef, F. Lupu, L. Moons, P. Carmeliet, and D. Colleen. 1998. Function of the plasminogen/plasmin and matrix metalloproteinase systems after vascular injury in mice with targeted inactivation of fibrinolytic system genes. Arterioscler. Thromb. Vasc. Biol. 18:1035–1045. [DOI] [PubMed] [Google Scholar]
- 8.Sukhova, G.K., G.-P. Shi, D.I. Simon, H.A. Chapman, and P. Libby. 1998. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J. Clin. Invest. 102:576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mason, D.P., R.D. Kenagy, D. Hasenstab, D.F. Bowen-Pope, R.A. Siefert, S. Coats, S.M. Hawkins, and A.W. Clowes. 1999. Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ. Res. 85:1179–1185. [DOI] [PubMed] [Google Scholar]
- 10.Kanda, S., M. Kuzuya, M.A. Ramos, T. Koike, K. Yoshino, S. Ikeda, and A. Iguchi. 2000. Matrix metalloproteinase and alphavbeta3 integrin-dependent vascular smooth muscle cell invasion through a type I collagen lattice. Arterioscler. Thromb. Vasc. Biol. 20:998–1005. [DOI] [PubMed] [Google Scholar]
- 11.Cho, A., and M.A. Reidy. 2002. Matrix metalloproteinase-9 is necessary for the regulation of smooth muscle cell replication and migration after arterial injury. Circ. Res. 91:845–851. [DOI] [PubMed] [Google Scholar]
- 12.Galis, Z.S., C. Johnson, D. Godin, R. Magid, J.M. Shipley, R.M. Senior, and E. Ivan. 2002. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ. Res. 91:852–859. [DOI] [PubMed] [Google Scholar]
- 13.Kuzuya, M., S. Kanda, T. Sasaki, N. Tamaya-Mori, X.W. Cheng, T. Itoh, S. Itohara, and A. Iguchi. 2003. Deficiency of gelatinase A suppresses smooth muscle cell invasion and development of experimental intimal hyperplasia. Circulation. 108:1375–1381. [DOI] [PubMed] [Google Scholar]
- 14.Johnson, C., and Z.S. Galis. 2004. Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler. Thromb. Vasc. Biol. 24:54–60. [DOI] [PubMed] [Google Scholar]
- 15.Ponticos, M., T. Partridge, C.M. Black, D.J. Abraham, and G. Bou-Gharios. 2004. Regulation of collagen type I in vascular smooth muscle cells by competition between Nkx2.5 and deltaEF1/ZEB1. Mol. Cell. Biol. 24:6151–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stegemann, J.P., and R.M. Nerem. 2003. Altered response of vascular smooth muscle cells to exogenous biochemical stimulation in two- and three-dimensional culture. Exp. Cell Res. 283:146–155. [DOI] [PubMed] [Google Scholar]
- 17.Cukierman, E., R. Pankov, D.R. Stevens, and K.M. Yamada. 2001. Taking cell-matrix adhesions to the third dimension. Science. 294:1708–1712. [DOI] [PubMed] [Google Scholar]
- 18.Pickering, J.G., S. Uniyal, C.M. Ford, T. Chau, M.A. Laurin, L.H. Chow, C.G. Ellis, J. Fish, and B.M. Chan. 1997. Fibroblast growth factor-2 potentiates vascular smooth muscle cell migration to platelet-derived growth factor: upregulation of alpha2beta1 integrin and disassembly of actin filaments. Circ. Res. 80:627–637. [DOI] [PubMed] [Google Scholar]
- 19.Sabeh, F., I. Ota, K. Holmbeck, H. Birkedal-Hansen, P. Soloway, M. Balbin, C. Lopez-Otin, S. Shapiro, M. Inada, S. Krane, et al. 2004. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase, MT1-MMP. J. Cell Biol. 167:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filippov, S., I. Caras, R. Murray, L.M. Matrisian, H.A. Chapman, S. Shapiro, and S.J. Weiss. 2003. Matrilysin-dependent elastolysis by human macrophages. J. Exp. Med. 198:925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lkhider, M., R. Castino, E. Bouguyon, C. Isidoro, and M. Ollivier-Bousquet. 2004. Cathepsin D released by lactating rat mammary epithelial cells is involved in prolactin cleavage under physiological conditions. J. Cell Sci. 117:5155–5164. [DOI] [PubMed] [Google Scholar]
- 22.Visse, R., and H. Nagase. 2003. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92:827–839. [DOI] [PubMed] [Google Scholar]
- 23.Howard, E.W., E.C. Bullen, and M.J. Banda. 1991. Preferential inhibition of 72- and 92-kDa gelatinases by tissue inhibitor of metalloproteinases-2. J. Biol. Chem. 266:13070–13075. [PubMed] [Google Scholar]
- 24.Lee, M.H., M. Rapti, V. Knauper, and G. Murphy. 2004. Threonine 98, the pivotal residue of tissue inhibitor of metalloproteinases (TIMP)-1 in metalloproteinase recognition. J. Biol. Chem. 279:17562–17569. [DOI] [PubMed] [Google Scholar]
- 25.Hotary, K., E. Allen, A. Punturieri, I. Yana, and S.J. Weiss. 2000. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J. Cell Biol. 149:1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chun, T.-H., F. Sabeh, I. Ota, H. Murphy, K. McDonagh, K. Holmbeck, H. Birkedal-Hansen, E.D. Allen, and S.J. Weiss. 2004. MT1-MMP-dependent neovessel formation within the confines of the 3-dimensional extracellular matrix. J. Cell Biol. 167:757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.d'Ortho, M.P., H. Will, S. Atkinson, G. Butler, A. Messent, J. Gavrilovic, B. Smith, R. Timpl, L. Zardi, and G. Murphy. 1997. Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable to many matrix metalloproteinases. Eur. J. Biochem. 250:751–757. [DOI] [PubMed] [Google Scholar]
- 28.Shimada, T., H. Nakamura, E. Ohuchi, Y. Fujii, Y. Murakami, H. Sato, M. Seiki, and Y. Okada. 1999. Characterization of a truncated recombinant form of human membrane type 3 matrix metalloproteinase. Eur. J. Biochem. 262:907–914. [DOI] [PubMed] [Google Scholar]
- 29.Hotary, K.B., I. Yana, F. Sabeh, X.Y. Li, K. Holmbeck, H. Birkedal-Hansen, E.D. Allen, N. Hiraoka, and S.J. Weiss. 2002. Matrix metalloproteinases (MMPs) regulate fibrin-invasive activity via MT1-MMP-dependent and -independent processes. J. Exp. Med. 195:295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmbeck, K., P. Bianco, J. Caterina, S. Yamada, M. Kromer, S.A. Kuznetsov, M. Mankani, P.G. Robey, A.R. Poole, I. Pidoux, et al. 1999. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 99:81–92. [DOI] [PubMed] [Google Scholar]
- 31.Cheng, K.S., D.P. Mikhailidis, G. Hamilton, and A.M. Seifalian. 2002. A review of the carotid and femoral intima-media thickness as an indicator of the presence of peripheral vascular disease and cardiovascular risk factors. Cardiovasc. Res. 54:528–538. [DOI] [PubMed] [Google Scholar]
- 32.Shofuda, K., Y. Nagashima, K. Kawahara, H. Yasumitsu, K. Miki, and K. Miyazaki. 1998. Elevated expression of membrane-type 1 and 3 matrix metalloproteinases in rat vascular smooth muscle cells activated by arterial injury. Lab. Invest. 78:915–923. [PubMed] [Google Scholar]
- 33.Rajavashisth, T.B., X.P. Xu, S. Jovinge, S. Meisel, X.O. Xu, N.N. Chai, M.C. Fishbein, S. Kaul, B. Cercek, B. Sharifi and P.K. Shah. 1999. Membrane type 1 matrix metalloproteinase expression in human atherosclerotic plaques: evidence for activation by proinflammatory mediators. Circulation. 99:3103–3109. [DOI] [PubMed] [Google Scholar]
- 34.Kumar, A., and V. Lindner. 1997. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler. Thromb. Vasc. Biol. 17:2238–2244. [DOI] [PubMed] [Google Scholar]
- 35.Lehti, K., E. Allen, H. Birkedal-Hansen, K. Holmbeck, Y. Miyake, T.-H. Chun, and S.J. Weiss. 2005. An MT1-MMP-PDGF receptor-β axis regulates mural cell investment of the microvasculature. Genes Dev. 19:979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf, K., I. Mazo, H. Leung, K. Engelke, U.H. von Andrian, E.I. Deryugina, A.Y. Strongin, E.B. Brocker, and P. Friedl. 2003. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 160:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Egeblad, M., and Z. Werb. 2002. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2:161–174. [DOI] [PubMed] [Google Scholar]
- 38.Corry, D.B., K. Rishi, J. Kanellis, A. Kiss, L. Song, J. Xu, L. Feng, Z. Werb, and F. Kheradmand. 2002. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat. Immunol. 3:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heissig, B., K. Hattori, S. Dias, M. Friedrich, B. Ferris, N.R. Hackett, R.G. Crystal, P. Besmer, D. Lyden, M.A. Moore, et al. 2002. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 109:625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kheradmand, F., K. Rishi, and A. Werb. 2002. Signaling through the EGF receptor controls lung morphogenesis in part by regulating MT1-MMP-mediated activation of gelatinase A/MMP2. J. Cell Sci. 115:839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiraoka, N., E. Allen, I.J. Apel, M.R. Gyetko, and S.J. Weiss. 1998. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 95:365–377. [DOI] [PubMed] [Google Scholar]
- 42.Baluk, P., W.W. Raymond, E. Ator, L.M. Coussens, D.M. McDonald, and G.H. Caughey. 2004. Matrix metalloproteinase-2 and -9 expression increases in mycoplasma-infected airways but is not required for microvascular remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 287:L307–L317. [DOI] [PubMed] [Google Scholar]
- 43.Masson, V., L.R. de la Ballina, C. Munaut, B. Wielockx, M. Jost, C. Maillard, S. Blacher, K. Bajou, T. Itoh, S. Itohara, et al. 2004. Contribution of host MMP-2 and MMP-9 to promote tumor vascularization and invasion of malignant keratinocytes. FASEB J. 19:234–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Owens, G.K., M.S. Kumar, and B.R. Wamhoff. 2004. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84:767–801. [DOI] [PubMed] [Google Scholar]
- 45.Hotary, K.B., E.D. Allen, P.C. Brooks, N.S. Datta, M.W. Long, and S.J. Weiss. 2003. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 114:33–45. [DOI] [PubMed] [Google Scholar]
- 46.Galis, Z.S., M. Muszynski, G.K. Sukhova, E. Simon-Morrissey, E.N. Unemori, M.W. Lark, E. Amento, and P. Libby. 1994. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ. Res. 75:181–189. [DOI] [PubMed] [Google Scholar]