

Abstract
Natural killer (NK) cells are known to reject certain tumors in vivo; however, the ability of NK cells to prevent metastasis of tumors into secondary lymphoid organs has not been addressed. Here, we report that in tumor-bearing hosts, NK cells are recruited to regional lymph nodes in wild-type mice, but not in mice deficient for L-selectin or L-selectin ligands. By adoptive transfer and complete Freund's adjuvant stimulation experiments, we demonstrated that L-selectin on NK cells and L-selectin ligands on endothelial cells are essential for NK cell recruitment to lymph nodes. Furthermore, freshly isolated resident lymph node NK cells lysed tumors efficiently, and metastasis of B16 melanoma cells to draining lymph nodes was suppressed in wild-type or Rag-1–deficient mice, but not when NK cells were depleted. Although L-selectin–deficient NK cells efficiently lysed tumor cells in vitro, NK cell–dependent suppression of tumor metastasis was diminished in mice deficient for L-selectin or L-selectin ligands because of insufficient NK cell recruitment to lymph nodes. Moreover, tumor metastasis was substantially inhibited in L-selectin–deficient mice reconstituted with wild-type NK cells. These findings indicate that L-selectin–mediated NK cell recruitment plays a crucial role in the control of tumor metastasis into secondary lymphoid organs.
In adults, NK cells develop from bone marrow–derived precursors, and the mature NK cells emigrate to peripheral tissues, including the spleen, liver, lung, and other organs (1). NK cells express a variety of adhesion molecules that may direct this migration (2). These molecules include integrins LFA-1/αLβ2, Mac-1/αMβ2, very late antigen (VLA)-4/α4β1, and VLA-2/α2β1. By using in vitro migration assays and blocking monoclonal antibodies, LFA-1 and VLA-4 have been reported to mediate the adhesion of NK cells to resting or IL-1–activated human umbilical vein endothelial cells (3, 4). The interaction between VLA-4 and vascular cell adhesion molecule–1 was shown to mediate the recruitment of NK cells to the liver and lung, as well as infiltration into an established subcutaneous melanoma, after activation by poly-I:C and IL-12 (5). In addition, LFA-1 mediates strong binding of NK cells to target cells, thereby permitting efficient target lysis, as shown in studies using CD11a/αL-deficient mice (6). Recently, the interaction between LFA-1 and intercellular adhesion molecule–1 has been shown to be sufficient to initiate NK cell activation and cytolysis of human intercellular adhesion molecule–1-transfected Drosophila SC2 cells (7).
In addition to integrins, NK cells also express L-selectin and selectin ligands that might be involved in adhesion and migration. L-selectin initiates the tethering and rolling of leukocytes over endothelial cells, the first step in a cascade toward the recruitment of leukocytes to inflammatory sites. The essential role of L-selectin in naive T lymphocyte homing to secondary lymphoid organs has been well documented (for reviews see references 8–10). L-selectin expression has been reported on resting human CD56bright NK cells, which efficiently bind peripheral lymph node (PLN) high endothelial venules in vitro, and this binding is blocked by an anti–L-selectin mAb (11). Although it is generally accepted that CD56bright NK cells develop in bone marrow and traffic to lymph nodes through the circulation, a human CD34dimCD45RA+ hematopoietic precursor cell has been identified within lymph nodes that differentiates into CD56bright NK cells when stimulated in vitro by IL-2, IL-15, or by cocultured, activated T cells (12). Both CD56bright NK cells and CD34dimCD45RA+ precursors express L-selectin along with other adhesion molecules, suggesting that L-selectin might mediate this trafficking. Sialyl Lewis x oligosaccharides, prototypic ligands for E-, L-, and P-selectins, are also present on both resting and IL-2–activated human NK cells (13). In vitro rolling experiments using blocking antibodies revealed that PSGL-1 expressed on human NK cells was essential for the binding to and rolling over Chinese hamster ovary cells expressing P-selectin, whereas sialyl Lewis x oligosaccharides on NK cells mediated the rolling over E-selectin–expressing Chinese hamster ovary transfectants (13). In addition, human NK cells isolated from peripheral blood were able to tether and roll on immobilized L-selectin under flow conditions (14). On mouse NK cells, however, the expression of sialyl Lewis x oligosaccharides has not been reported.
NK cells play a critical role in tumor immunosurveillance (15–17). They express both activating and inhibitory receptors, and the integration of signals derived from these two receptor types determines the outcome (i.e., to kill or not to kill target cells) (18). Once activated, NK cells release granzymes and perforin, thereby eliminating transformed cells. In addition, NK cells secrete IFN-γ, which plays critical roles in antiviral defense as well as in tumor surveillance (19, 20). In humans, these two functions can be ascribed to two distinct subsets (21–24): (a) CD56dim NK cells are predominantly distributed in the spleen and peripheral blood and are strongly cytolytic, but have less cytokine production activity; and (b) CD56bright NK cells are enriched in lymph nodes and express only low levels of perforin, but secrete abundant IFN-γ when activated by IL-2 and IL-12 or by dendritic cells, as shown in in vitro coculture experiments (22). A low frequency of NK cells is also found in mouse lymph nodes (25), but the mechanism underling NK cell trafficking to lymph nodes is not fully understood. A recent report showed that mouse NK cells were recruited to lymph nodes in a CXCR3- but not CCR7-dependent manner, and that injection of an anti–L-selectin antibody greatly reduced this recruitment (25). Whether or not mouse lymph node NK cells are a phenotypically or functionally distinct subset of NK cells, as was found for human lymph node NK cells, remains unknown.
Here, we show that a subset of NK cells selectively traffics to lymph nodes in mice, and that NK cells are recruited to regional lymph nodes by stimulation in vivo with complete Freund's adjuvant or by metastatic B16 melanoma cells. By using L-selectin–deficient and L-selectin ligand–deficient mice, we demonstrate that NK cell migration to lymph nodes under both resting and activated states is facilitated by L-selectin expressed on NK cells, as well as by L-selectin ligands expressed on endothelial cells. Defects in this recruitment results in aggressive tumor formation in lymph nodes.
Results
Distinct NK cell subsets in mouse lymph nodes
Single cell suspensions of PLNs and mesenteric lymph nodes (MLNs) from C57BL/6 mice were analyzed for the presence of CD3−NK1.1+ lymphocytes, which are the prototypic phenotype of NK cells in C57BL/6 mice (Fig. 1 A). NK cells represented an average of 0.5% of total lymphocytes in PLNs (pool of inguinal, axillary, brachial, and cervical nodes) and in MLNs. NK cells comprised ∼3.8 and 8.5% of total lymphocytes in the spleen and the peripheral blood, respectively.
Figure 1.

Antigenic phenotype and function of NK cells in lymph nodes. (A) NK cells in mouse lymph nodes. Single cell suspensions were prepared from peripheral lymph nodes (PLN), mesenteric lymph nodes (MLN), spleen, and peripheral blood of wild-type C57BL/6 mice. The frequency of CD3−NK1.1+ cells as a percentage of total lymphocytes (set by an electronic gate based on the light scattering profile) represents the average calculated based on the analysis of 25 mice. (B) Secretion of IFN-γ by splenic and lymph node NK cells. The numbers inside the dot plots, which were gated on CD3−NK1.1+ cells, indicate the percentage of NK cells that produced IFN-γ. One representative experiment out of three is shown. (C) Cytolysis of YAC-1 and RMA/S tumors by lymph node (•) and splenic (Δ) NK cells. Lymph node and splenic NK cells were isolated from 10 C57BL/6 mice that received an i.p. injection of 200 μg of poly-I:C 1 d earlier. One representative experiment out of two is shown.
We further found that NK cells in lymph nodes are phenotypically distinct from those in the spleen or blood (Table I). As in the spleen and blood, ∼15% of the CD3−NK1.1+ cells in lymph nodes expressed the Ly49A receptor. The percentage of NK cells that expressed NKG2A/C/E (∼45%) was virtually the same in lymph nodes, spleen, and blood. Alternately, CD3−NK1.1+ cells that coexpressed Ly49C/I were less abundant in the PLN and MLN than in the spleen and blood. Similarly, fewer Ly49D- or Ly49H-positive NK cells were present in the PLN and MLN compared with those in the spleen and blood. Virtually all NK cells expressed NKG2D receptor in these four tissues, and the majority of NK cells expressed L-selectin. Using mAb KM93 (26), we detected the expression of sialyl Lewis x oligosaccharides on 15.9 and 25.6% of CD3−NK1.1+ cells in the PLN and MLN, respectively. Comparing the CD3−NK1.1+ cells in the spleen and peripheral blood, 7 and 5.2% reacted with the KM93 mAb, respectively.
Table I.
Percentage of CD3−NK1.1+ cells that also express Ly49A, Ly49C/I, Ly49D, Ly49H, NKG2A/C/E, NKG2D, sialyl Lewis x oligosaccharides, and L-selectin
| CD3−NK1.1+ | MLN | PLN | Spleen | Blood |
|---|---|---|---|---|
| % | % | % | % | |
| Ly49A+ | 15.4 ± 1.1 | 16.8 ± 0.9 | 15.5 ± 0.4 | 13.2 ± 0.1 |
| Ly49C/I+ | 26.2 ± 3.6a | 29.6 ± 4.9a | 50.3 ± 3.5 | 63.8 ± 3.9 |
| Ly49D+ | 33.6 ± 4.1a | 37.6 ± 5.6a | 50.7 ± 3.2 | 52.3 ± 1.6 |
| Ly49H+ | 33.8 ± 0.8a | 40.3 ± 1.3a | 50.5 ± 0.9 | 53.0 ± 1.0 |
| NKG2A/C/E+ | 50.6 ± 3.7 | 45.2 ± 2.3 | 43.4 ± 1.2 | 44.9 ± 1.7 |
| NKG2D+ | 100 | 100 | 100 | 100 |
| sialyl Lewis x+ | 25.6 ± 3.9a | 15.9 ± 0.7a | 7.0 ± 0.4 | 5.2 ± 0.9 |
| L-selectin+ | 72.2 ± 4.2 | 81.0 ± 3.2 | 87.5 ± 1.9 | 92.2 ± 1.4 |
Data represent the means and SD of at least 10 mice obtained from at least four independent experiments. The percentage of sialyl Lewis x–expressing cells among CD3−NK1.1+ cells was determined after removing macrophages and B cells that adhere to nylon fibers.
P < 0.001 relative to blood samples.
IFN-γ secretion and cytolysis of tumors by lymph node NK cells
The presence of a distinct NK cell subset in lymph nodes suggested that they might have substantial functional differences. Therefore, we tested IFN-γ production and cytotoxicity of lymph node NK cells. Consistent with a recent report (25), ∼17% (16.8 ± 5.8) of lymph node NK cells secreted IFN-γ in response to LPS stimulation (Fig. 1 B). In contrast, after LPS stimulation, >70% (74.5 ± 5.7) of splenic NK cells were positive for intracellular IFN-γ staining. Moreover, we found that freshly isolated mouse lymph node NK cells had cytolytic activity toward YAC-1 and RMA/S tumors equivalent to splenic NK cells (Fig. 1 C). These results indicate that splenic and lymph node–derived NK cells have equivalent lytic activity, but splenic NK cells produce more INF-γ in mice stimulated with endotoxin.
L-selectin mediates the recruitment of NK cells to resting lymph nodes
Because the majority of mouse NK cells express L-selectin (Table I), a well-characterized adhesion molecule that mediates T and B lymphocyte homing to lymph nodes, we tested if it is also required for NK cell trafficking to lymph nodes. To compare the migration of wild-type and L-selectin–deficient (L-selectin−/−) NK cells, we labeled wild-type (C57BL/6 or Rag-1 −/−) NK cells with the vital dye (SNARF-1) carboxylic acid acetate succinimidyl ester and L-selectin−/− NK cells with the vital dye carboxyfluorescein diacetate succinimidyl ester (CFSE), respectively, or vice versa. A mixture containing an approximately equal number of each cell type was injected into the tail vein of a C57BL/6 mouse. After 3, 18, or 42 h, recipient tissues were collected and stained with PE-labeled anti-NK1.1 antibody. NK1.1+, NK1.1+CFSE+, and NK1.1+ SNARF-1+ cells were readily detected by flow cytometry (Fig. 2 A). The relative efficiency of migration to peripheral organs was expressed as the ratio of L-selectin−/− NK cells over wild-type NK cells, which was further normalized by this ratio in peripheral blood because it serves as a reservoir for NK cells to enter different organs. At all time points tested, L-selectin−/− NK cells were as abundant as wild-type NK cells in the spleen, liver, lung, and bone marrow (Fig. 2, B and C). In contrast, very few L-selectin−/− NK cells entered into PLNs (ratio = 0.04–0.12) or MLNs (ratio = 0.18–0.35). Fluorescent labeling apparently had no effect on NK cell migration because similar results were also obtained when fluorescent dyes were switched in labeling of cells (Fig. 2, B and C). The effects of fluorescent dyes in NK cell migration could be further ruled out by the observation that CFSE-labeled Core2GnT-I–deficient (27) NK cells and SNARF-1–labeled wild-type NK cells had the same distribution in all tissues tested (unpublished data). The results indicate that L-selectin on NK cells facilitates the recruitment of NK cells into resting lymph nodes.
Figure 2.
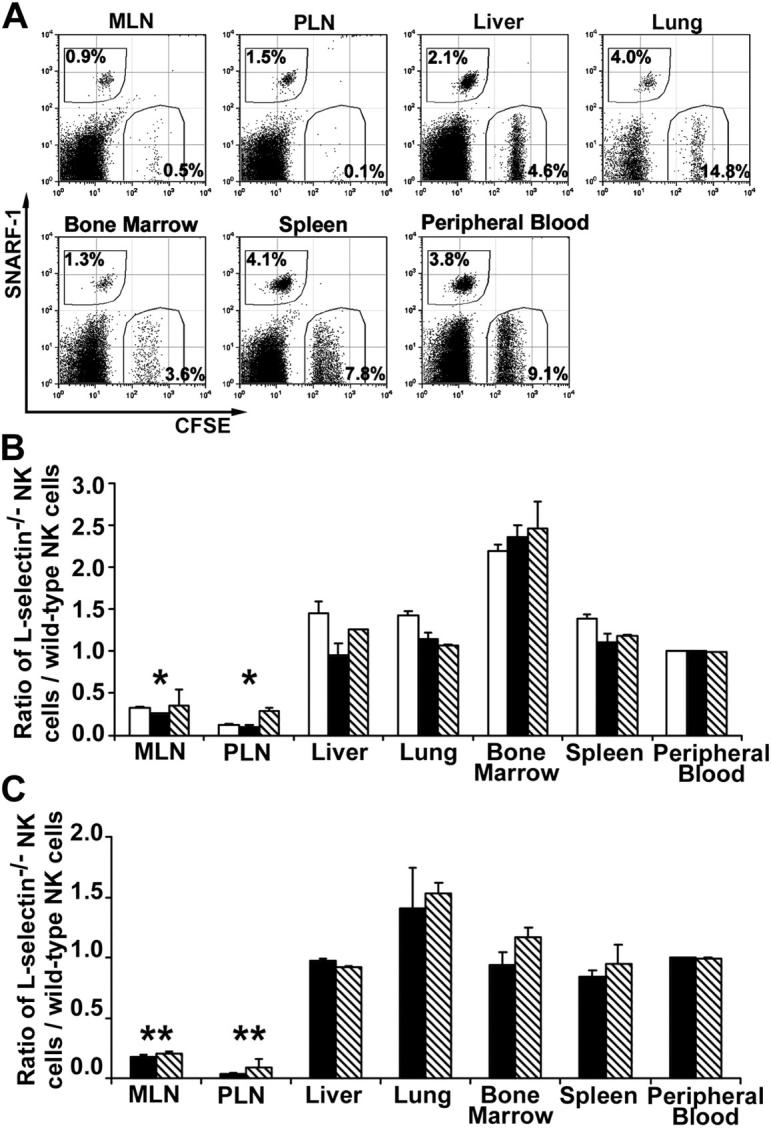
L-selectin–mediated NK cell trafficking to lymph nodes. (A) Wild-type and L-selectin–deficient NK cells were labeled with SNARF-1 and CFSE, respectively, or vice versa. A mixture was injected i.v. into a wild-type C57BL/6 mouse. After 3, 18, or 42 h, various tissues were harvested and single cell suspensions were stained with anti-NK1.1 mAb and subjected to flow cytometric analysis. Only NK1.1+ (gated) lymphocytes are shown 42 h after the transfer. The numbers in dot plots indicate the percentages of transferred NK cells among all NK1.1+ cells. (B) Wild-type NK cells were labeled with CFSE and L-selectin–deficient NK cells were labeled with SNARF-1. The efficiency of NK cells migrating into the tissues was expressed by the ratio of L-selectin–deficient NK cells to wild-type NK cells. White bars, black bars, and diagonally striped bars represent the ratios of 3, 18, and 42 h after the transfer, respectively. The data shown represent the average ± SD of two independent experiments. (C) Wild-type NK cells were labeled with SNARF-1 and L-selectin–deficient NK cells were labeled with CFSE. Black and diagonally striped bars represent the ratios of 18 and 42 h after the transfer, respectively. The data shown represent the average ± SD of three independent experiments. *P < 0.01 and ** P < 0.001 versus the peripheral blood.
L-selectin mediates the recruitment of NK cells to activated regional lymph nodes
We then investigated whether L-selectin is involved in the trafficking of NK cells into lymph nodes during immune stimulation in which a rapid recruitment of lymphocytes occurs. Immediately after i.v. injection of fluorescent-labeled wild-type and L-selectin−/− NK cells as described before, C57BL/6 mice were injected with CFA into the right hind footpad. The right (draining) popliteal lymph nodes were collected 22 or 42 h later. As a control, we also collected the left popliteal lymph nodes. We found that significantly fewer adoptively transferred L-selectin−/− NK cells were recruited to the draining nodes compared with wild-type NK cells (Fig. 3 A). The efficiency of L-selectin−/− NK cell recruitment was only ∼27 and 46% (P < 0.001) of wild-type NK cells at 22 or 42 h after CFA stimulation, respectively. It is noteworthy that only ∼0.1% of adoptively transferred wild-type NK cells were recruited to the draining lymph node, and the great majority remained in the spleen and the peripheral blood.
Figure 3.
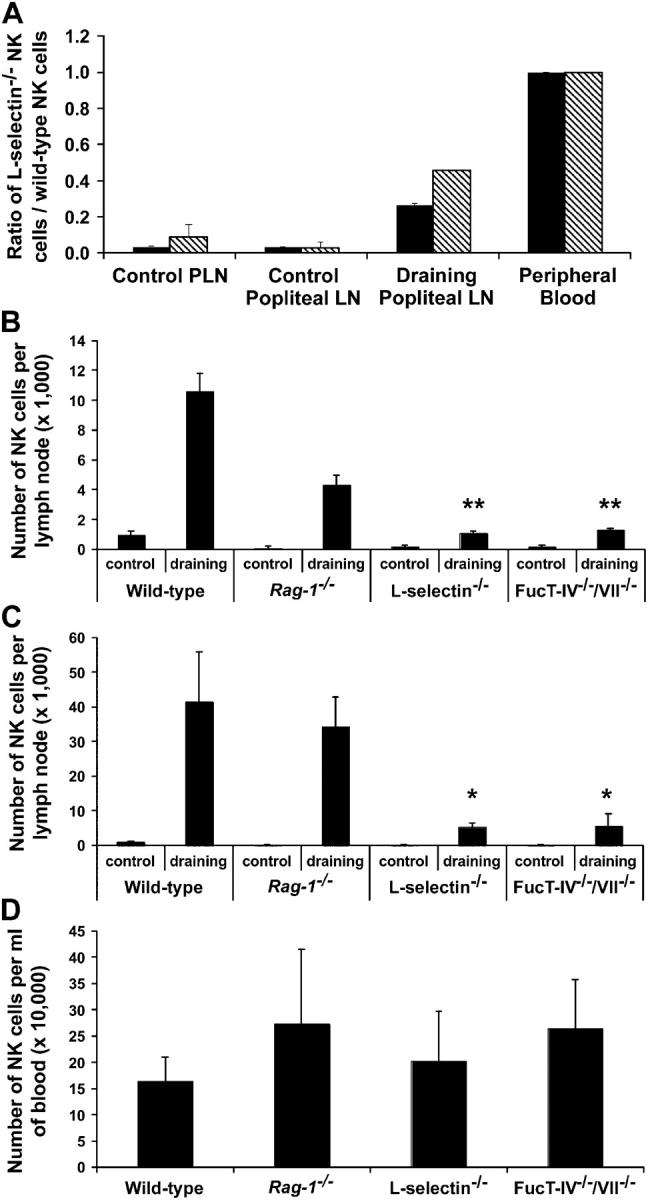
CFA-induced recruitment of NK cells. (A) A mixture of SNARF-1–labeled wild-type NK cells and CFSE-labeled L-selectin−/− NK cells was injected i.v. into wild-type C57BL/6 mice. Immediately after the i.v. injection, the mice received an injection of 50 μl CFA into the right hind footpad. Recipient popliteal lymph nodes were collected and analyzed for the presence of the adoptively transferred NK cells at 22 and 42 h after CFA stimulation. The efficiency of NK cells migrating into lymph nodes was expressed by the ratio of L-selectin−/− NK cells to wild-type NK cells. Black bars and diagonally striped bars represent the ratios of 22 and 42 h after the transfer, respectively. The data shown represent the average ± SD of two independent experiments. PLN refers to a pool of inguinal, axillary, and cervical nodes, representing unstimulated nodes. (B and C) 50 μl CFA was injected into the right hind footpads of wild-type and mutant mice and the absolute number of NK cells that were recruited to the right (draining) or left (control) popliteal lymph nodes was determined at 24 h (B) or 48 h (C) after CFA stimulation. (D) The number of NK cells was also determined in the peripheral blood (per milliliter). The data are represented as the mean and SD of at least five mice for each group determined in two to six separate experiments. *P < 0.02 and ** P < 0.001 versus wild-type draining lymph nodes.
We further examined the absolute number of NK cells that can be recruited to the draining lymph nodes in CFA-stimulated L-selectin−/− mice. In wild-type animals, CFA induced an 11-fold increase in the absolute number of CD3−NK1.1+ cells in the draining lymph node at ∼24 h (Fig. 3 B) and a 41-fold increase at ∼48 h (Fig. 3 C). Without stimulation, L-selectin−/− popliteal lymph nodes had significantly fewer NK cells (200 ± 60 per node), compared with wild-type popliteal lymph nodes (1,000 ± 230 per node). Notably, the number of NK cells recruited to popliteal lymph nodes 48 h after CFA stimulation was dramatically reduced in L-selectin−/− mice (5,300 ± 1,070 per node) compared with wild-type mice (42,000 ± 14,000 per node), although L-selectin−/− mice had a similar number of NK cells in the blood (203,000 ± 94,000/ml) compared with wild-type mice (164,000 ± 46,000/ml) (Fig. 3 D). These results indicate that reduced NK cell recruitment to lymph nodes is not due to the low availability of NK cells in the circulation, but rather that it is caused by the absence of L-selectin.
L-selectin ligands mediate the recruitment of NK cells to activated regional lymph nodes
We next tested whether L-selectin ligands expressed on endothelial cells are also involved in NK cell migration into lymph nodes. L-selectin ligands, sialyl Lewis x, and its sulfated derivatives are synthesized by concerted actions of fucosyltransferase-IV and -VII, a sulfotransferase (LSST), and α2,3-sialyltransferase (28). In previous studies, it has been demonstrated that mice doubly defective in fucosyltransferase-IV and -VII (FucT-IV−/−/VII−/−) did not express L-selectin ligands on high endothelial venules of PLNs (29). As expected, we found that the total number of NK cells per node was significantly less in the mutant mice at both 24 h (P < 0.001) and 48 h (P < 0.02) after CFA stimulation (Fig. 3, B and C). Because FucT-IV−/−/VII−/−-deficient NK cells migrated normally to lymph nodes when adoptively transferred to a wild-type recipient (unpublished data), our results suggest that fucosyltransferase-IV– and -VII–generated L-selectin ligands in lymph nodes are crucial for the recruitment of NK cells.
Compared with wild-type C57BL/6 mice, lymph nodes are smaller in both L-selectin−/− and FucT-IV−/−/VII−/− mutants; however, the overall nodal architecture is normal. To examine whether the size of lymph nodes affects NK cell recruitment, we challenged Rag-1 −/− mice, which also have very small lymph nodes due to the lack of B and T cells, with CFA. We found that the number of NK cells in unstimulated popliteal lymph nodes (150 ± 100 per node) in Rag-1 −/− mice was approximately sevenfold fewer than that in unstimulated nodes in wild-type C57BL/6 mice (Fig. 3, B and C). However, 48 h after CFA stimulation, the number of NK cells in the Rag-1 −/− mice was dramatically increased to a level similar to wild-type C57BL/6 mice (Fig. 3 C), indicating that NK cells could be efficiently recruited to regional lymph nodes in Rag-1 −/− mice, and that this recruitment does not require the presence of B or T cells.
NK cells control tumor formation in lymph nodes
Because NK cells are known to eliminate certain tumors, we examined whether NK cells could be recruited into lymph nodes of tumor-bearing hosts. To address this question, we injected 2 × 106 B16-F10 melanoma cells, which aggressively form tumors in syngeneic mice (30), into the right hind footpad and monitored the development of tumor foci and the number of tumor cells and NK cells in the draining popliteal lymph nodes 10 d later. To confirm that NK cells are indeed involved in the control of tumor formation in lymph nodes, we selectively depleted NK cells with anti-NK1.1 monoclonal antibody in C57BL/6 mice and compared the growth of tumors in lymph nodes. As controls, mice were injected i.p. with mouse IgG purified from pooled normal serum. Very few NK1.1+ cells were detected in the spleens and lymph nodes of mice treated with anti-NK1.1 antibody, whereas control IgG treatment had no effect on the NK cell population (Fig. 4 A). Small tumor foci were found in control IgG-treated mice (Fig. 4 B), whereas more extensive tumors were found in anti-NK1.1–treated lymph nodes (Fig. 4 C). The median of relative tumor burden, assessed by the size of tumor mass, in control IgG-treated animals was 7%, whereas it was 40% in anti-NK1.1–treated animals (Fig. 4 D). Consistently, the number of tumor cells in the draining lymph nodes of anti-NK1.1–treated mice was significantly higher than control IgG-treated lymph nodes (P = 0.02; unpublished data). These results indicate a critical role of NK cells in the inhibition of tumor metastasis. However, we presently do not know which types of NK cells are selectively recruited by B16 tumors because the frequency of Ly49D- and Ly49H-expressing NK cell subsets was similar between tumor-free and tumor-bearing lymph nodes (unpublished data).
Figure 4.
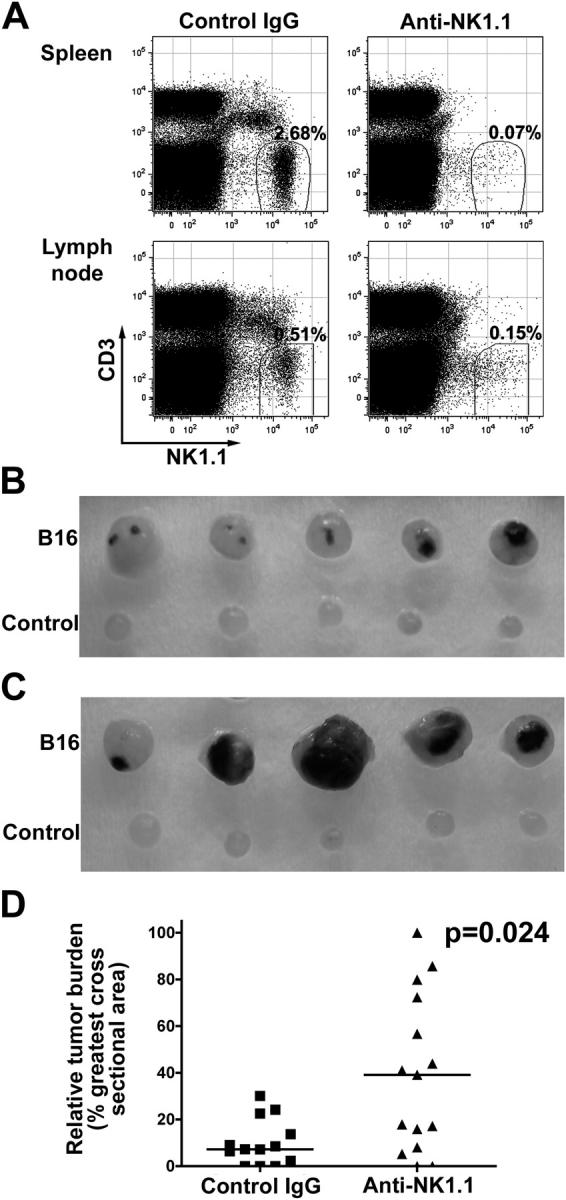
Tumor metastasis in NK cell–depleted C57BL/6 mice. (A) NK cells were depleted by repeated i.p. injection of anti-NK1.1 (PK136) ascites, and B16-F10 metastasis was monitored as described in Materials and methods. Splenocytes and lymph node lymphocytes from each mouse were stained for CD3 and NK1.1. Significantly fewer NK1.1+ cells were detected in the anti-NK1.1–treated mice compared with the control IgG-treated mice because of either depletion of the NK1.1+ cells or coating of the cells with the anti-NK1.1 antibody in vivo. The result for one representative mouse is shown. The numbers in the dot plots represent the percentages of CD3−NK1.1+ cells detected among all lymphocytes. (B and C) Larger tumor foci were detected in anti-NK1.1–treated lymph nodes (C), compared with control IgG-treated lymph nodes (B). B16 refers to B16-F10 melanoma-draining popliteal lymph nodes, whereas Control refers to the other popliteal lymph node. 13 mice were treated with control IgG, and 15 mice were injected with anti-NK1.1 antibody. Only five representative mice from each group are shown. (D) The relative tumor burden for each group. Symbols represent individual mice. Bars indicate the median values.
To evaluate the potential contribution of NK/T cells, we performed similar experiments in Rag-1 −/− mice, which lack T, B, and NK/T cells. Aggressive tumors were formed in anti-NK1.1–treated, but not in control IgG-treated, lymph nodes of Rag-1 −/− mice (Fig. 5, A and B). Anti-NK1.1 treatment resulted in a dramatic reduction in number of NK cells that were recruited to the B16-bearing lymph nodes (median = 100 NK cells), compared with a median of 15,600 NK cells in control IgG-treated lymph nodes (Fig. 5 C). As a consequence, the relative tumor burden was significantly (P = 0.001) higher in anti-NK1.1–treated lymph nodes (median = 76%) than in control IgG-treated lymph nodes (median = 7%) (Fig. 5 D). The results indicate that NK cells are the major immune cells to inhibit tumor metastasis under these conditions, most likely by killing B16 tumor cells after metastasis to lymph nodes.
Figure 5.
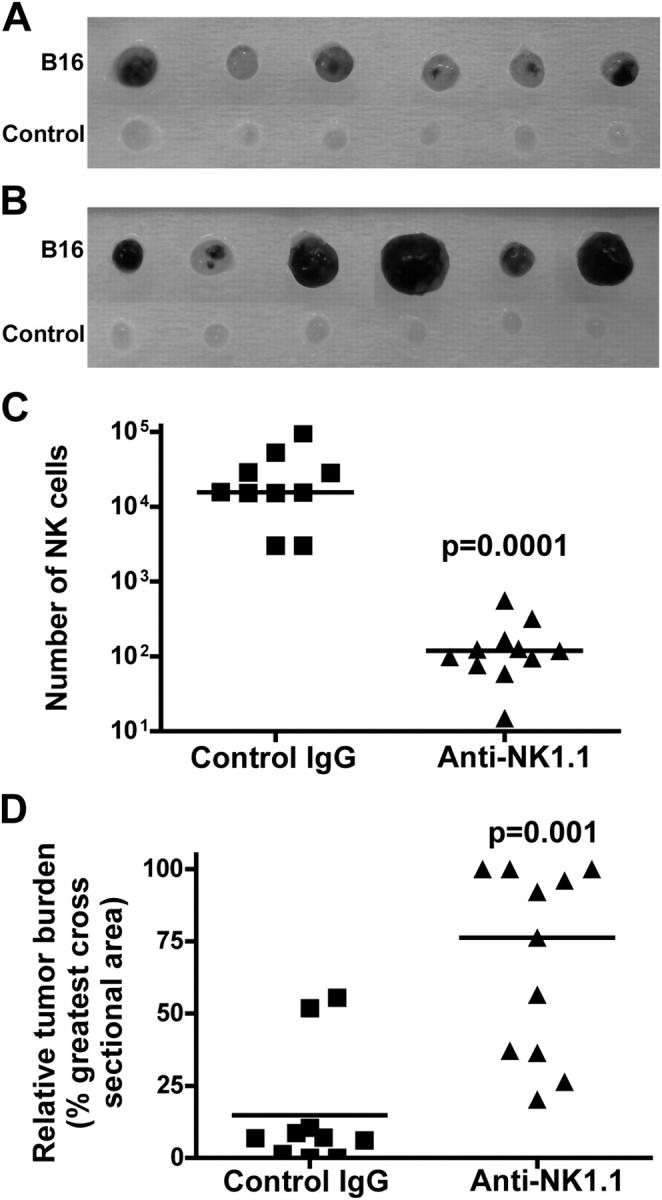
Tumor metastasis in NK cell-depleted Rag-1 −/− mice. (A and B) See Fig. 4 for details. The development of tumor foci in control IgG-treated lymph nodes (A) and anti-NK1.1–treated lymph nodes (B). 6 out of 11 anti-NK1.1–treated and 6 out of 10 control IgG-treated lymph nodes are shown. (C) The number of NK cells and (D) the relative tumor burden in the popliteal lymph nodes bearing B16 tumors. Symbols in C and D represent individual mice. Bars in C and D indicate the median values. p-values are relative to control IgG-treated mice.
L-selectin–mediated NK cell recruitment suppresses tumor formation in lymph nodes
Having established that NK cells inhibited B16 melanoma metastasis to lymph nodes, we evaluated the role of L-selectin and L-selectin ligands in the control of tumor formation. In the majority of wild-type mice, we found no tumors or only small tumor foci in the lymph nodes (Fig. 6 A). Similar results were also found in Rag-1 −/− mice (Fig. 6 B). In contrast, the popliteal lymph nodes were full of tumors in the majority of L-selectin−/− mice (Fig. 6 C) and in FucT-IV−/−/ VII−/− mutant mice (Fig. 6 D). The number of NK cells recruited to the tumor-bearing nodes was significantly less in L-selectin−/− (median = 2,000) (P < 0.001) and FucT-IV−/−/ VII−/− (median = 4,600) (P = 0.001) mutant mice compared with wild-type (median = 8,100) and Rag-1 −/− (median = 15,000) animals (Fig. 6 E). Alternately, the relative tumor burden was significantly higher in L-selectin−/− (median = 56%, P < 0.001) and FucT-IV−/−/VII−/− (median = 88%, P < 0.001) mutant mice compared with wild-type (median = 6%) and Rag-1 −/− (median = 3%) animals (Fig. 6 F). The difference in tumor burden between wild-type and L-selectin−/− or FucT-IV−/−/VII−/− mice was statistically significant (P < 0.001).
Figure 6.

Tumor metastasis in L-selectin−/− and FucT-IV−/−/VII−/− mice. (A–D) B16-F10 melanoma cells were inoculated s.c. into the right hind footpads and the development of tumor foci was examined 10 d later. Tumor formation was examined in 22 C57BL/6 (A), 11 Rag-1 −/− (B), 26 L-selectin−/− (C), and 14 FucT-IV−/−/VII−/− (D) mice. Representative results for six mice from each group are shown. (E) The number of NK cells in the tumor-bearing popliteal lymph nodes. Bars indicate the median values. (F) The relative tumor burden in the popliteal lymph nodes bearing B16 tumors. Bars indicate the median values. (G) In vitro cytotoxicity of L-selectin−/− (•) and wild-type (Δ) NK cells against B16-F10 tumors. One out of four independent experiments is shown. p-values are relative to C57BL/6 mice.
To exclude the possibility that L-selectin−/− NK cells are intrinsically deficient for tumor recognition, we performed in vitro cytotoxicity assays to determine the lytic activity of L-selectin−/− NK cells. Our result revealed no difference between wild-type versus L-selectin−/− NK cells against B16 melanoma in vitro (Fig. 6 G). Collectively, these results indicate that L-selectin–mediated interactions facilitate the mobilization of NK cells to regional lymph nodes in response to tumor challenge, and a defect in this trafficking leads to aggressive tumor formation in lymph nodes.
Adoptively transferred NK cells reduce tumor metastasis in L-selectin–deficient mice
To further demonstrate that L-selectin on NK cells is indeed crucial for tumor suppression, we examined whether adoptively transferred wild-type NK cells could reduce tumor metastasis in L-selectin–deficient mice. Consistent with the aforementioned results, extensive tumors were found in the lymph nodes of L-selectin–deficient mice (Fig. 7, A and C). In contrast, tumor formation was substantially reduced when wild-type NK cells were adoptively transferred to L-selectin–deficient mice (Fig. 7, B and C). However, statistical analysis of the medians of the relative tumor burdens was not significant (P = 0.11), most likely due to the fact that only a small fraction (< 0.1%) of intravenously introduced NK cells were recruited to the lymph nodes.
Figure 7.
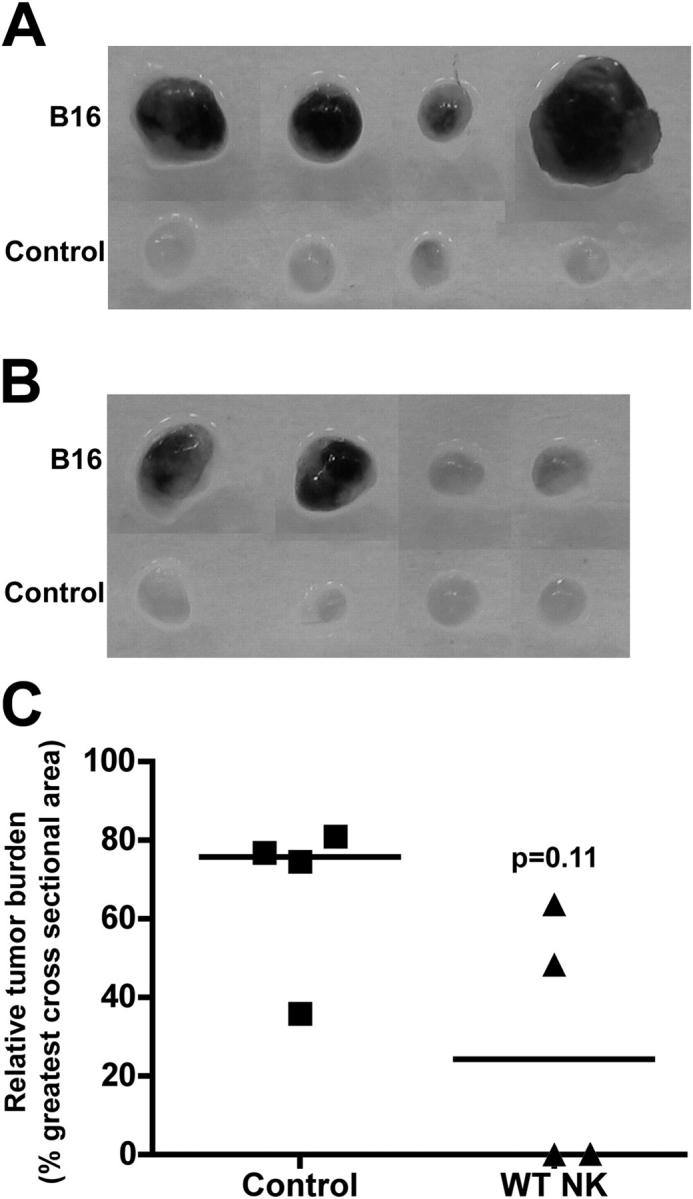
Reduced tumor formation by adoptively transferred wild-type NK cells in L-selectin–deficient mice. (A and B) B16-F10 melanoma cells were inoculated s.c. into the right hind footpads, and the development of tumor foci was examined 10 d later. The development of tumor foci in L-selectin−/− mice (A) and in L-selectin−/− mice reconstituted with wild-type NK cells (B), and the relative tumor burden in the popliteal lymph nodes bearing B16 tumors (C). Four mice were tested for each group. Horizontal bars in C indicate the median values.
Discussion
NK cells were originally discovered because of their ability to kill certain tumor cells in vitro without prior sensitization (31). The involvement of NK cells in antitumor immunity in vivo was demonstrated subsequently in several mouse tumor models by using NK cell–depleting antibodies and genetically modified mutant mice (17). In these models, the involvement of NK cells was demonstrated by suppression of spontaneous or chemically induced primary tumors (32–35), or suppression of intraperitoneally or subcutaneously inoculated transplantable tumors (36–39). In models of metastatic cancer, tumors were often injected i.v. and metastases were monitored in the liver or lung (40, 41). Here, we show that NK cells are also responsible for the control of tumor metastasis into lymph nodes.
This work and a previous study (25) indicated that a small number of NK cells are constitutively present in lymph nodes in mice. We further showed that the cell surface antigenic phenotype of lymph node NK cells is different from splenic or blood NK cells. In agreement with previous reports (22, 25), we found that mouse lymph node NK cells are capable of producing IFN-γ. However, unlike human lymph node NK cells that require long-term (1 wk) stimulation by IL-2 to become cytolytic (21), freshly isolated mouse lymph node NK cells (poly-I:C-stimulated for 24 h in vivo) were as effective as splenic NK cells in cytolysis of YAC-1 and RMA/S tumors.
Lymphocyte trafficking to lymph nodes, in particular to the PLNs, depends on L-selectin (8, 9). Lymphocyte migration into PLNs, including popliteal lymph nodes, is almost completely abolished in fucosyltransferase-IV and -VII double-deficient mice due to the loss of L-selectin ligands on the PLNs (29). We showed here that L-selectin and fucosyltransferase-IV– and -VII–generated L-selectin ligands are also crucial for the recruitment of NK cells to lymph nodes at both resting and activated states. These results indicate that NK cells are recruited to the PLNs or MLNs through the same mechanisms responsible for the recruitment of T and B lymphocytes. A very low number of NK cells were still present in resting lymph nodes of L-selectin–deficient, E-, L-, and P-selectin triple-deficient (unpublished data), or fucosyltransferase-IV and -VII double-deficient mice, suggesting that molecules other than selectins are also involved. Consistent with prior reports (5, 42), we found that the migration of NK cells to the liver and lung does not require L-selectin. More recently, P-selectin on thymic epithelial cells was shown to interact with ligand PSGL-1 on lymphoid progenitors, and this interaction regulated the recruitment of T cell progenitors to the adult thymus (43). Together, these observations suggest that different carbohydrate–protein interactions may function in various recruitment systems.
Rapid recruitment of lymphocytes is crucial to mount an efficient immune response. In lymph nodes undergoing an immune response, lymphocytes accumulate dramatically primarily due to the trafficking of cells from peripheral blood (44). We found here that NK cells were also rapidly recruited to lymph nodes in response to stimulation with CFA and by the presence of metastatic tumor cells, and this recruitment was facilitated by L-selectin–mediated interactions. NK cells recruited to lymph nodes by treatment with adjuvants have been shown to provide IFN-γ that is in some cases necessary for the priming of TH1 cells (25). In addition, CFA immunization augments NK cell–mediated cytotoxicity and induces the secretion of IFN-γ (45). Recently, CFA-stimulated NK cells have been shown to prevent diabetes in NOD mice (46). The recruitment of NK cells to regional lymph nodes by infiltrating tumors implied that they might play a role in the control of tumor metastasis and growth. Indeed, NK cells were recruited to the peritoneal cavity by MHC class I–deficient tumor cells and tumors expressing NKG2D ligands, and then became activated at this site (36, 47, 48). The requirement of NK cells in tumor immunity has also been demonstrated in the rejection of RMA/S (a T cell lymphoma), EL4-S3 (a β2-microglobulin–deficient thymoma), and RM-1 (a prostate carcinoma) (38, 39). However, other immune cells, including NK/T and CD8+ T cells, also reject tumors, depending on the model used and the tumor dose inoculated (36, 39).
In the present study, we found that a decrease in NK cell number correlated with an increase in tumor burden in lymph nodes of L-selectin−/− and FucT-IV−/−/VII−/− mice. Because L-selectin−/− NK cells effectively lysed tumors in vitro, the observed aggressive tumor formation is therefore likely attributable to defects in the recruitment of NK cells, rather than their lytic capacity. Reduced tumor formation by adoptive transfer of wild-type NK cells into L-selectin–deficient host mice provides direct evidence for the role of L-selectin–mediated NK cell recruitment. In our experimental model, T and NK/T lymphocytes do not seem to significantly contribute to the inhibition of B16 growth in lymph nodes because Rag-1 −/− mice suppressed tumor formation equivalently to wild-type animals. A predominant NK cell contribution was further demonstrated by more extensive tumor growth in NK cell–depleted C57BL/6 and Rag-1 −/− mice. Together, our findings show a mechanism by which NK cells are recruited to regional lymph nodes and may provide a basis for novel approaches to treat metastatic malignancies.
MATERIALS AND METHODS
Mice.
Wild-type C57BL/6 mice were purchased from Harlan Sprague Dawley, Inc. FucT-IV−/−/VII−/− mice were obtained from the Functional Glycomics core facility at The Scripps Research Institute and were generated as described previously (29). L-selectin−/− and Rag-1 −/− mice were purchased from The Jackson Laboratory. Mice were maintained according to National Institutes of Health (NIH) and other institutional regulations and guidelines.
Lymphoid organ cell isolation.
PLNs (cervical, brachial, axillary, and inguinal) and MLNs were collected in RPMI 1640 medium (Irvine Scientific) supplemented with 5% FCS, 10−5 M β-mercaptoethanol (β-ME, Sigma-Aldrich), 100 μg/ml streptomycin, and 100 U/ml of penicillin (Invitrogen), 1× sodium pyruvate, and 1× nonessential amino acids. Single cell suspensions were prepared by mechanical dissociation of lymph nodes and filtered through a 70-μm cell strainer (BD Labware). Splenocytes and peripheral blood were treated with 17 mM Tris-HCl buffer (pH 7.4) containing 0.83% NH4Cl to lyse red blood cells. For mAb KM93 staining, single cell suspensions were further passed through a nylon fiber (Wako) column to remove macrophages and B cells (49).
Antibodies and flow cytometry.
Single cell suspensions were first blocked with 4 μg/ml of anti-FcR mAb 2.4G2 (mouse BD Fc Block; BD Biosciences) and stained with the following antibodies (BD Biosciences): FITC-conjugated anti-CD3 (1A17) or peridin chlorophyll protein (PerCP)–conjugated or allophycocyanin-conjugated anti-CD3ɛ (145-2C11), PE-conjugated anti-NK1.1 (PK136), PE-conjugated DX5 mAb, FITC-conjugated anti-Ly49A (A1), FITC-conjugated anti-Ly49C/I (5E6), FITC-conjugated anti-Ly49D (4E5), FITC-conjugated anti-NKG2A/C/E (20d5), and FITC-conjugated anti–L-selectin (Mel14). PE-conjugated anti-NKG2D (CX5) was purchased from eBioscience. FITC-conjugated anti-Ly49H (3D10) was provided by W. Yokoyama (Washington University, St. Louis, MO). To determine the expression of sialyl Lewis x oligosaccharides, mAb 2.4G2-treated cells were first stained with mAb KM93 (CalBiochem), followed by FITC-conjugated F(ab′)2 fragments of goat anti–mouse IgM (Cappel and ICN Biomedicals), PerCP-labeled anti-CD3ɛ, and PE-labeled anti-NK1.1. Stained cells were analyzed on a FACSort or FACScanto flow cytometer (BD Biosciences). Data were analyzed with FlowJo software.
Detection of intracellular IFN-γ.
Mice were injected i.p. with 200 μg of Escherichia coli–derived LPS (Sigma-Aldrich), and spleens and lymph nodes were harvested 24 h later. NK cells were prepared from nylon fiber nonadherent fractions and further negatively sorted by using a mouse NK cell isolation kit (Miltenyi Biotec) according to manufacturer's protocols. In brief, 10 μl of a cocktail of biotin-labeled anti-CD3, -CD14, -CD19, -CD36, and anti-IgE antibodies was added to 107 cells resuspended in 40 μl of PBS containing 0.5% BSA and 2 mM EDTA (Buffer A). After incubation for 10 min at 6–12°C, 20 μl of antibiotin-conjugated magnetic beads were added and incubated for another 15 min. Cells were washed once with the addition of 1–2 ml of Buffer A and subjected to magnetic sorting. NK cells were collected in the nonbound fraction, and cultured in RPMI 1640 medium containing 10% FCS and Golgi-Stop (4 μl/6 ml medium) (BD Biosciences) for 4 h. Cells were next stained for CD3 and NK1.1 and permeabilized with Cytofix/Cytoperm reagent (BD Biosciences). Permeabilized cells were stained with allophycocyanin-conjugated anti–IFN-γ and analyzed by flow cytometry.
Adoptive transfer.
NK cells were isolated from donor spleens as described before and resuspended in PBS with 2% FCS at 107 cells/ml. Cells were labeled either with 1 μM CFSE or 2.5 μM SNARF-1 (GE Healthcare) at 37°C for 10 min. After three washes with RPMI 1640 medium containing 10% FCS, cells were resuspended in ice-cold, serum-free RPMI 1640 medium. A mixture containing an approximately equal number of wild-type C57BL/6 and mutant NK cells (1–5 × 106) was injected i.v. (tail vein) into wild-type C57BL/6 mice. In some cases, wild-type NK cells were isolated from Rag-1 −/− spleens by passage through a nylon fiber column without further purification. After 3, 18, or 42 h, peripheral blood was collected from the posterior vena cava, and the mouse was perfused with PBS containing 10 U/ml heparin (Sigma-Aldrich). Single cell suspensions of MLNs, PLNs, spleen, and peripheral blood were prepared as described before. Bone marrow cells from one femur and one tibia were flushed with RPMI 1640 medium containing 10% FCS. The liver and lung were minced and digested with 50 U/ml collagenase (Roche Applied Sciences) and 0.01% DNase I (Sigma-Aldrich) at 37°C for 1 h. Tissue homogenates were forced through a 70-μm filter and centrifuged at 300 g for 5 min. Cell pellets were resuspended in 20 ml (for liver) or 8 ml (for lung) of 35% Percoll (Invitrogen) in PBS containing 200 U/ml heparin and overlaid onto 67% Percoll in PBS (50). Samples were centrifuged at 600 g for 20 min at room temperature without braking. Mononuclear cells were collected at the interface and washed three times with PBS before antibody staining. The frequency of adoptively transferred NK cells (NK1.1+CFSE+ or NK1.1+ SNARF-1+) in various organs of the recipients was determined by flow cytometry.
CFA stimulation.
50 μl CFA was injected s.c. into the right hind footpad. At the indicated times, single cell suspensions were prepared from the draining popliteal lymph nodes and stained with FITC-labeled anti-CD3 and PE-labeled anti-NK1.1 mAb. Left popliteal lymph nodes were also collected as unstimulated controls. The total cell number was determined by using a Cell Viability Kit (BD Biosciences).
In vitro cytotoxicity assay.
Splenic and lymph node NK cells were isolated as described before from mice that received i.p. 200 μg of poly-I:C 1 d earlier. Freshly prepared NK cells were tested for their ability to lyse YAC-1 and RMA/S tumors in a standard 6 h 51Cr-release assay as described previously (19). The cytolysis of B16 tumors by IL-2–activated L-selectin−/− NK cells, cultured in RPMI 1640 medium containing 10% FCS and 1,000 U/ml IL-2 (Roche Applied Sciences) for 5 d, was tested in a standard 4 h 51Cr-release assay.
Tumor metastasis.
The mouse melanoma cell line B16-F10 was purchased from American Type Culture Collection and cultured in DME high glucose medium (Irvine Scientific) supplemented with 10% FCS. B16-F10 cells were harvested in cell dissociation buffer (Specialty Media), washed three times with PBS, and resuspended in serum-free DME high glucose medium. 2 × 106 B16-F10 cells were inoculated s.c. into the right hind footpad. Right (tumor-draining) and left (control) popliteal lymph nodes were collected 10 d later. NK cells were identified as CD3−NK1.1+ lymphocytes. Lymph node metastases are melanotic, and occur preferentially in marginal sinuses and spread superficially to subcortical regions (51). Lymph nodes were photographed. The relative tumor burden was expressed as the percentage of tumor areas over the greatest cross-sectional area of the lymph nodes. Area was measured by using NIH ImageJ software.
To deplete NK cells, C57BL/6 or Rag-1−/− mice were injected i.p. with 500 μg of mouse control IgG (Sigma-Aldrich) or anti-NK1.1 (PK136) ascites made from nude mice (equivalent to 500 μg of mouse IgG2a) 2 d (day −2) before inoculation of B16-F10 tumors. On day 0, B16-F10 tumors (2 × 106) were inoculated s.c. at footpads. Meanwhile, mice were also given an i.p. injection of control IgG or anti-NK1.1 ascites. The antibody administration was repeated on days 3, 6, and 9. Mice were killed on day 10. Popliteal lymph nodes were removed to determine tumor formation and NK cell recruitment. It has been shown that anti-NK1.1 antibody (PK136) treatment effectively depletes NK cells (52, 53). In prior experiments, we confirmed the effectiveness of the NK cell depletion by flow cytometry, as indicated by the absence of α2β1 integrin-reacting DX5 mAb positively staining cells in the peripheral blood of anti-NK1.1 mAb-treated mice (54).
To suppress tumor formation in L-selectin–deficient mice, NK cells were isolated from 48 wild-type C57BL/6 mice as describe before, and purified NK cells (6–10 × 106 cells per mouse) were injected i.v. into 4 L-selectin–deficient mice 1 day (day 1) after B16 tumors were inoculated. Similar number of NK cells was adoptively transferred to the same mice on days 4 and 7, and the popliteal lymph nodes were monitored for tumor formation on day 10.
Statistical analysis.
Significant values were determined by using the Microsoft Excel Student's t test program or Prism Mann Whitney test. P < 0.05 was considered statistically significant.
Acknowledgments
We gratefully acknowledge the experimental assistance from Y. Altman (High Throughput Cell Analysis core facility) and the members of Animal Facility at the Burnham Institute. We also thank Dr. J. Hamerman (University of California San Francisco) for helpful suggestions and discussion.
This work was supported by National Institutes of Health (NIH) grant nos. PO1 CA71932 (M. Fukuda and J.B. Lowe) and CA89294 (L.L. Lanier). A Cancer Research Institute Postdoctoral Fellowship supported S. Chen. L.L. Lanier is an American Cancer Society Research Professor. Functional Glycomics Consortium is supported by NIH grant no. GM62116.
The authors have no conflicting financial interests.
Abbreviations used: CFSE, carboxyfluorescein diacetate succinimidyl ester; MLN, mesenteric lymph node; PLN, peripheral lymph node; VLA, very late antigen.
L.L. Lanier and M. Fukuda contributed equally to this work.
References
- 1.Colucci, F., M.A. Caligiuri, and J.P. Di Santo. 2003. What does it take to make a natural killer? Nat. Rev. Immunol. 3:413–425. [DOI] [PubMed] [Google Scholar]
- 2.Morris, M.A., and K. Ley. 2004. Trafficking of natural killer cells. Curr. Mol. Med. 4:431–438. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi, G., M. Sironi, E. Ghibaudi, C. Selvaggini, M. Elices, P. Allavena, and A. Mantovani. 1993. Migration of natural killer cells across endothelial cell monolayers. J. Immunol. 151:5135–5144. [PubMed] [Google Scholar]
- 4.Allavena, P., G. Bianchi, D. Zhou, J. van Damme, P. Jilek, S. Sozzani, and A. Mantovani. 1994. Induction of natural killer cell migration by monocyte chemotactic protein-1, -2 and -3. Eur. J. Immunol. 24:3233–3236. [DOI] [PubMed] [Google Scholar]
- 5.Fogler, W.E., K. Volker, K.L. McCormick, M. Watanabe, J.R. Ortaldo, and R.H. Wiltrout. 1996. NK cell infiltration into lung, liver, and subcutaneous B16 melanoma is mediated by VCAM-1/VLA-4 interaction. J. Immunol. 156:4707–4714. [PubMed] [Google Scholar]
- 6.Schmits, R., T.M. Kundig, D.M. Baker, G. Shumaker, J.J. Simard, G. Duncan, A. Wakeham, A. Shahinian, A. van der Heiden, M.F. Bachmann, et al. 1996. LFA-1–deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J. Exp. Med. 183:1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barber, D.F., M. Faure, and E.O. Long. 2004. LFA-1 contributes an early signal for NK cell cytotoxicity. J. Immunol. 173:3653–3659. [DOI] [PubMed] [Google Scholar]
- 8.von Andrian, U.H., and T.R. Mempel. 2003. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 3:867–878. [DOI] [PubMed] [Google Scholar]
- 9.Rosen, S.D. 2004. Ligands for L-selectin: homing, inflammation, and beyond. Annu. Rev. Immunol. 22:129–156. [DOI] [PubMed] [Google Scholar]
- 10.Ley, K., and G.S. Kansas. 2004. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 4:325–335. [DOI] [PubMed] [Google Scholar]
- 11.Frey, M., N.B. Packianathan, T.A. Fehniger, M.E. Ross, W.C. Wang, C.C. Stewart, M.A. Caligiuri, and S.S. Evans. 1998. Differential expression and function of L-selectin on CD56bright and CD56dim natural killer cell subsets. J. Immunol. 161:400–408. [PubMed] [Google Scholar]
- 12.Freud, A.G., B. Becknell, S. Roychowdhury, H.C. Mao, A.K. Ferketich, G.J. Nuovo, T.L. Hughes, T.B. Marburger, J. Sung, R.A. Baiocchi, et al. 2005. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity. 22:295–304. [DOI] [PubMed] [Google Scholar]
- 13.Yago, T., M. Tsukuda, H. Fukushima, H. Yamaoka, K. Kurata-Miura, T. Nishi, and M. Minami. 1998. IL-12 promotes the adhesion of NK cells to endothelial selectins under flow conditions. J. Immunol. 161:1140–1145. [PubMed] [Google Scholar]
- 14.Andre, P., O. Spertini, S. Guia, P. Rihet, F. Dignat-George, H. Brailly, J. Sampol, P.J. Anderson, and E. Vivier. 2000. Modification of P-selectin glycoprotein ligand-1 with a natural killer cell-restricted sulfated lactosamine creates an alternate ligand for L-selectin. Proc. Natl. Acad. Sci. USA. 97:3400–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cerwenka, A., and L.L. Lanier. 2001. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 1:41–49. [DOI] [PubMed] [Google Scholar]
- 16.Diefenbach, A., and D.H. Raulet. 2002. The innate immune response to tumors and its role in the induction of T-cell immunity. Immunol. Rev. 188:9–21. [DOI] [PubMed] [Google Scholar]
- 17.Smyth, M.J., Y. Hayakawa, K. Takeda, and H. Yagita. 2002. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer. 2:850–861. [DOI] [PubMed] [Google Scholar]
- 18.Lanier, L.L. 2005. NK cell recognition. Annu. Rev. Immunol. 23:225–274. [DOI] [PubMed] [Google Scholar]
- 19.Arase, H., E.S. Mocarski, A.E. Campbell, A.B. Hill, and L.L. Lanier. 2002. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 296:1323–1326. [DOI] [PubMed] [Google Scholar]
- 20.Smith, H.R., J.W. Heusel, I.K. Mehta, S. Kim, B.G. Dorner, O.V. Naidenko, K. Iizuka, H. Furukawa, D.L. Beckman, J.T. Pingel, et al. 2002. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA. 99:8826–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferlazzo, G., D. Thomas, S.L. Lin, K. Goodman, B. Morandi, W.A. Muller, A. Moretta, and C. Munz. 2004. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J. Immunol. 172:1455–1462. [DOI] [PubMed] [Google Scholar]
- 22.Ferlazzo, G., M. Pack, D. Thomas, C. Paludan, D. Schmid, T. Strowig, G. Bougras, W.A. Muller, L. Moretta, and C. Munz. 2004. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc. Natl. Acad. Sci. USA. 101:16606–16611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fehniger, T.A., M.A. Cooper, G.J. Nuovo, M. Cella, F. Facchetti, M. Colonna, and M.A. Caligiuri. 2003. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood. 101:3052–3057. [DOI] [PubMed] [Google Scholar]
- 24.Cooper, M.A., T.A. Fehniger, and M.A. Caligiuri. 2001. The biology of human natural killer-cell subsets. Trends Immunol. 22:633–640. [DOI] [PubMed] [Google Scholar]
- 25.Martin-Fontecha, A., L.L. Thomsen, S. Brett, C. Gerard, M. Lipp, A. Lanzavecchia, and F. Sallusto. 2004. Induced recruitment of NK cells to lymph nodes provides IFN-γ for T(H)1 priming. Nat. Immunol. 5:1260–1265. [DOI] [PubMed] [Google Scholar]
- 26.Jacob, G.S., C. Kirmaier, S.Z. Abbas, S.C. Howard, C.N. Steininger, J.K. Welply, and P. Scudder. 1995. Binding of sialyl Lewis x to E-selectin as measured by fluorescence polarization. Biochemistry. 34:1210–1217. [DOI] [PubMed] [Google Scholar]
- 27.Ellies, L.G., S. Tsuboi, B. Petryniak, J.B. Lowe, M. Fukuda, and J.D. Marth. 1998. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 9:881–890. [DOI] [PubMed] [Google Scholar]
- 28.Yeh, J.C., N. Hiraoka, B. Petryniak, J. Nakayama, L.G. Ellies, D. Rabuka, O. Hindsgaul, J.D. Marth, J.B. Lowe, and M. Fukuda. 2001. Novel sulfated lymphocyte homing receptors and their control by a Core1 extension β 1,3-N-acetylglucosaminyltransferase. Cell. 105:957–969. [DOI] [PubMed] [Google Scholar]
- 29.Homeister, J.W., A.D. Thall, B. Petryniak, P. Maly, C.E. Rogers, P.L. Smith, R.J. Kelly, K.M. Gersten, S.W. Askari, G. Cheng, et al. 2001. The α(1,3)fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity. 15:115–126. [DOI] [PubMed] [Google Scholar]
- 30.Streit, M., and M. Detmar. 2003. Angiogenesis, lymphangiogenesis, and melanoma metastasis. Oncogene. 22:3172–3179. [DOI] [PubMed] [Google Scholar]
- 31.Trinchieri, G. 1989. Biology of natural killer cells. Adv. Immunol. 47:187–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van den Broek, M.E., D. Kagi, F. Ossendorp, R. Toes, S. Vamvakas, W.K. Lutz, C.J. Melief, R.M. Zinkernagel, and H. Hengartner. 1996. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med. 184:1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shankaran, V., H. Ikeda, A.T. Bruce, J.M. White, P.E. Swanson, L.J. Old, and R.D. Schreiber. 2001. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 410:1107–1111. [DOI] [PubMed] [Google Scholar]
- 34.Street, S.E., Y. Hayakawa, Y. Zhan, A.M. Lew, D. MacGregor, A.M. Jamieson, A. Diefenbach, H. Yagita, D.I. Godfrey, and M.J. Smyth. 2004. Innate immune surveillance of spontaneous B cell lymphomas by natural killer cells and γδ T cells. J. Exp. Med. 199:879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda, K., M.J. Smyth, E. Cretney, Y. Hayakawa, N. Kayagaki, H. Yagita, and K. Okumura. 2002. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 195:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diefenbach, A., E.R. Jensen, A.M. Jamieson, and D.H. Raulet. 2001. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 413:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerwenka, A., J.L. Baron, and L.L. Lanier. 2001. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA. 98:11521–11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smyth, M.J., K.Y. Thia, S.E. Street, E. Cretney, J.A. Trapani, M. Taniguchi, T. Kawano, S.B. Pelikan, N.Y. Crowe, and D.I. Godfrey. 2000. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 191:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smyth, M.J., M. Taniguchi, and S.E. Street. 2000. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J. Immunol. 165:2665–2670. [DOI] [PubMed] [Google Scholar]
- 40.Takeda, K., M.J. Smyth, E. Cretney, Y. Hayakawa, N. Yamaguchi, H. Yagita, and K. Okumura. 2001. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-γ-dependent suppression of subcutaneous tumor growth. Cell. Immunol. 214:194–200. [DOI] [PubMed] [Google Scholar]
- 41.Smyth, M.J., J. Swann, J.M. Kelly, E. Cretney, W.M. Yokoyama, A. Diefenbach, T.J. Sayers, and Y. Hayakawa. 2004. NKG2D recognition and perforin effector function mediate effective cytokine immunotherapy of cancer. J. Exp. Med. 200:1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salazar-Mather, T.P., J.S. Orange, and C.A. Biron. 1998. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1α (MIP-1α)-dependent pathways. J. Exp. Med. 187:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rossi, F.M.V., S.Y. Corbel, J.S. Merzaban, D.A. Carlow, K. Gossens, J. Duenas, L. So, L. Yi, and H.J. Ziltener. 2005. Recruitment of adult thymic progenitors is regulated by P-selectin and its ligand PSGL-1. Nat. Immunol. 6:626–634. [DOI] [PubMed] [Google Scholar]
- 44.Mackay, C.R., W. Marston, and L. Dudler. 1992. Altered patterns of T cell migration through lymph nodes and skin following antigen challenge. Eur. J. Immunol. 22:2205–2210. [DOI] [PubMed] [Google Scholar]
- 45.Shi, F.D., H.B. Wang, H. Li, S. Hong, M. Taniguchi, H. Link, L. Van Kaer, and H.G. Ljunggren. 2000. Natural killer cells determine the outcome of B cell-mediated autoimmunity. Nat. Immunol. 1:245–251. [DOI] [PubMed] [Google Scholar]
- 46.Lee, I.F., H. Qin, J. Trudeau, J. Dutz, and R. Tan. 2004. Regulation of autoimmune diabetes by complete Freund's adjuvant is mediated by NK cells. J. Immunol. 172:937–942. [DOI] [PubMed] [Google Scholar]
- 47.Glas, R., L. Franksson, C. Une, M.L. Eloranta, C. Ohlen, A. Orn, and K. Karre. 2000. Recruitment and activation of natural killer (NK) cells in vivo determined by the target cell phenotype. An adaptive component of NK cell–mediated responses. J. Exp. Med. 191:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smyth, M.J., J.M. Kelly, A.G. Baxter, H. Korner, and J.D. Sedgwick. 1998. An essential role for tumor necrosis factor in natural killer cell–mediated tumor rejection in the peritoneum. J. Exp. Med. 188:1611–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Julius, M.H., E. Simpson, and L.A. Herzenberg. 1973. A rapid method for the isolation of functional thymus-derived murine lymphocytes. Eur. J. Immunol. 3:645–649. [DOI] [PubMed] [Google Scholar]
- 50.Masopust, D., V. Vezys, A.L. Marzo, and L. Lefrancois. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 291:2413–2417. [DOI] [PubMed] [Google Scholar]
- 51.Moore, A., N. Sergeyev, S. Bredow, and R. Weissleder. 1998. A model system to quantitate tumor burden in locoregional lymph nodes during cancer spread. Invasion Metastasis. 18:192–197. [DOI] [PubMed] [Google Scholar]
- 52.Koo, G.C., F.J. Dumont, M. Tutt, J. Hackett Jr., and V. Kumar. 1986. The NK-1.1(−) mouse: a model to study differentiation of murine NK cells. J. Immunol. 137:3742–3747. [PubMed] [Google Scholar]
- 53.Seaman, W.E., M. Sleisenger, E. Eriksson, and G.C. Koo. 1987. Depletion of natural killer cells in mice by monoclonal antibody to NK-1.1. Reduction in host defense against malignancy without loss of cellular or humoral immunity. J. Immunol. 138:4539–4544. [PubMed] [Google Scholar]
- 54.Takaki, R., Y. Hayakawa, A. Nelson, P.V. Sivakumar, S. Hughes, M.J. Smyth, and L.L. Lanier. 2005. IL-21 enhances tumor rejection through a NKG2D-dependent mechanism. J. Immunol. 175:2167–2173. [DOI] [PubMed] [Google Scholar]