

Abstract
Systemic onset juvenile idiopathic arthritis (SoJIA) encompasses ∼10% of cases of arthritis that begin in childhood. The disease is unique in terms of clinical manifestations, severity of joint involvement, and lack of response to tumor necrosis factor blockade. Here, we show that serum from SoJIA patients induces the transcription of innate immunity genes, including interleukin (IL)-1 in healthy peripheral blood mononuclear cells (PBMCs). Upon activation, SoJIA PBMCs release large amounts of IL-1β. We administered recombinant IL-1 receptor antagonist to nine SoJIA patients who were refractory to other therapies. Complete remission was obtained in seven out of nine patients and a partial response was obtained in the other two patients. We conclude that IL-1 is a major mediator of the inflammatory cascade that underlies SoJIA and that this cytokine represents a target for therapy in this disease.
Juvenile idiopathic arthritis (JIA), which affects an estimated 250,000 children in the United States alone, is an important cause of short- and long-term disability. The term JIA encompasses a heterogeneous group of diseases that is classified according to three major types of presentation: (a) oligoarthritis, (b) polyarthritis, and (c) systemic onset JIA (SoJIA). Each of these groups has a different prognosis and responds differently to available therapies (1, 2); this suggests that their pathogenesis also is unique.
Children who have SoJIA present with systemic symptoms, fever, and/or rash, which may precede the development of arthritis by months or even years. Fever, anemia, leukocytosis, and elevated erythrocyte sedimentation rate (ESR) are the main initial features of the disease. Because these symptoms are nonspecific, patients often undergo extensive diagnostic tests and hospitalizations. Although the disease outcome is highly variable, the overall prognosis seems to correlate with the persistence of systemic symptoms and the number of joints that is involved 6 mo into the disease course (3–6). Overall, up to 50% of SoJIA patients continue to have active arthritis 5–10 yr after diagnosis (2, 7, 8). Because long-term disability is correlated directly with duration of active disease, this group has the most severe outcome, and thus, represents the most serious challenge to pediatric rheumatologists. The pathogenesis of SoJIA remains an enigma, but increased levels of IL-6 seem to correlate with the systemic activity of the disease and with the development of arthritis (9).
Multi-drug treatment of SoJIA patients depends on the phase (systemic versus arthritic) of the disease and the extent of involvement. Although a minority of patients do well with nonsteroidal anti-inflammatory drugs, most patients require oral and/or systemic corticosteroids (10) and methotrexate (MTX) for prolonged periods to treat the systemic manifestations and arthritis, respectively. Steroid treatment results in significant morbidity, including vertebral compression fractures, cataracts, and severe growth retardation. Other medications that are used in recalcitrant cases include intravenous gamma globulin, cyclosporine, and thalidomide (11, 12). Anti-TNF therapy is effective against some types of JIA (13, 14), but most SoJIA patients do not respond to this treatment (15, 16). Here we show data which indicate that IL-1 is a major mediator of the inflammatory cascade that underlies SoJIA, and that IL-1Ra is an effective treatment for this disease.
RESULTS
Incubation of healthy PBMCs with SoJIA serum up-regulates transcription of innate immunity genes
We have previously shown that interferon-α, which is present in the serum of patients who have systemic lupus erythematosus (SLE), induces the differentiation of healthy monocytes into dendritic cells (17) and that all active SLE PBMCs display an interferon signature (18). After a similar strategy, we cultured healthy PBMCs with the serum of four active SoJIA patients and examined the induced gene transcription pattern using Affymetrix oligonucleotide microarrays (accession nos. are provided in Table S1, available at http://www.jem.org/cgi/content/full/jem.20050473/DC1). Each PBMC sample was processed: (a) fresh without culture; (2) after 6 h incubation with autologous serum; and (3) after 6 h incubation with SoJIA serum. Fig. 1 a shows 46 genes whose expression increased more than twofold in healthy PBMCs that were cultured with SoJIA serum. Up-regulated genes included several members of the IL-1 cytokine/cytokine receptor family. IL-1b transcription was induced by 4/4 SoJIA sera from 4- to 40-fold (median, 8.2-fold). IL-1a was up-regulated by 3/4 of the SoJIA sera (median 13-fold), as were their receptors, IL-1R1 and IL-1R2 (median four- and twofold, respectively). In contrast, IL-6 was up-regulated by only one of the SoJIA sera. RT-PCR analysis of IL-1b transcription confirmed the microarray data (unpublished data). SoJIA sera also induced the transcription of chemokines that are involved in the chemotaxis of stem cells (CXCL2), neutrophils (CXCL1, CXCL3, CXCL5, CXCL6), monocyte/macrophages (CCL2, CCL7), and lymphocyte/dendritic cells (CCL20; reference 19). CCR1, a receptor for numerous chemokines which is considered to be a target in autoimmunity (20), also was increased. Among receptors that are associated with pathogen recognition, fibronectin was the most significantly up-regulated (17-fold), followed by the diphtheria toxin receptor (9.6-fold); and the lectins, ANGPTL4 and MDL1/CLECSF5 (greater than fivefold). Pentraxin-3, the scavenger macrophage receptor with collagenous structure (MARCO), toll-like receptor 2, and the C1q receptor were up-regulated two- to fourfold by 3/4 SoJIA sera. Triggering receptor expressed on myeloid cells (TREM1) was up-regulated significantly only by the sera from the two untreated patients. Transcripts which encode molecules that are involved in cell adhesion and/or motility, as well as a variety of enzymes, including the proinflammatory cyclooxygenase-2, were up-regulated. Other up-regulated molecules are listed in Fig. 1 and Tables S1 and S2 (available at http://www.jem.org/cgi/content/full/jem.20050473/DC1).
Figure 1.
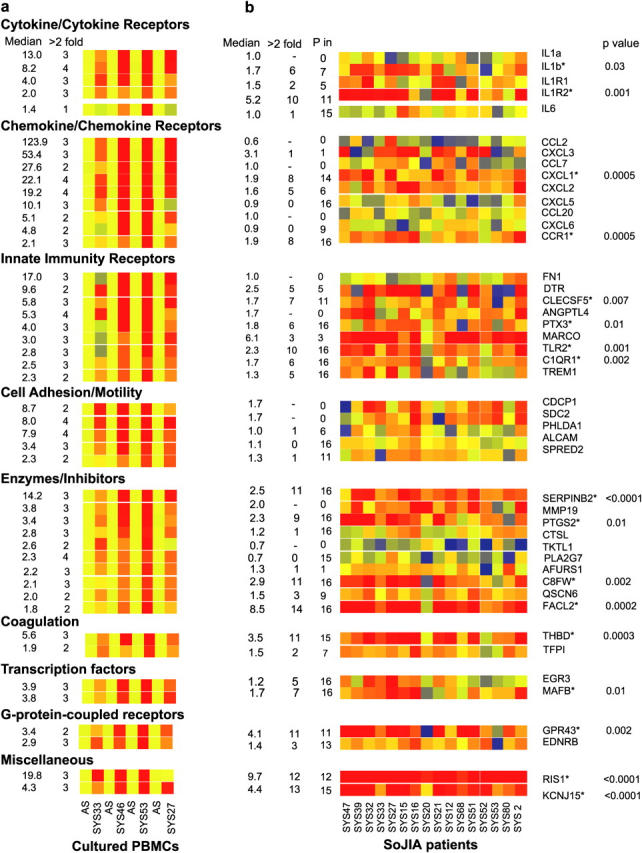
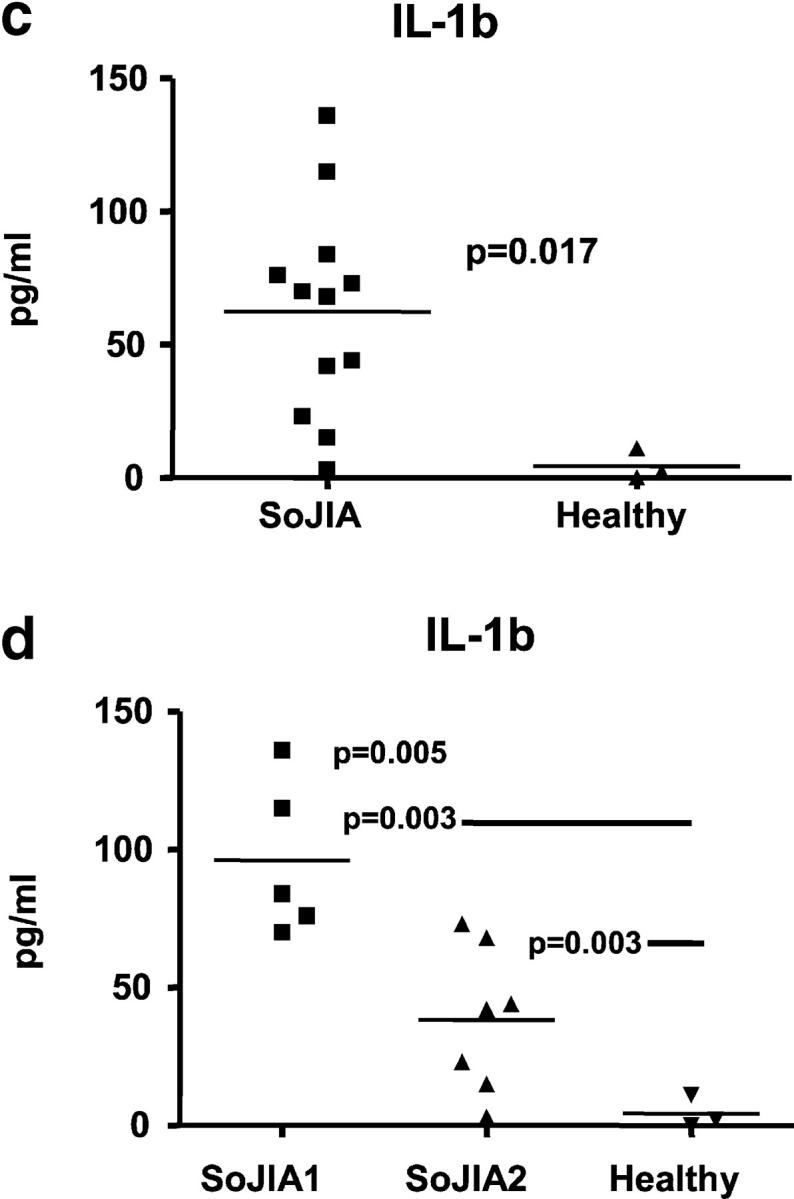
(a) Effects of SoJIA sera on healthy PBMCs. Incubation of healthy PBMCs with autologous sera and sera from four patients who had active SoJIA (SYS33, SYS46, SYS53, and SYS27) induced up-regulation of 46 genes. The microarray data of healthy PBMCs that were incubated with patients' sera were normalized to the same PBMCs that were cultured with autologous serum. Probes that did not differ significantly between unincubated PBMCs and PBMCs that were cultured with autologous serum were selected. Of those, only gene probes whose expression was up-regulated more than twofold by at least two SoJIA sera were selected further. IL-6 did not fulfill this criterion but is shown for comparison. Median fold up-regulation by the four SoJIA sera incubation is depicted in the left column. Number of SoJIA sera that induced greater than twofold up-regulation is shown in the next column. (b) Expression of the gene probes selected in Fig. 1 a in the PBMCs of 16 active SoJIA patients. The patient's PBMCs expression data were normalized to the median expression of the same gene probes in the PBMCs of 12 healthy children. Median gene expression and number of samples with greater than twofold up-regulation are depicted in the first two columns. Third column represents the number of samples with a P (present) flag according to Affymetrix MAS 5.0 scaled gene expression data. Asterisks on the gene denominations mean that the expression of those genes is significantly different (Mann-Whitney < 0.05) in patients compared with controls. p-values are given next to these genes. (c) Induction of IL-1b protein secretion in healthy PBMCs incubated with SoJIA sera. Supernatants from 6 h incubation of healthy PBMCs with 12 SoJIA sera and 3 healthy controls were assayed for IL-1b production by Luminex. (d) Induction of IL-1b protein secretion in SoJIA patients who did and did not have systemic symptoms. IL-1b protein secretion is induced preferentially by sera from SoJIA patients who experience systemic symptoms (SoJIA1, n = 5) over those patients who only had active arthritis (SoJIA2, n = 7). All results were analyzed using nonparametric tests (Mann-Whitney).
The remarkable transcription induction capacity of SoJIA serum led us to consider that the observed genes also may be up-regulated in SoJIA patients. Microarray analysis showed that >800 transcripts were overexpressed in the PBMCs of 16 patients, compared with 12 healthy controls (unpublished data). Of the 46 gene transcripts that were induced by SoJIA sera, 30 were present in more than one third of the SoJIA PBMCs; 17 were increased significantly in SoJIA compared with control PBMCs (Mann-Whitney P < 0.05; Fig. 1 b and Tables S2 and S4, available at http://www.jem.org/cgi/content/full/jem.20050473/DC1). These included IL-1b and the IL-1 decoy receptor (IL-1R2), CXCL1, and CCR1; this may explain the neutrophilia and increased monocyte numbers in these patients. C1qR; toll-like receptor 2; PTX3; and the proinflammatory enzyme, prostaglandin G/H synthase-2/cyclooxygenase-2, also were increased significantly. KCNJ15, an inward-rectifier–type potassium channel that is linked to the lysosome-mediated control of IL-1b secretion (21), was among the most significantly up-regulated genes in PBMCs from SoJIA patients. Conversely, a group of gene transcripts that was induced after 6 h of incubation of healthy PBMCs with SoJIA sera in vitro were not detected in SoJIA patients. These included IL-1a, CCL2, CCL7, CCL20, fibronectin, SDC2, and others (Fig. 1 b).
SoJIA sera induce healthy PBMCs to secrete IL-1b
The increased expression of IL-1b was analyzed further at the protein level. SoJIA sera induced healthy PBMCs to produce 63 ± 39 pg/ml of IL-1b (mean ± SD; range <10–136 pg/ml; n = 10; P = 0.017), whereas healthy sera did not (<10 pg/ml; n = 3; Fig. 1 c). Sera from febrile SoJIA patients were more efficient at inducing IL-1b secretion than were sera from afebrile patients (P = 0.005), which, in turn, were more efficient than healthy sera (P = 0.003; Fig. 1 d). Taken together, these data indicate that SoJIA serum induces transcription and translation of IL-1b in healthy PBMCs.
Activated SoJIA PBMCs produce high levels of IL-1b
Recently, monocytes from patients who had autoinflammatory diseases were shown to produce higher levels of IL-1b than control monocytes (22). Thus, we tested the capacity of SoJIA patients' PBMCs to secrete IL-1b upon activation with PMA-ionomycin. After 24 h, PBMCs from five SoJIA patients secreted 333 ± 204 pg/ml (range, 154–569 pg/ml), whereas PBMCs from five healthy controls secreted 21 ± 4 pg/ml IL-1b (mean ± SD; P = 0.007; Fig. 2 a). In the same cultures, IL-6 and TNF production was not significantly different between patients and controls (Fig. 2 b).
Figure 2.
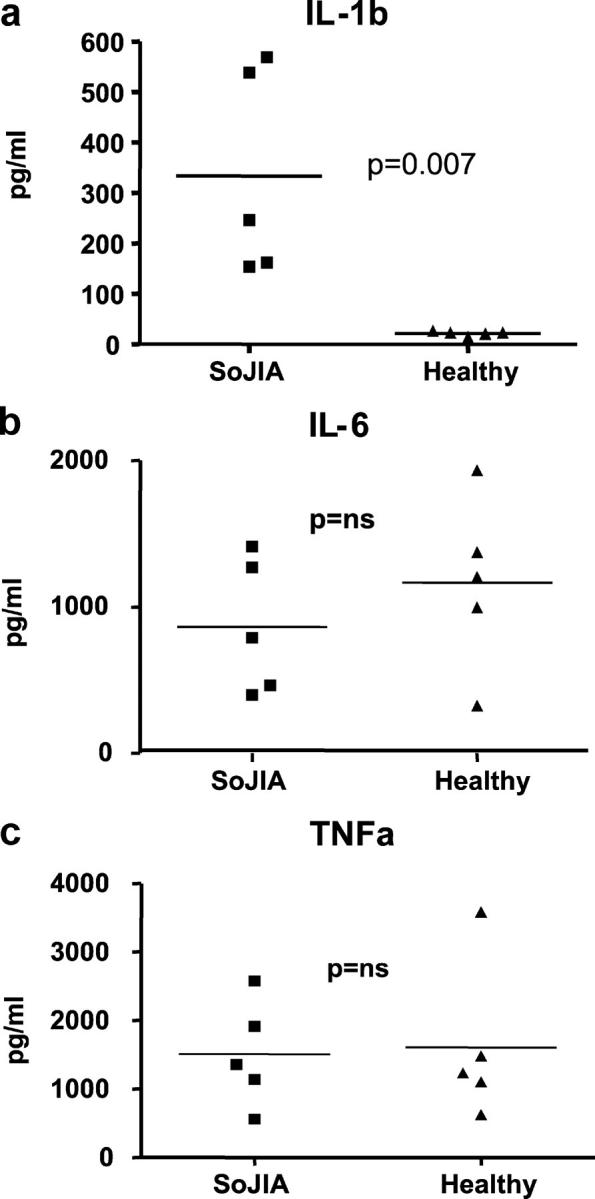
(a) Production of IL-1b by activated SoJIA PBMCs. PBMCs from SoJIA patients (n = 5) and healthy controls (n = 5) were activated with PMA/Ionomycin for 24 h. Release of IL-1b into the supernatants was assayed with Luminex. Production of IL-6 (b) and TNF (c) by the same activated SoJIA PBMCs. p-values were calculated by nonparametric (Mann-Whitney) tests.
Administration of IL-1Ra resolves SoJIA clinical symptoms
The above findings suggested that SoJIA patients might suffer from a dysregulation of IL-1 production, as opposed to other forms of chronic inflammatory arthritis (e.g., rheumatoid arthritis) that depend on TNF. Therefore, we hypothesized that SoJIA patients would benefit from therapy with an IL-1 antagonist. Nine patients who had active disease that was resistant to conventional aggressive treatment were treated with a commercially available rIL-1Ra (Anakinra, Amgen Inc.). The clinical characteristics of the patients at treatment initiation are summarized in Table I. Seven of nine patients had systemic symptoms and eight had active, uninterrupted arthritis that had lasted for 5–125 mo. All patients were receiving oral prednisone, 7/9 were on i.v. methylprednisolone, 7/9 were on MTX, and 4/9 had received anti-TNF (infliximab) therapy for 12–25 mo without improvement. rIL-1Ra was initiated as a daily subcutaneous injection at 2 mg/kg, up to 100 mg. Patients were followed every 2 mo with a complete physical examination and laboratory tests for an average of 6.6 mo (range 2–12 mo) after therapy initiation. All nine patients responded to therapy. All patients who experienced systemic symptoms (7/9) became afebrile within the first week after initiation of therapy, and remained afebrile for the follow-up period of 2–12 mo (Fig. 3 a). Arthritis score completely resolved in 6/8 patients who had active arthritis at the time of therapy initiation, and it improved in the remaining two patients (Sys2 and Sys15) who had had a persistent course without remissions for 12 and 3 yr, respectively. i.v. methylprednisolone was discontinued during the herein described follow-up period in 5/7 patients who had been on this therapy for months. This medication was tapered and eventually was discontinued in the remaining two patients. Oral prednisone was stopped in one patient and tapered in 6/7 patients (Fig. 4). Although the patient who had the longest unremitting disease showed persistent response of her systemic symptoms, her arthritis responded only initially; she had two active joints at the 4-mo follow-up (Fig. 3 b). Complications included two episodes of hypotension and vomiting with negative viral and bacterial cultures in one patient who had underlying myocardial dysfunction. Therapy was restarted after resolution of the symptoms without complications. All patients experienced mild erythema at the local injection sites. The core set criteria for the time Anakima treatment was started and the last follow-up are summarized in Tables S7–S15 (http://www.jem.org/cgi/content/full/jem.20050473/DC1).
Table I.
Clinical characteristics of the patients at the time of initiation of anakinra therapy
| Patient | Age | Sex | Disease duration |
Flare duration |
Fever/ rash |
Active arthritis |
Oral prednisone |
i.v. solumedrol |
MTX | Previous anti-TNF |
Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|
| mo | mo | (frequency) | mo | ||||||||
| Sys33 | 17 | F | 36 | 6 | Yes | Yes | Yes | Yes (q. 4 wk) | Yes | Yes | 12 |
| Sys47 | 9 | M | 29 | 12 | Yes | Yes | Yes | Yes (q. 2 wk) | No | No | 12 |
| Sys56 | 4 | F | 23 | 13 | Yes | Yes | Yes | Yes (q. 1 wk) | Yes | No | 8 |
| Sys2 | 12 | F | 125 | 125 | Yes | Yes | Yes | Yes (q. 4 wk) | Yes | Yes | 6 |
| Sys12 | 6 | F | 35 | 10 | Yes | No | Yes | Yes (q. 4 wk) | Yes | Yes | 6 |
| Sys15 | 4 | F | 30 | 30 | No | Yes | Yes | Yes (q. 4 wk) | No | Yes | 6 |
| Sys21 | 6 | F | 40 | 5 | Yes | Yes | Yes | Yes (q. 2 wk) | Yes | No | 4 |
| Sys38 | 4 | M | 38 | 18 | Yes | Yes | Yes | No | Yes | No | 4 |
| Sys25 | 14 | F | 144 | 30 | No | Yes | Yes | No | Yes | No | 2 |
Figure 3.
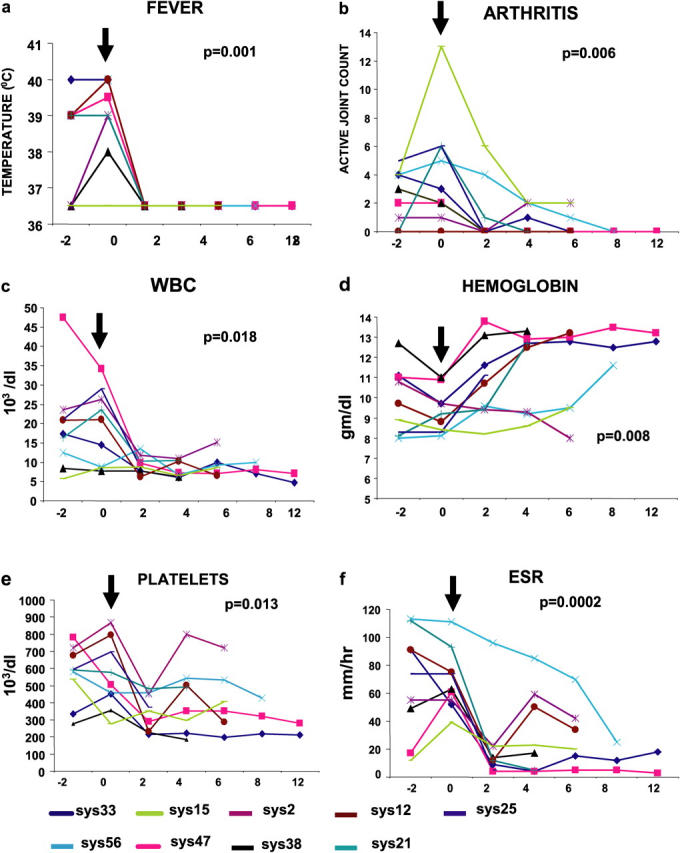
Values of (a) temperature; (b) active joint count; (c) white blood cells (WBC); (d) hemoglobin; (e) platelet count; and (f) ESR in 9 SoJIA patients. Values on the x axis represent months before (−2) initiation of Anakinra treatment (0) and up to 2–12 mo of follow up (average, 6.6 mo). Arrows indicate the time of treatment initiation. p-values were calculated at time 0 and at 2-mo follow-up (paired, two-tailed t test). Color codes for individual patients (from Table I) are represented at the bottom.
Figure 4.

Oral, daily prednisone dose in nine patients treated with anakinra at initiation of therapy (time 0) and at last follow-up. p-value was obtained by paired Student's t test.
Administration of IL-1Ra normalizes SoJIA laboratory findings
The striking improvement in clinical symptoms upon IL-1Ra treatment was associated with a parallel improvement in the inflammatory markers. Leukocytosis resolved in 9/9 patients (Fig. 3 c). The chronic persistent anemia improved in all but one patient. This was demonstrated by an increase of hemoglobin levels (Fig. 3 d) and erythrocyte counts (not depicted). Thrombocytosis resolved in 9/9 patients. Only one patient (Sys2) showed a rebound increase at the 4–6 mo follow-up (Fig. 3 e). Most significant (P = 0.0002) was the reduction of ESR, with 8/9 patients reaching a normal value within 2 mo. One patient (Sys58) who had a slower resolution of arthritis symptoms required a longer time (8 mo) to normalize the ESR. Two patients (Sys2 and Sys12) had a mild rebound in ESR at the 4-mo visit; this coincided with temporary withdrawal of rIL-1Ra for viral-like symptoms (Fig. 3 f). Thus, treatment of SoJIA patients with daily rIL-1Ra results in prompt clinical and biologic improvement.
DISCUSSION
Three sets of findings lead us to conclude that dysregulated production of IL-1 plays a critical role in the pathogenesis of SoJIA. First, the serum of SoJIA patients up-regulates the expression of IL-1a, IL-1b, and other innate immunity genes by healthy PBMCs. Second, the patient's PBMCs produce an excess of IL-1b upon activation. Third, treatment with IL-1Ra efficiently treats the disease.
Although SoJIA serum induces IL-1b secretion by healthy PBMCs in vitro, IL-1b serum levels in our patients were as low as those of healthy individuals. Serum cytokine levels, however, may not reflect a cytokine's role in disease pathogenesis. For example, virtually all PBMCs from children who have SLE display an interferon-α signature, although this cytokine is detectable in the serum in less than 50% of patients (18). Furthermore, IL-1b is involved in the pathogenesis of familial autoinflammatory syndromes (22, 23), and blocking IL-1 with IL-1Ra resolves the clinical symptoms that are associated with mutations in the NALP3/cryopyrin gene (Muckle-Wells syndrome and neonatal onset multisystem inflammatory disease/chronic, infantile, neurologic, cutaneous and articular syndrome) (24). However, the serum IL-1 concentration and the cytokine mRNA in PBMCs from these patients are not raised either (25). Although SoJIA normally is a sporadic disease, de novo mutations and/or subtle polymorphisms of genes within the IL-1 pathway could contribute to its pathogenesis. The IL-1 family includes nine members (26), eight of which were not up-regulated using real-time PCR in patients' PBMCs (unpublished data).
The central role of IL-1 in SoJIA is illustrated by the remarkable clinical response of the patients to IL-1Ra. Nine out of nine patients who had long-standing disease that was resistant to other forms of treatment responded to this therapy. Seven of nine cleared symptoms and laboratory abnormalities within days to weeks of therapy initiation; one patient showed a clear trend toward improvement. One patient who had had active disease for 12 uninterrupted years had a quick response and remained afebrile for the 6-mo follow-up period. Her arthritis and laboratory abnormalities, however, improved only transiently. Microarray analysis showed that this patient had a modest (2.7-fold) up-regulation of the IL-1b gene (Table S2). Complete characterization of her IL-1b response to PBMC activation is underway.
IL-1b was shown to be a key mediator of synovial inflammation in experimental arthritis models. IL-1b–deficient mice fail to develop chronic, erosive arthritis in response to arthritogenic stimuli (27) and IL-1Ra–deficient mice develop autoimmunity and arthritis spontaneously (28). IL-1 blockade with recombinant IL-1Ra, however, did not offer significant advantages over anti-TNF blockade in rheumatoid arthritis patients, and did not show efficacy in patients who were failing anti-TNF treatment (29, 30). Increased serum level of soluble TNF receptors have been described in SoJIA (31), and it has been suggested that TNF probably is more important than IL-1 in the systemic inflammation that characterizes this disease (32). However, although anti-TNF agents are efficacious in children within other JIA subgroups (14), most SoJIA patients do not respond to this therapy. Furthermore, a subgroup of these patients progresses to an unremitting course that is unresponsive to any combination of therapies. These patients also experience significant morbidity from treatment.
Based on our observation that IL-1 production is dysregulated in SoJIA, we treated nine SoJIA patients who were failing other therapies with IL-1 blockade and obtained complete disease remission in seven of them. Lack of full response in two patients may suggest a level of disease heterogeneity that needs to be addressed further. Two other SoJIA patients who had prolonged, therapy-resistant disease recently were reported to respond to IL-1 blockade (33). The unique pattern of cytokine abnormalities and response to biologic therapy that we describe suggests that SoJIA may be classified better as an autoinflammatory syndrome, rather than as an autoimmune disease under the broad spectrum of JIA.
The availability of IL-1Ra may represent a major step toward preventing the development of severe, deforming arthritis and avoiding the use of prolonged corticosteroids and their devastating side effects in this group of patients. Controlled clinical trials are necessary to delineate clinical response, remission duration, and the use of IL-1Ra to substitute eventually for corticosteroids as the first-line treatment for this disease. The current findings should encourage the development of other IL-1 antagonists with improved potency and pharmacokinetics.
MATERIALS AND METHODS
Patient population
PBMCs and sera from 23 SoJIA patients (15 females, 8 males; average age, 7.1 yr) who fulfilled the American College of Rheumatology diagnostic criteria (34) were collected on repeated occasions. The disease duration, clinical characteristics, and treatment at the time of analysis are summarized in Table I and Tables S3–S7 (available at http://www.jem.org/cgi/content/full/jem.20050473/DC1). Patients were classified as active if they had systemic symptoms (fever and/or rash) and/or active arthritis (swollen and/or tender and limited joints). The control population consisted of 12 children (average age, 14 yr) and 7 adults (average age, 35 yr). The sera from healthy controls were cultured on repeated occasions with autologous and heterologous PBMCs. Patients and pediatric controls were recruited at Texas Scottish Rite Hospital for Children in Dallas. The study was approved by the Institutional Review Boards (IRBs) of UT Southwestern Medical Center, Texas Scottish Rite Hospital, and Baylor Health Care System (IRB no. 0199017, 0701–513). Informed consent was obtained from parents or legal guardians.
PBMC cultures and RNA extraction
PBMCs were obtained by Ficoll-Histopaque gradient centrifugation of 20 ml of blood. PBMCs were cultured (106/ml) in RPMI 1640 supplemented with 20% autologous or SoJIA patient serum for 6 h. RNA was extracted using RNAeasy kit (QIAGEN) and assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies). Supernatants were frozen at −80°C. PBMCs were cultured with RPMI 1640 and 10% FCS with PMA (50 ng/ml)–Ionomycin (1 μg/ml) for 24 h. Cells were harvested and the RNA was extracted as above. Supernatants were frozen at −80°C.
Microarrays and real-time PCR
Samples for microarray analysis were processed as described previously (18) and hybridized to the HG U133A Affymetrix GeneChip array containing 22,283 probe sets (Affymetrix, Inc.) at 45°C for 16 h. GeneChip arrays were washed, stained, and scanned according to protocols that are described in the GeneChip Expression Analysis Technical Manual (Affymetrix Inc.). Scanned GeneChips were inspected visually for abnormalities or irregularities.
Data analysis
Intensity values were scaled to 500 using global scaling in MAS 5.0 and data were exported in MS Excel for import into GeneSpring software (Silicon Genetics) for gene expression analyses. No “per chip” normalization was performed because global scaling had been applied in MAS 5.0. Global scaling adjusts for chip-to-chip variations in hybridization intensities. Subsequent samples were normalized to pediatric healthy controls and/or to the median of all samples. Statistical comparisons were performed in GeneSpring using parametric (Welch's approximate t test) and nonparametric (Mann-Whitney U-test) methods. Unsupervised hierarchical clustering was performed to visualize transcripts that had a control signal of 50 or greater (above the background intensity) and that were identified as “present” according to MAS 5.0 in 75% of all samples. Statistical comparisons were performed in GeneSpring using parametric (Welch's approximate t test) and nonparametric (Mann-Whitney U-test) methods. Unsupervised hierarchical clustering was performed to visualize transcript/sample relationships using standard correlation, Pearson correlation, or Euclidian distance where indicated.
The GenBank/EMBL/DDBJ accession numbers of the genes presented in Fig. 1 are (in order of appearance on the figure): M15329; NM_000576; NM_000877; NM_004633; NM_000600; S69738; NM_002090; NM_006273; NM_001511; M57731; AK026546; NM_004591; NM_002993; AI421071; BC005858; NM_001945; NM_013252; NM_016109; NM_002852; NM_006770; NM_003264; NM_012072; NM_018643; NM_022842; AI380298; AA576961; AA156721; H97931; NM_002575; U38321; NM_000963; NM_001912; X91817; NM_005084; NM_024524; NM_025195; NM_002826; NM_001995; NM_000361; BF511231; NM_004430; NM_005461; NM_005306; M74921; BF062629; U73191.
Two-step RT-PCR was performed using Applied Biosystems TaqMan Assays on Demand probe and primer sets according to the manufacturer's instructions and the ABI Prism 7700 Sequence detection System (Applied Biosystems). The endogenous GAPDH gene and/or 18S RNA were used for correcting the results with the comparative threshold cycle method for relative quantification as described by the manufacturer.
Multiplex analysis
Culture supernatants were analyzed for six cytokines and chemokines using the FluorikineMAP cytokine assay kit (R&D Systems) as per the manufacturer's protocol.
Online supplemental material
Table S1 provides a description of the genes that were up-regulated by SoJIA serum (from Fig. 1, a and b). Table S2 displays the microarray data corresponding to the 46 probes from Fig. 1 (a and b). For each gene probe, raw (top) and normalized (bottom) values are provided. Normalization was done to the value of the corresponding autologous serum cultures (for the in vitro experiments), and to the median of 12 healthy pediatric controls (for the patient's data). Tables S3–S6 show the demographics, clinical symptoms, and treatment of the patients whose sera was used in the experiments that are depicted in Figs. 1, a–d and Fig. 2, a–c, respectively. Tables S7–S15 summarize the core set criteria for visits 0 (time of initiation of Anakinra treatment and 1 (2-mo follow-up), and the last clinic visit (2–12-mo follow-up). Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050473/DC1.
Acknowledgments
We thank our patients and their parents/guardians for agreeing to participate in the study. We also thank Drs. R. Cimaz, K. Madson, K. Palucka, M. Stone, O. Ramilo, and G. Zurawski for critically reading the text and Drs. L. Bennett and J. Connolly for help with microarray and Luminex analyses, respectively.
This work was supported by Baylor Health Care System Foundation and the National Institutes of Health grant nos. R01 AR050770-01 (to V. Pascual) and CA78846 and U19A1057234-02 (to J. Banchereau). J. Banchereau holds the Caruth Chair for Transplantation Immunology.
The authors have no conflicting financial interests.
Abbreviations used: ESR, erythrocyte sedimentation rate; IL-1Ra, IL-1R antagonist; JIA, juvenile idiopathic arthritis; MTX, methotrexate; SLE, systemic lupus erythematosus; SoJIA, systemic onset juvenile idiopathic arthritis.
V. Pascual and F. Allantaz contributed equally to this work.
References
- 1.Cassidy, J.T., and R.E. Petty. 2001. Textbook of Pediatric Rheumatology. W.B. Saunders, Philadelphia, PA. 976 pp.
- 2.Wallace, C.A., and J.E. Levinson. 1991. Juvenile rheumatoid arthritis: outcome and treatment for the 1990s. Rheum. Dis. Clin. North Am. 17:891–905. [PubMed] [Google Scholar]
- 3.Ravelli, A., and A. Martini. 2003. Early predictors of outcome in juvenile idiopathic arthritis. Clin. Exp. Rheumatol. 21:S89–S93. [PubMed] [Google Scholar]
- 4.Modesto, C., P. Woo, J. Garcia-Consuegra, R. Merino, M. Garcia-Granero, C. Arnal, and A.M. Prieur. 2001. Systemic onset juvenile chronic arthritis, polyarticular pattern and hip involvement as markers for a bad prognosis. Clin. Exp. Rheumatol. 19:211–217. [PubMed] [Google Scholar]
- 5.Ravelli, A. 2004. Toward an understanding of the long-term outcome of juvenile idiopathic arthritis. Clin. Exp. Rheumatol. 22:271–275. [PubMed] [Google Scholar]
- 6.Spiegel, L.R., R. Schneider, B.A. Lang, N. Birdi, E.D. Silverman, R.M. Laxer, D. Stephens, and B.M. Feldman. 2000. Early predictors of poor functional outcome in systemic-onset juvenile rheumatoid arthritis: a multicenter cohort study. Arthritis Rheum. 43:2402–2409. [DOI] [PubMed] [Google Scholar]
- 7.Lomater, C., V. Gerloni, M. Gattinara, J. Mazzotti, R. Cimaz, and F. Fantini. 2000. Systemic onset juvenile idiopathic arthritis: a retrospective study of 80 consecutive patients followed for 10 years. J. Rheumatol. 27:491–496. [PubMed] [Google Scholar]
- 8.Bowyer, S.L., P.A. Roettcher, G.C. Higgins, B. Adams, L.K. Myers, C. Wallace, R. Rennebohm, T.L. Moore, P.H. Pepmueller, C. Spencer, et al. 2003. Health status of patients with juvenile rheumatoid arthritis at 1 and 5 years after diagnosis. J. Rheumatol. 30:394–400. [PubMed] [Google Scholar]
- 9.de Benedetti, F., M. Massa, P. Robbioni, A. Ravelli, G.R. Burgio, and A. Martini. 1991. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 34:1158–1163. [DOI] [PubMed] [Google Scholar]
- 10.Adebajo, A.O., and M.A. Hall. 1998. The use of intravenous pulsed methylprednisolone in the treatment of systemic-onset juvenile chronic arthritis. Br. J. Rheumatol. 37:1240–1242. [DOI] [PubMed] [Google Scholar]
- 11.Lehman, T.J. 2000. Clinical trials for the treatment of systemic onset juvenile rheumatoid arthritis-juvenile idiopathic arthritis. Curr. Rheumatol. Rep. 2:313–315. [DOI] [PubMed] [Google Scholar]
- 12.Lehman, T.J., K.H. Striegel, and K.B. Onel. 2002. Thalidomide therapy for recalcitrant systemic onset juvenile rheumatoid arthritis. J. Pediatr. 140:125–127. [DOI] [PubMed] [Google Scholar]
- 13.Lovell, D.J., E.H. Giannini, A. Reiff, G.D. Cawkwell, E.D. Silverman, J.J. Nocton, L.D. Stein, A. Gedalia, N.T. Ilowite, C.A. Wallace, et al. 2000. Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric Rheumatology Collaborative Study Group. N. Engl. J. Med. 342:763–769. [DOI] [PubMed] [Google Scholar]
- 14.Lovell, D.J., E.H. Giannini, A. Reiff, O.Y. Jones, R. Schneider, J.C. Olson, L.D. Stein, A. Gedalia, N.T. Ilowite, C.A. Wallace, et al. 2003. Long-term efficacy and safety of etanercept in children with polyarticular-course juvenile rheumatoid arthritis: interim results from an ongoing multicenter, open-label, extended-treatment trial. Arthritis Rheum. 48:218–226. [DOI] [PubMed] [Google Scholar]
- 15.Horneff, G., H. Schmeling, T. Biedermann, I. Foeldvari, G. Ganser, H.J. Girschick, T. Hosbach, H.I. Huppertz, R. Keitzer, R.M. Kuester, et al. 2004. The German etanercept registry for treatment of juvenile idiopathic arthritis (JIA). Ann Rheum Dis. 63:1638–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quartier, P., P. Taupin, F. Bourdeaut, I. Lemelle, P. Pillet, M. Bost, J. Sibilia, I. Kone-Paut, S. Gandon-Laloum, M. LeBideau, et al. 2003. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum. 48:1093–1101. [DOI] [PubMed] [Google Scholar]
- 17.Blanco, P., A.K. Palucka, M. Gill, V. Pascual, and J. Banchereau. 2001. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 294:1540–1543. [DOI] [PubMed] [Google Scholar]
- 18.Bennett, L., A.K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, and V. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moser, B., M. Wolf, A. Walz, and P. Loetscher. 2004. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 25:75–84. [DOI] [PubMed] [Google Scholar]
- 20.Saeki, T., and A. Naya. 2003. CCR1 chemokine receptor antagonist. Curr. Pharm. Des. 9:1201–1208. [DOI] [PubMed] [Google Scholar]
- 21.Andrei, C., P. Margiocco, A. Poggi, L.V. Lotti, M.R. Torrisi, and A. Rubartelli. 2004. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: implications for inflammatory processes. Proc. Natl. Acad. Sci. USA. 101:9745–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shoham, N.G., M. Centola, E. Mansfield, K.M. Hull, G. Wood, C.A. Wise, and D.L. Kastner. 2003. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. USA. 100:13501–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aksentijevich, I., M. Nowak, M. Mallah, J.J. Chae, W.T. Watford, S.R. Hofmann, L. Stein, R. Russo, D. Goldsmith, P. Dent, et al. 2002. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 46:3340–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawkins, P.N., H.J. Lachmann, E. Aganna, and M.F. McDermott. 2004. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 50:607–612. [DOI] [PubMed] [Google Scholar]
- 25.Hoffman, H.M., S. Rosengren, D.L. Boyle, J.Y. Cho, J. Nayar, J.L. Mueller, J.P. Anderson, A.A. Wanderer, and G.S. Firestein. 2004. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 364:1779–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinarello, C.A. 2002. The IL-1 family and inflammatory diseases. Clin. Exp. Rheumatol. 20:S1–13. [PubMed] [Google Scholar]
- 27.Saijo, S., M. Asano, R. Horai, H. Yamamoto, and Y. Iwakura. 2002. Suppression of autoimmune arthritis in interleukin-1–deficient mice in which T cell activation is impaired due to low levels of CD40 ligand and OX40 expression on T cells. Arthritis Rheum. 46:533–544. [DOI] [PubMed] [Google Scholar]
- 28.Horai, R., S. Saijo, H. Tanioka, S. Nakae, K. Sudo, A. Okahara, T. Ikuse, M. Asano, and Y. Iwakura. 2000. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist–deficient mice. J. Exp. Med. 191:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genovese, M.C., S. Cohen, L. Moreland, D. Lium, S. Robbins, R. Newmark, and P. Bekker. 2004. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 50:1412–1419. [DOI] [PubMed] [Google Scholar]
- 30.Buch, M.H., S.J. Bingham, Y. Seto, D. McGonagle, V. Bejarano, J. White, and P. Emery. 2004. Lack of response to anakinra in rheumatoid arthritis following failure of tumor necrosis factor alpha blockade. Arthritis Rheum. 50:725–728. [DOI] [PubMed] [Google Scholar]
- 31.Muller, K., E.B. Herner, A. Stagg, K. Bendtzen, and P. Woo. 1998. Inflammatory cytokines and cytokine antagonists in whole blood cultures of patients with systemic juvenile chronic arthritis. Br. J. Rheumatol. 37:562–569. [DOI] [PubMed] [Google Scholar]
- 32.Muzaffer, M.A., J.M. Dayer, B.M. Feldman, W. Pruzanski, P. Roux-Lombard, R. Schneider, R.M. Laxer, and E.D. Silverman. 2002. Differences in the profiles of circulating levels of soluble tumor necrosis factor receptors and interleukin 1 receptor antagonist reflect the heterogeneity of the subgroups of juvenile rheumatoid arthritis. J. Rheumatol. 29:1071–1078. [PubMed] [Google Scholar]
- 33.Verbsky, J.W., and A.J. White. 2004. Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J. Rheumatol. 31:2071–2075. [PubMed] [Google Scholar]
- 34.Cassidy, J.T., J.E. Levinson, J.C. Bass, J. Baum, E.J. Brewer Jr., C.W. Fink, V. Hanson, J.C. Jacobs, A.T. Masi, J.G. Schaller, et al. 1986. A study of classification criteria for a diagnosis of juvenile rheumatoid arthritis. Arthritis Rheum. 29:274–281. [DOI] [PubMed] [Google Scholar]