

Abstract
The soluble methane monooxygenase (sMMO; EC 1.14.13.25) from the pseudothermophile Methylococcus capsulatus (Bath) is a three-component enzyme system that catalyzes the selective oxidation of methane to methanol. We have used NMR spectroscopy to produce a highly refined structure of MMOB, the 16-kDa regulatory protein of this system. This structure has a unique and intricate fold containing seven β-strands forming two β-sheets oriented perpendicular to each other and bridged by three α-helices. The rate and efficiency of the methane hydroxylation by sMMO depend on dynamic binding interactions of the hydroxylase with the reductase and regulatory protein components during catalysis. We have monitored by NMR the binding of MMOB to the hydroxylase in the presence and absence of the reductase. The results of these studies provide structural insight into how the regulatory protein interacts with the hydroxylase.
Methanotrophic bacteria abound at the interface of aerobic and anaerobic sediments, where they rely on methane as their primary source of carbon and energy. In the first step of metabolism, these organisms employ either soluble or membrane-bound methane monooxygenase (MMO; EC 1.14.13.25) systems to oxidize methane selectively to methanol (1, 2):
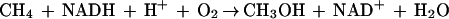 |
1 |
The soluble methane monooxygenase system (sMMO) isolated from the pseudothermophile Methylococcus capsulatus (Bath) functions optimally at 45°C. It is composed of a 251-kDa dinuclear iron-containing dimeric hydroxylase (MMOH), a 39-kDa NADH-dependent [2Fe–2S]- and FAD-containing reductase (MMOR), and a 16-kDa regulatory protein (MMOB).
The regulatory protein (MMOB) exists in solution as a monomer (3, 4) and binds to the hydroxylase dimer at two weakly interacting sites (3). When present at low concentration, MMOB converts MMOH from an oxidase to a hydroxylase (3, 5) and stabilizes intermediates required for the activation of dioxygen (6, 7). As the MMOB concentration is increased to saturate both binding sites, the hydroxylase activity is attenuated (3, 7).
The crystal structure of MMOH reveals two juxtaposed canyon regions formed by its α and β subunits (8). The dinuclear iron centers of the hydroxylase reside just 12 Å below the canyon floor, and it has been suggested that the binding sites of MMOR and MMOB may be located in these deep recesses (8). The results of spectroscopic (9, 10), chemical crosslinking (11), and steady-state kinetic and isothermal calorimetric (3) studies support this proposal.
We have used high-resolution NMR spectroscopy to determine the solution structure of the regulatory protein and to identify residues on its surface involved in hydroxylase binding. The effects of reductase on the MMOB/H interaction were also investigated.
MATERIALS AND METHODS
Sample Preparation.
MMOB was prepared from a recombinant expression system in Escherichia coli (ref. 12; D. Coufal, J. L. Blazyk, and S.J.L., unpublished work) and purified as described previously (3). NMR experiments were typically run at 25°C with 1.0 mM samples in 50 mM sodium phosphate buffer (pH 6.5) containing 4 mM Pefabloc (Boehringer Mannheim) and 0.1% sodium azide. Isotopically enriched samples were isolated from cells grown on M-9 medium supplemented with 15NH4Cl, 13C-glucose, and/or 2H2O (D2O).
NMR Spectroscopy.
All spectra were acquired at 750 MHz (Varian INOVA), 600 MHz (Bruker DMX), 500 MHz (Varian Unity or Varian INOVA), or 400 MHz (Varian Unity Plus). Backbone resonance assignments were obtained by using standard three-dimensional heteronuclear experiments and selective labeling techniques (13, 14). Distance restraints were obtained by using two 15N- (150- and 120-ms mixing time), one 13C-dispersed (80-ms mixing time), and two-dimensional (80- and 120-ms mixing time) nuclear Overhauser effect spectroscopy (NOESY) spectra. Spectra were processed with felix software packages (Hare Research/Biosym) and analyzed by using xeasy (15) on Silicon Graphics work stations.
Dihedral φ and χ1 angle constraints were obtained by using HNHA (16) and HNHB (17) and double quantum filtered COSY spectra (18), respectively. Additional dihedral φ and ψ angles were obtained with the talos program (19).
Structure Calculations.
Structure calculations were performed with the use of simulated annealing (20) in xplor 3.851 on R10000 IndigoII Silicon Graphics workstations (21). A total of 20 random structures were subjected to 50,000 simulated annealing and cooling steps of 0.005 ps at 3000 K. Coordinate and restraint files have been submitted to the Brookhaven protein database.
MMOB/H Titration.
To characterize the interaction with hydroxylase, a NMR titration study was performed in which the [15N,1H]-heteronuclear single-quantum coherence (HSQC) spectrum of MMOB was monitored upon addition of increasing concentrations of MMOH. Because significant decreases in hydroxylase activity occur after freezing and thawing cycles, fresh samples were prepared prior to each experiment. Concentrations were calculated by using extinction coefficients based on amino acid analysis (3). In all cases, MMOB was 15N-labeled and at 0.2 mM concentration. Ratios of 10:1, 6:1, 3:1, and 2:1 MMOB to hydroxylase were studied at both 25°C and 45°C with [15N,1H]-HSQC spectra. These experiments were then repeated in the presence of reductase. In these binding titration experiments, we observed differential line broadening of HSQC cross-peaks, indicating that it would be possible to locate the MMOB-binding site. To rationalize the use of differential line broadening in the slow chemical exchange limit, and to evaluate such an interaction with the hydroxylase, we conducted the analysis described below.
Analysis of Binding Data.
In the presence of hydroxylase the system examined by NMR spectroscopy contains the small, 16-kDa, MMOB protein in the free state and a large, 267-kDa, complex undergoing a binding equilibrium characterized by the lifetimes of the two states. Fast transverse relaxation rates in the bound state and large chemical shift differences between the two states can cause line broadening in free MMOB. With the use of an empirical approximation based on molecular weight (22), rotational correlation times of each species can be estimated. Thus, proton line widths for the complex are expected to be on the order of 250 Hz, which causes signals arising from bound MMOB to be absent from the spectra. When MMOB is present in excess, the lines of the free form are broadened because of exchange with the bound state. The amount of line broadening depends on kon and koff, the relaxation rates in the bound and free forms and, under favorable conditions, on chemical shift differences between the free and bound forms. Since resonances at the binding interface are expected to experience the largest chemical shift changes, this effect can be used to map binding interactions. To verify that chemical shift changes can produce broadened lines in the observable free MMOB signal, we have simulated the expected line shapes by using well-known formalisms derived previously (23). The line shape I(ω) is given by Eqs. 2–4:
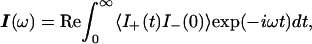 |
2 |
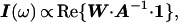 |
3 |
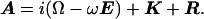 |
4 |
1 is a unity vector and E is an identity matrix. Matrix R contains the transverse relaxation rates in s−1 for free and bound MMOB where relaxation is assumed to be described by a single exponential. Matrices K and Ω contain the chemical exchange rates and chemical shifts, respectively, whereas W is a matrix containing the probability of occurrence at each of the bound and free frequencies. To simplify the calculation of the line shape, ωF was set equal to −ωB, where ωF and ωB are the chemical shifts of the free and bound states, respectively. Finally, the line shape was simulated by using Eq. 5:
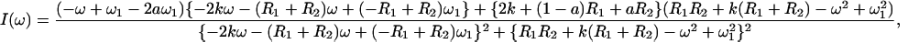 |
5 |
where a and k are the fraction of the free state and the inverse of the lifetime (koff) of the bound state, respectively. R1 and R2 are the transverse relaxation rates of the free and bound states, respectively, and ω1 is the chemical shift in the free state.
Simulations of the line width of MMOB in a 10:1 ratio over the complex are presented in Fig. 1. These calculations (Eq. 5) were performed with koff values of 3.2 s−1 (Fig. 1A) and 25.6 s−1 (Fig. 1B) and chemical shift differences, ω1, of 0 Hz (black) and 500 Hz (red). For clarity, the axis of each plot is offset such that ωfree = 0 Hz. These koff values represent experimentally determined rate constants for the dissociation of MMOB from MMOH at 25°C and 45°C, respectively (3). The chemical shift differences are conservative estimates of the anticipated changes in amide proton resonance frequencies upon binding as discussed below. It is clear from these simulations that peak heights decrease as values for the dissociation rate constants and chemical shift increase. A peak height reduction of greater than 20% results from a chemical shift difference of 0.8 ppm when koff is 25.6 s−1 (Fig. 1B). This reduction is consistent with our observations.
Figure 1.

Line shape simulations of MMOB titrations with MMOH by using Eq. 5 and koff values of 3.2 s−1 (A) or 25.6 s−1 (B) for chemical shift differences of 0 Hz (black) and 500 Hz (red). R1, R2, and a are 23 Hz, 250 Hz, and 0.8, respectively. In C is presented a comparison of observed peak height differences from [15N,1H]-HSQC spectra of MMOB in the absence of hydroxylase and at a 10 MMOB to 1 MMOH ratio. Differences are normalized according to Eq. 6, and the homology conservation color scheme of Fig. 3 is used to highlight the correlation between sequence conservation and binding data.
Simulations of Eq. 5 using a dissociation rate constant of 3.2 s−1 produce line shapes that are less sensitive to the chemical shift differences between the free and bound states than observed in the binding data (Fig. 1 A and B). MMOB binds MMOH at two interacting sites, which differ in affinity by approximately 4-fold at 25°C, and the dissociation rate constant of 3.2 s−1 corresponds to the higher-affinity site (3). A dissociation rate constant has not been reported for the low-affinity site; however, the decreased affinity may be the consequence of a significantly larger koff than the value of 3.2 s−1 reported for the high-affinity site. Substitution of a 4-fold larger koff value of 12.8 s−1 into Eq. 5 resulted in simulated line shapes that were representative of the low-temperature binding data. We conclude that the changes we observe in the NMR titration study at 25°C most likely arise from binding at the low-affinity site. The interacting sites binding behavior encountered at 25°C is not observed at 45°C (3). Correspondingly, at high temperatures, the NMR titration data reflect only a single type of binding interaction. It is possible that the subtle differences we observe in the binding data recorded at 25°C and 45°C are related to a temperature-dependent conformational change, which results in a higher affinity complex at 45°C.
Differential broadening of resonances was observed as the ratio of MMOB to MMOH was decreased. Because determining accurate half-heights was technically difficult, we measured the maximum peak height, which is inversely proportional to the line width. This parameter was obtained for each resonance of the [15N,1H]-HSQC spectra.
RESULTS
Structure Determination.
A summary of data used for the structural calculations is provided in Table 1. Of 20 random starting structures, 14 had no NOE or dihedral angle violation greater than 0.5 Å and 5°, respectively. Within regions of the defined secondary structural elements of residues 35–131, the 14 converged structures have an average backbone rmsd of 0.46 Å to the average structure (Fig. 2A).
Table 1.
Structural statistics for NMR structure calculations
| Parameter | Value |
|---|---|
| NOE distance restraints (Total) | 1,628 |
| Intraresidue | 707 |
| Interresidue | 912 |
| Medium | 211 |
| i, i + 2 | 88 |
| i, i + 3 | 65 |
| i, i + 4 | 58 |
| Long (|i − j| > 4) | 247 |
| Hydrogen bonds | 76 |
| Dihedral angle restraints, deg | |
| φ (C′(i−1)–Ni–Cαi–C′i) | 60 |
| ψ (Ni–Cαi–C′i–Ni+1) | 39 |
| χ1 (Ni–Cαi–Cβi–Cγi) | 30 |
| Ramachandran plot | |
| Most favorable region, % | 60.6 |
| Additionally allowed region, % | 32.7 |
| Generously allowed region, % | 5.8 |
| Disallowed region, % | 1.0 |
| Average rmsd for distance restraints, Å | 3.97 × 10−2 |
| Average rmsd for dihedral restraints, deg | 0.93 |
| Average rmsd from idealized covalent geometry | |
| Bonds, Å | 2.8 × 10−3 |
| Angles, deg | 0.43 |
| Improper angles | 0.29 |
| Average rmsd to average structure, Å | 0.45 |
rmsd, rms deviation.
Figure 2.

Stereoview of 14 converged structures (A) and representative ribbon diagram (B) of MMOB. Restraints are given in Table 1. In A, strictly conserved residues and those with conservative mutations are colored in cyan and purple, respectively. Residues W78 and Y117 are colored orange. Residues 1–30 and 135–141 are excluded, and the backbone of residues 37–44, 48–58, 68–72, 77–82, 85–87, 92–97, 109–113, 115–119, and 122–128 is used for alignment. (B) Ribbon diagram of the structure including terminal regions, the orientations of which are not defined. This figure was made by using molmol (28).
Description of the Structure.
A stereoview of the best-fit superposition of the backbone atoms of residues 35–131 displays the structural core containing two β-sheets and three α-helices (Fig. 2A). Residues 1–34 contain two partially folded helices (residues I12–K16 and A21–F24) but their orientation relative to the protein core is not defined. The C-terminal region is unstructured.
A secondary structure diagram of MMOB (Fig. 2B) reveals a remarkable, nearly perpendicular arrangement of the two β-sheets, composed of antiparallel β-strands, bridging three α-helices. β1 and β2 are separated by a long, well-defined helix, α1. The tight turn between β2 and β3 arches back, away from α1, such that R73 covers W78. W77 forms close contacts with residues within α1. After a sharp bend, β3 becomes β4, which forms the first strand of the second sheet. Between β4 and β5 are two helices, α2 and α3. α2 comes in close contact with α1, with 20 NOEs defining their relative orientation. Residues 98–106 are not well defined, but there is a small helix containing residues 102–106. Helix α3 contains two aromatic residues, one of which, F105, forms a stacking interaction with F123 of β6. β5 is the final strand of the first sheet and turns at a 90° angle to become β6. β7, the last β-strand, forms hydrogen bonds to β4 and β6.
The MMOB structural core is formed through numerous long-range hydrophobic interactions between the β-sheets and helices and between the helices with each other. Residues located on the interior side of the four-stranded β-sheet come in close contact with α1. The C-terminal residues of α1 facing the four-stranded β-sheet, including residues I55, L56, and Y60, come in close contact with V68, while the N-terminal residues of α1, including residues I48, N49, and I52, come in close contact with V70, W77, I79, and T112. The opposite side of α1 is close to α2, and 20 NOEs define their relative orientations. Helix α3 comes in close contact with the three-stranded β-sheet as F105 stacks with F123.
MMOB Has a Protein Fold Not Previously Described.
We submitted the structure of MMOB to the dali server (24), which did not find similar folds. The closest related folds are for the metalloexozymogen procarboxypeptidase A (PCPA) alone and in complex with proproteinase E and chymotrypsinogen C, the Z-values of which are 2.1 and 2.5, respectively. The globular part of PCPA contains a four-stranded antiparallel β-sheet covered on one surface with two helices. Whereas the orientation of the helices relative to the β-sheet is similar to that of MMOB, this protein contains only one β-sheet and thereby lacks the more complicated β-sheet orientation of MMOB. Thus, MMOB can be considered to have a novel fold.
Homologous Proteins.
Seven methane and aromatic multicomponent monooxygenases have protein components with sequence similarities (Fig. 3) (4). There are 10 residues that are conserved in at least six of these sequences: S2, V38, I52, E53, I79, G83, E94, L96, G97, and G114, and it is likely that some of these are involved in binding interactions of MMOB with the hydroxylase. Four residues (V38, I52, I79, and L96) are involved in critical tertiary interactions. The strictly conserved glycine residues, G83 and G114, adopt φ (C′(i−1)−Ni–Cαi–C′i) and ψ (Ni–Cαi–C′i–N(i+1)) dihedral angles inaccessible to other residues, which allows the orthogonal orientation of the β-sheets. Conserved residue G97 at the end of α2 similarly adopts otherwise inaccessible dihedral angles to make a well-ordered abrupt turn.
Figure 3.

Amino acid sequence comparisons for MMOB and related hydroxylase cofactor proteins. MMOB proteins labeled B-caps, B-trich, and B-st.M are from M. capsulatus (Bath), Methylosinus trichosporium, and Methylocystis sp. strain M. Cofactor proteins for the toluene monooxygenases systems include TMOD, protein D of the toluene-4-monooxygenase system from Pseudomonas mendocina; T3MOD, cofactor protein of the toluene-3-monooxygenase system from Pseudomonas picketti; and TbmC, cofactor protein of the toluene/benzene-2-monooxygenase system from Pseudomonas sp. CF600. The two phenol hydroxylase cofactor proteins include PH3_ac of Acinetobacter calcoaceticus and P2 from Pseudomonas putida. Helices and β-strands of MMOB (B-caps) are provided and labeled above the primary sequence as arrows and bars, respectively. Residues conserved in at least six of the seven sequences are shaded cyan, while those with conservative mutations are shaded purple. Remaining residues that are identical in all MMOB species are shaded gray. This alignment is described in more detail elsewhere (4).
Fig. 4A shows a mapping of the conserved residues onto the MMOB surface. L96, G97, and F100 form a cluster of conserved residues with D108 in close vicinity. E94 and E53 are similarly surface exposed and lack an obvious structural role. The implications of the sequence homology in the binding of MMOB to MMOH are discussed below.
Figure 4.

Mapping of homology (A), binding (B), and electrostatic (C) data onto a surface representation of MMOB. Structures on the right are rotated by 180° relative to those on the left. The terminal regions are shown, but their orientations relative to the structural core are undefined. In A, the homology color scheme follows that of Fig. 3. In B, the critical residues involved in hydroxylase binding at 25°C are mapped onto the surface by evaluating the change in peak height of each residue at a concentration of 10 MMOB to 1 MMOH according to Eq. 6. Blue and red regions correspond to residues with backbone amides experiencing greater and less than average broadening, respectively. In C, basic residues are shown in blue and acidic residues in red. White regions indicate uncharged surfaces. Parameters used to generate this figure are −70 to −35 and 70 to 35 kT. These structures were produced with grasp (29).
MMOB/H Binding Interaction.
Line widths and cross-peak heights of [1H,15N]-HSQC spectra of MMOB at various ratios of MMOB to MMOH were determined and plotted according to Eq. 6:
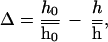 |
6 |
where h0 and h are the peak heights of free MMOB in the absence of hydroxylase and at a given MMOB to MMOH ratio, respectively. The symbols  and
and  represent the average peak heights at the corresponding MMOB to MMOH ratios. As discussed earlier, bound MMOB is unobservable because of its long rotational correlation time and cross-peaks corresponding to free MMOB at ratios greater than 2 MMOB to 1 MMOH are exchange broadened. Such a line shape analysis at a ratio of 10 MMOB to 1 MMOH for data collected at 25°C is provided in Fig. 1C.
represent the average peak heights at the corresponding MMOB to MMOH ratios. As discussed earlier, bound MMOB is unobservable because of its long rotational correlation time and cross-peaks corresponding to free MMOB at ratios greater than 2 MMOB to 1 MMOH are exchange broadened. Such a line shape analysis at a ratio of 10 MMOB to 1 MMOH for data collected at 25°C is provided in Fig. 1C.
When these data are mapped onto a surface diagram (Fig. 4B), the “northern half” of MMOB in this representation appears to form hydroxylase interactions, in contrast to the less affected “southern half” containing the terminal regions (Fig. 4B). Furthermore, residues that are conserved in the primary sequence of MMOB (Fig. 3) are among those most significantly affected by the hydroxylase binding interactions (Fig. 4 A and B). In particular, the conserved amino acids L96, G97, F100, and D108 are within a cluster of residues having cross-peaks that are significantly broadened upon binding. The residues in β6, including G114, R115, and Y117, are similarly affected by the presence of hydroxylase. β6, which is conserved among the hydroxylase regulatory protein family, contains a conserved hydrophobic residue at position 116 (Fig. 3). Residues D22 through A26 of the N-terminal region experience greater than average broadening upon binding. These residues form a short helix and residues F24, F25, and A26 are strictly conserved in all three MMO component proteins.
This analysis was repeated for experiments conducted at 45°C with only subtle differences. First, residues E54, L57, K61, and K62, which form a cluster on the surface of MMOB (Fig. 4B Left), are less affected. Second, the significant broadening of residues D22 through A26 is absent. A more detailed statement awaits the structure determination of the regulatory protein at 45°C.
The Interaction Face Is Hydrophobic.
An electrostatic surface diagram of MMOB reveals that, although some charged residues appear on the surface, MMOB is largely hydrophobic (Fig. 4C). The aromatic residues, Y117, F100, and W78, are surface exposed and significantly broadened, indicating that they may form hydrophobic interactions with the hydroxylase. The general conclusion that the interaction of the hydroxylase and MMOB protein involves hydrophobic contacts is consistent with the observed increase in entropy upon binding (3) and the finding that binding affinity increases with salt concentration (4).
Reductase Does Not Affect MMOB/H Interactions.
Comparing NMR line shapes and chemical shifts of the [15N,1H]-HSQC spectrum of MMOB upon addition of the reductase reveals that these two proteins do not interact in the absence of hydroxylase (data not shown). To determine whether MMOR and MMOB compete for binding sites on the hydroxylase, the MMOB/H titrations were repeated in the presence of excess MMOR. No significant changes in the line shape or chemical shifts of the MMOB protein were observed upon addition of MMOR, indicating that MMOR and MMOB do not interact in either the presence or the absence of hydroxylase (data not shown).
Extended Termini.
Our titration data suggest that the long unstructured N terminus of MMOB is less involved in binding at 45°C. At 25°C, however, cross-peaks corresponding to residues D22 through A26 experience greater than average broadening (Fig. 4B). Many N- and C-terminal residues appear weakly in the 25°C spectra at MMOB to MMOH ratios of 2:1, however, where all other residues are absent because of the binding. 15N relaxation experiments on free MMOB clearly indicate that the termini do experience greater flexibility than the core (data not shown), and this property appears to be maintained in the presence of the hydroxylase. The extended structure of the N terminus, which we observe in our NMR structure, is consistent with anomalous behavior of MMOB as a dimer in gel filtration studies (3, 4).
MMOB/H Model.
It has been proposed that MMOB binds in the canyon regions formed by the α and β subunits of the hydroxylase (8). We manually docked MMOB into the canyon region of the oxidized hydroxylase in the orientation best satisfying our binding data (Fig. 5). The MMOB structure can be easily positioned into the large canyon, and its mainly hydrophobic surface is ideally suited for binding the very hydrophobic cleft at the α2β2 dimer interface of the hydroxylase.
Figure 5.

A surface diagram model for docking MMOB (top) into the canyon of MMOH (bottom). Each subunit of MMOH is distinguished by color, whereas MMOB is colored according to our binding data. Residues of MMOB most affected by binding are colored blue and those least affected are red. For clarity, MMOB has been translated away from its proposed docking site on the surface of the hydroxylase and rotated clockwise about the y-axis by 90° to expose residues most involved in binding. This figure was produced by using grasp (29).
DISCUSSION
The compact structural core of MMOB forms an unusual fold containing two orthogonal β-sheets bridging three α-helices. Whereas the termini possess a helical tendency, they are quite disordered, and we were unable to orient them relative to the core. The functional significance of the termini is unclear. The N terminus of MMOB is quite susceptible to proteolysis (25), and the active forms of the regulatory proteins from related hydroxylases are N-terminally truncated relative to MMOB (Fig. 3). Mutant versions of MMOB that lack the N-terminal regions have significantly lower activity than the native protein (4). Proteolytically truncated MMOB binds to MMOH with only slightly lower affinity than the native protein, but loses its ability to tune the redox potential of the dinuclear iron centers of sMMO (26). The three sMMO regulatory protein sequences share high sequence conservation in their N termini, and our binding data suggest that D22 to A26 interact with the hydroxylase at 25°C.
Conserved Residues Convey Structural and Functional Information.
The above analysis of the sequence homology alignment of MMOB with related hydroxylase cofactor proteins indicates the presence of many conserved residues that are critical in forming the MMOB structure. This conservation suggests that the structures of the seven proteins are similar. In particular, the conservation of G83 and G114 supports a similar orthogonal arrangement of two β-sheets in the homologous proteins. Furthermore, conserved residue I79 of β3 comes into juxtaposition with conserved residue I52 of α1, suggesting that the close proximity of α1 to the four-stranded β-sheet is common to all proteins of this family.
A preliminary NMR structure of the homologous phenol hydroxylase protein component P2 has been reported (27). The secondary structure of P2 is slightly different from that of MMOB and the tertiary structure is much less compact. The angle between the β-sheets is obtuse in the P2 structure, and α1 and α2 are relatively far apart. As predicted, however, the homologous residues for I52 and I79 are in close proximity.
Highly conserved residues lacking roles in maintaining the MMOB structure are likely to participate in hydroxylase binding. The sequence alignment (Fig. 3) contains 10 strictly and 12 moderately conserved residues. A cluster of conserved residues, namely L96, G97, F100, V107, and D108, appears on the back surface of MMOB (Fig. 4 A and B Right), and this same set was hypothesized to form interactions with the hydroxylase by our binding data.
The surface diagram of MMOB contains a large number of exposed hydrophobic residues, including L96, F100, and V107. It has been previously suggested that hydrophobic interactions dominate MMOB/H binding (4). In addition to these three residues, MMOB/H binding data indicate interactions between the hydroxylase and W78, A116, and Y117. Homology considerations combined with the binding data reveal good candidates for site-directed mutagenesis studies.
Line Broadening Can Be Correlated with Hydroxylase Interactions.
Solving the structure of MMOB provides an opportunity to understand the interaction of MMOB with the hydroxylase. Adding MMOH to a solution of MMOB produces differential line broadening in the HSQC spectrum of MMOB. Free MMOB exchanges in the slow to intermediate time regime with a bound state having very broad line widths. Our simulations predict that, upon addition of hydroxylase, the HSQC cross-peaks of free MMOB can be sensitive to chemical shift differences between the bound and free states and that this information can be used to identify the binding site on the “northern” face of the protein core (Fig. 4B). The terminal regions have much less than average broadening. These regions are highly flexible in both the free and bound state of MMOB, and their cross-peaks are still visible in HSQC spectra at concentrations of MMOH where all core resonances are absent from broadening.
Although we can simulate our data well by using Eq. 5 with a change in chemical shift of 500 Hz between the free and bound states, we have not included relaxation of MMOB caused by the paramagnetic iron centers of MMOH. Contacts with the paramagnetic hydroxylase could cause chemical shift changes much larger than those expected for interaction with a diamagnetic protein and could broaden the lines in the bound state well beyond the estimated 250 Hz. When the residues most broadened in the presence of hydroxylase are mapped onto a surface diagram, they do not follow an obvious r−6 distance-dependent broadening characteristic of paramagnetic relaxation. Instead, residues most broadened are located in regions remote from one another. Hence, although one such region on the surface diagram could be experiencing the effects of the paramagnetism, paramagnetic relaxation is clearly not the only phenomenon contributing to broadening. Furthermore, the Fe atoms of the hydroxylase are rather buried and, when positioned in the hydroxylase canyon, MMOB is >10 Å away from these centers.
We have used our binding data to predict residues of MMOB that form critical interactions with the hydroxylase. Our predictions are supported by sequence conservation among homologous hydroxylase cofactor proteins, but our analysis assumes that chemical shift changes arising from structural changes of MMOB upon complex formation are negligible. Without structural data on MMOB in the complexed form, we cannot be certain that such structural perturbations of the protein cause broadening unrelated to interactions with the hydroxylase.
MMOB Is Easily Positioned in the Canyon Formed by the MMOH Dimer Interface.
Steric considerations suggest that MMOB binds in the canyon formed by the α and β subunits of the dimeric hydroxylase, and crosslinking studies suggest that MMOB binds to the α subunit of the hydroxylase (11). The space within this canyon does not get completely filled after manual insertion of MMOB. The structure and binding location of the reductase are currently unknown, although crosslinking experiments indicate that it binds to the β subunit of the hydroxylase (11). Our NMR binding and other (3) data indicate that the reductase and MMOB do not compete for binding sites on the hydroxylase and do not interact with each other in the absence or presence of hydroxylase.
In conclusion, we have solved the structure of the sMMO hydroxylase regulatory protein. MMOB forms an intricate and unusual fold. The largely hydrophobic surface is ideally suited for binding the hydrophobic cleft formed at the α2β2 interface of the hydroxylase. Whereas MMOB couples NADH consumption by MMOR to methane hydroxylation by MMOH, it does not appear to interact directly with the reductase. Instead, this regulatory function can be explained by an induced conformational change on the hydroxylase upon MMOB binding. The present structure and titration data are important contributions in understanding the highly complex sMMO systems.
Acknowledgments
We are grateful to Dr. Hiroshi Matsuo for helpful discussions regarding analysis of the binding data and to Greg Heffron for technical assistance. This work was supported by grants from the National Institute of General Medical Sciences (GM32134 to S.J.L. and GM47467 to G.W.) and the National Science Foundation (MCB 9527181 to G.W.). G.T.G. is a National Institutes of Health Postdoctoral Fellow.
ABBREVIATIONS
- MMO
methane monooxygenase
- sMMO
soluble methane monooxygenase
- MMOH
hydroxylase protein of MMO isolated from Methylococcus capsulatus (Bath)
- MMOB
MMO regulatory protein B
- MMOR
MMO reductase
- HSQC
heteronuclear single-quantum coherence
- NOE
nuclear Overhauser effect
- NOESY
NOE spectroscopy
- rmsd
rms deviation
Note Added in Proof
We call the reader’s attention to a recently published (30) article describing the NMR structure of protein B from Methylosinus trichosporium OB3b.
Footnotes
Data deposition: The coordinate and restraint files have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (PDB ID code 1ckv for coordinates and r1ckvmr for restraints).
References
- 1.Higgins I J, Best D J, Hammond R C, Scott D. Microbiol Rev. 1981;45:556–590. doi: 10.1128/mr.45.4.556-590.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whittenbury R, Phillips K C, Wilkinson J F. J Gen Microbiol. 1970;61:205–218. doi: 10.1099/00221287-61-2-205. [DOI] [PubMed] [Google Scholar]
- 3.Gassner, G. T. & Lippard, S. J. (1999) Biochemistry, in press. [DOI] [PubMed]
- 4.Brandstetter H, Whittington D A, Lippard S J, Frederick C A. Chem Biol. 1999;6:441–449. doi: 10.1016/s1074-5521(99)80062-3. [DOI] [PubMed] [Google Scholar]
- 5.Lund J, Woodlund M P, Dalton H. Eur J Biochem. 1985;147:297–305. doi: 10.1111/j.1432-1033.1985.tb08750.x. [DOI] [PubMed] [Google Scholar]
- 6.Lee S-K, Nesheim J C, Lipscomb J D. J Biol Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 7.Liu K E, Valentine A M, Wang D, Huynh B H, Edmondson D E, Salifoglou A, Lippard S J. J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 8.Rosenzweig A C, Frederick C A, Lippard S J, Nordlund P. Nature (London) 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 9.Pulver S, Froland W A, Fox B G, Lipscomb J D, Solomon E I. J Am Chem Soc. 1993;115:12409–12422. [Google Scholar]
- 10.Davydov R, Valentine A M, Komar-Panicucci S, Hoffman B M, Lippard S J. Biochemistry. 1999;38:4188–4197. doi: 10.1021/bi982391o. [DOI] [PubMed] [Google Scholar]
- 11.Fox B G, Liu Y, Dege J E, Lipscomb J D. J Biol Chem. 1991;266:540–550. [PubMed] [Google Scholar]
- 12.DeWitt J G, Rosenzweig A C, Salifoglou A, Hedman B, Lippard S J, Hodgson K O. Inorg Chem. 1995;34:2505–2515. [Google Scholar]
- 13.Matsuo H, Li H, McGuire A M, Fletcher C M, Gringras A-C, Sonenberg N, Wagner G. Nat Struct Biol. 1997;4:717–724. doi: 10.1038/nsb0997-717. [DOI] [PubMed] [Google Scholar]
- 14.Muchmore D C, McIntosh L P, Russell C B, Anderson D E, Dahlquist F W. Methods Enzymol. 1989;177:44–73. doi: 10.1016/0076-6879(89)77005-1. [DOI] [PubMed] [Google Scholar]
- 15.Bartels C, Xia T-H, Billeter M, Güntert P, Wüthrich K. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 16.Vuister G W, Bax A. J Am Chem Soc. 1993;115:7772–7777. [Google Scholar]
- 17.Archer S J, Ikura M, Torchia D A, Bax A. J Magn Reson. 1991;95:636–641. [Google Scholar]
- 18.Piantini U, Sørensen O W, Ernst R R. J Am Chem Soc. 1982;104:6800–6801. [Google Scholar]
- 19.Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 20.Nilges M, Clore G M, Gronenborn A M. FEBS Lett. 1988;239:129–136. doi: 10.1016/0014-5793(88)80559-3. [DOI] [PubMed] [Google Scholar]
- 21.Brünger A T. xplor Version 3.1: A System for X-Ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1993. [Google Scholar]
- 22.Wagner G. Nat Struct Biol. 1997;4:841–844. [PubMed] [Google Scholar]
- 23.Abragam A. Principles of Nuclear Magnetism. New York: Oxford Univ. Press; 1961. [Google Scholar]
- 24.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 25.Lloyd J S, Bhambra A, Murrell J C, Dalton H. Eur J Biochem. 1997;248:72–79. doi: 10.1111/j.1432-1033.1997.t01-1-00072.x. [DOI] [PubMed] [Google Scholar]
- 26.Kazlauskaite J, Hill H A O, Wilkins P C, Dalton H. Eur J Biochem. 1996;241:552–556. doi: 10.1111/j.1432-1033.1996.00552.x. [DOI] [PubMed] [Google Scholar]
- 27.Qian H, Edlund U, Powlowski J, Shingler V, Sethson I. Biochemistry. 1997;36:495–504. doi: 10.1021/bi9619233. [DOI] [PubMed] [Google Scholar]
- 28.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 29.Nicholls A J. grasp Manual. New York: Columbia Univ.; 1993. [Google Scholar]
- 30.Chang S-L, Wallar B J, Lipscomb J D, Mayo K H. Biochemistry. 1999;38:5799–5812. doi: 10.1021/bi982992f. [DOI] [PubMed] [Google Scholar]