

Abstract
Aspirin (ASA) triggers a switch in the biosynthesis of lipid mediators, inhibiting prostanoid production and initiating 15-epi-lipoxin generation through the acetylation of cyclooxygenase II. These aspirin-triggered lipoxins (ATL) may mediate some of ASA’s beneficial actions and therefore are of interest in the search for novel antiinflammatories that could manifest fewer unwanted side effects. Here, we report that design modifications to native ATL structure prolong its biostability in vivo. In mouse whole blood, ATL analogs protected at carbon 15 [15(R/S)-methyl-lipoxin A4 (ATLa1)] and the omega end [15-epi-16-(para-fluoro)-phenoxy-LXA4 (ATLa2)] were recoverable to ≈90 and 100% at 3 hr, respectively, compared with a ≈40% loss of native lipoxin A4. ATLa2 retains bioactivity and, at levels as low as ≈24 nmol/mouse, potently inhibited tumor necrosis factor-α-induced leukocyte recruitment into the dorsal air pouch. Inhibition was evident by either local intra-air pouch delivery (≈77% inhibition) or systemic delivery by intravenous injection (≈85% inhibition) and proved more potent than local delivery of ASA. Rank order for inhibiting polymorphonuclear leukocyte infiltration was: ATLa2 (10 μg, i.v.) ≈ATLa2 (10 μg, local) ≈dexamethasone (10 μg, local) >ASA (1.0 mg, local). Applied topically to mouse ear skin, ATLa2 also inhibited polymorphonuclear leukocyte infiltration induced by leukotriene B4 (≈78% inhibition) or phorbol ester (≈49% inhibition), which initiates endogenous chemokine production. These results indicate that this fluorinated analog of natural aspirin-triggered lipoxin A4 is bioavailable by either local or systemic delivery routes and is a more potent and precise inhibitor of neutrophil accumulation than is ASA.
Aspirin (acetylsalicylic acid; ASA) has been available for use as an analgesic-antipyretic for almost a century (1) and novel therapeutic applications for this drug, for example in lowering the risk of myocardial infarction (2) or as a prophylaxis against colorectal cancer (3), continue to be uncovered. The acetylation of cyclooxygenases I and II (COX I and II) and the subsequent irreversible inhibition of prostaglandin and thromboxane biosyntheses are well understood mechanisms of some of ASA’s pharmacological actions (4, 5). More recently, ASA was found to cause a switch in eicosanoid biosynthesis as the acetylation of COX II changes the enzyme’s activity to produce 15R-hydroxyeicosatetraenoic acid from agonist-released arachidonic acid (5). Human neutrophils and other cells possessing 5-lipoxygenase use this substrate via transcellular biosynthetic routes to produce 15-epi-lipoxin A4 (15-epi-LXA4) and 15-epi-lipoxin B4 (15-epi-LXB4) (6, 7). These aspirin-triggered lipoxins (ATL) are the endogenous 15R enantiomeric counterparts of lipoxin A4 (LXA4) and lipoxin B4 (LXB4) respectively, and share their bioactivities (reviewed in ref. 6).
Unlike other eicosanoids (e.g., leukotrienes, prostaglandins, etc.), which are generally considered local proinflammatory mediators, lipoxins (LX) display potent inhibitory actions in several key events in inflammation, such as polymorphonuclear cell (PMN) chemotaxis, transmigration across endothelial and epithelial cells, and diapedesis from postcapillary venules (6). LX are generated in several pathogenic scenarios in vivo, for example: in lung tissue of patients with severe pulmonary disease (8) and by PMN from patients with asthma (9, 10) or rheumatoid arthritis (11), where their presence is proposed to be linked to long-term clinical improvement. Interestingly, ATL show an even greater level of inhibition than native LX in preventing neutrophil adhesion, where they are approximately twice as potent (6). ATL are also more potent inhibitors of microbial induction of cytokine release. Specifically, 15-epi-LXA4 showed greater inhibition than LXA4 of Salmonella typhimurium-induced secretion and gene regulation of the potent leukocyte chemoattractant IL-8, generated by intestinal epithelial cells (12). It is therefore likely that, in addition to the inhibition of prostaglandin formation, the benefits of ASA therapy also result from the triggering of antiinflammatory lipid mediators that act locally to down-regulate leukocytes.
PMN accumulation and activation play central roles in the pathogenesis of a wide range of disease states as diverse as rheumatoid arthritis (13), atherosclerosis (14), ulcerative colitis (15, 16), and psoriasis (17). Hence the elucidation of endogenous regulatory mechanisms that can control neutrophil functions is of considerable therapeutic interest. Because they are small lipophilic compounds amenable to total organic synthesis, the natural lipoxins, and specifically their endogenous isoform ATL, are well suited as potential leads for novel small molecule therapeutics as well as pharmacological tools for uncovering endogenous counter-regulatory and/or antiinflammatory signaling pathways.
Design modifications that enhance biostability are advantageous because the lipoxins are autacoids that are rapidly biosynthesized in response to stimuli, in turn elicit counter-regulatory responses, and then are rapidly enzymatically inactivated (6). 15-Hydroxy-prostaglandin dehydrogenase, which catalyzes the reversible oxidation of the carbon-15 position alcohol group of prostaglandins (18), also catalyzes the first step of lipoxin inactivation (Fig. 1A Inset) (19, 20). In view of these findings, several stable analogs of ATL and LXA4 were designed that resist oxidation at carbon-15 by recombinant dehydrogenase in vitro (21). Both LXA4 and 15-epi-LXA4 act at common LXA4 receptors on leukocytes and are active within the nanomolar range, inhibiting PMN transmigration, diapedesis, and vascular permeability (21–23). The stable LXA4 and ATL analogs also compete at this LXA4 receptor (23). Here, we report that design modifications to native ATL biostabilize these mediators in whole blood to resist rapid inactivation. Moreover, the fluorinated ATL analog, namely 15-epi-16-(para-fluoro)-phenoxy-LXA4 (ATLa2), is a potent inhibitor of PMN recruitment in murine in vivo models when administered through both local and systemic routes.
Figure 1.

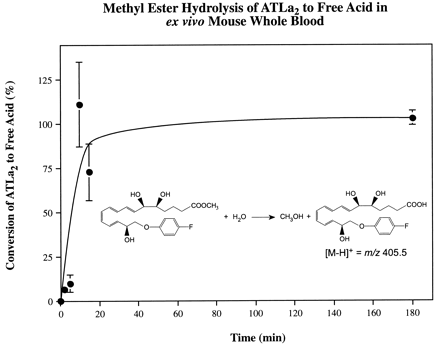
(A) Initial metabolic step of LXA4 inactivation in mouse whole blood and 15-oxo-LXA4 MS/MS spectrum. LXA4 (21 μM) was incubated ex vivo in mouse whole blood for 3 hr. The MS/MS spectrum of the major oxo-product is indicative of 15-oxo-LXA4, with diagnostic product ions at m/z: 349 (a = [M-H]−), 331 (a - H2O), 313 (a - 2H2O), 305 (b = [M-H]− - CO2), 287 (b - H2O), 269 (b - 2H2O), 233 (c), and 217 (c - O). (B) Biostability of LXA4 and stable analogs in mouse whole blood. LXA4, ATLa1, which carries a racemic methyl group at C-15, and 15-epi-16-(para-fluoro)-phenoxy-LXA4 [ATLa2, in which a bulky (para-fluoro)-phenoxy group replaces the ω-chain at C-16] were added (see Materials and Methods) to heparinized mouse whole blood and incubated at 37°C for 0 and 3 hr. After centrifugation at 800 × g and 0°C, the plasma supernatants were drawn off and stopped in two volumes of ice-cold methanol. The lipoxins were extracted by solid phase methodology and quantitated by LC/MS/MS. Values represent mean ± SEM (n = 3–4).
MATERIALS AND METHODS
Biostability.
The analogs 15(R/S)-methyl-LXA4 (ATLa1) and ATLa2 were prepared by total organic synthesis (21) and their structures confirmed by NMR. Male BALB/c mice (6- to 8-wk old) (Harlan—Sprague–Dawley) were anesthetized with pentobarbital (70 mg/kg), and whole blood was drawn via cardiac puncture into heparin (500 units/ml). LXA4, ATLa1, and ATLa2 (2.4 μM) were incubated in 250 μl of blood (37°C) for either 0 or 3 hr. For time zero (T = 0), the blood aliquots were placed in an ice bath for 1 min and, immediately after the addition of LXA4 or ATLa, were centrifuged at 800 × g at 0°C for 20 min. The plasma supernatants were collected, stopped in 400 μl of ice cold methanol, and stored at −20°C before solid phase extraction. For T = 3 hr, the blood aliquots were incubated with ATLa and gently mixed by shaking at 37°C. After each incubation period, the plasma was collected and stopped as above. Prostaglandin B2 (Oxford Biomedical Research, Oxford, MI) was added to the blood samples immediately before centrifugation as an internal standard for extraction recovery. Denatured protein precipitates were pelleted from the stopped plasma samples and were washed twice with 200 μl of methanol. The plasma supernatant and washes were pooled and extracted with Extract-Clean solid phase extraction cartridges (500 mg C18, Alltech Associates). The methyl formate fractions were taken to dryness with a gentle stream of nitrogen and suspended in methanol for injection and quantitative analyses by UV spectrophotometry and liquid chromatography tandem mass spectrometry (LC/MS/MS).
LC/MS/MS Analyses.
LC/MS/MS was performed using an LCQ (Finnigan-MAT, San Jose, CA) quadrupole ion trap mass spectrometer system equipped with an electrospray atmospheric pressure ionization probe. Samples were suspended in methanol and injected into the HPLC component, which consisted of a SpectraSYSTEM P4000 (Thermo Separation Products, San Jose, CA) quaternary gradient pump, a Prodigy octadecylsilane-3 (100 × 2 mm, 5 μm) column (Phenomenex, Belmont, CA) or a LUNA C18–2 (150 × 2 mm, 5 μm) column, and a rapid spectra scanning SpectraSYSTEM UV2000 (Thermo Separation Products) UV/VIS absorbance detector. The column was eluted isocratically with methanol/water/acetic acid (65:35:0.01, vol/vol/v) at 0.2 ml/min into the electrospray probe. The spray voltage was set to 5–6 kV and the heated capillary to 250°C. LXA4 and the ATLa were quantitated by selected ion monitoring for analyte molecular anions (e.g., [M-H]− = m/z 351.5 for LXA4, m/z 365.5 for ATLa1, and m/z 405.5 for ATLa2 free acid) or by UV absorbance at 300 nm. Product ion mass spectra (MS/MS) were also acquired for definitive identification of the compounds.
PMN Infiltration into Mouse Air Pouch.
While male BALB/c mice (6–8 wk) were anesthetized with isoflurane, dorsal air pouches were raised by injecting 3 ml of sterile air subcutaneously on days 0 and 3 (as in ref. 24). On day 6 and while the mice were anesthetized with isoflurane, 10 μg of ATLa2 was delivered as a bolus injection either into the tail vein in 100 μl of sterile 0.9% saline or locally into the air pouch in 900 μl of PBS −/− (Dulbecco’s Phosphate Buffered Saline without magnesium or calcium ions, BioWhittaker). Dexamethasone and ASA (Sigma) were delivered locally as 10 μg and 1.0 mg doses in 900 μl of PBS −/−, respectively. Inflammation in the air pouch was induced by local injection of recombinant murine tumor necrosis factor α (TNF-α) (20 ng) (Boehringer Mannheim) dissolved in 100 μl of sterile PBS. While the mice were anesthetized with isoflurane, the air pouches were lavaged twice with 3 ml of sterile PBS 4 hr after the initial TNF-α injection. Aspirates were centrifuged at 2,000 rpm for 15 min at 23°C. The supernatants were removed, and the cells were suspended in 500 μl of PBS. Aliquots of the cell suspension were stained with Trypan blue and enumerated by light microscopy. Resuspended aspirate cells (50 μl) were added to 150 μl of 30% BSA and centrifuged onto microscope slides at 2,200 rpm for 4 min by using a Cytofuge (Statspin, Norwood, MA). Slides were allowed to air dry and were stained with Wright Giemsa stain (Sigma) for determination of differential leukocyte counts. For microscopic analysis, tissues were obtained with a 6-mm tissue biopsy punch (Acu-Punch, Acuderm, Ft. Lauderdale, FL) and fixed in 10% buffered formaldehyde. Samples were then embedded in paraffin, sliced and stained with hematoxylin-eosin.
Arterial Pressure.
Male BALB/c mice (6- to 8-wk old, 20 g) were anesthetized with pentobarbital (80 mg/kg). The trachea was isolated and a small polyethylene catheter (PE50) was introduced to maintain a patent airway. The right carotid artery was isolated and cannulated with PE10 tubing filled with heparinized (10 units/ml) normal saline. The arterial catheter was connected to a pressure transducer (World Precision Instruments, Sarasota, FL), and the arterial pressure tracing was recorded continuously (Astromed MT95K2, West Warwick, RI). All surgical manipulations were performed by using a surgical microscope (Zeiss).
PMN Infiltration into Ear Skin.
The mouse ear inflammation model (22) was used to evaluate the impacts of i.v. and topical deliveries of ATLa2 on leukotriene B4 (LTB4)- and phorbol 12-myristate 13-acetate (PMA)-induced PMN infiltration. Briefly, ATLa2 either was applied topically (20 μg in 10 μl acetone) to the inner side of the left mouse ear with vehicle applied contralaterally or was delivered as a bolus injection (10 μg in 100 μl of 0.9% sterile saline) through the tail vein. Five to seven minutes later, inflammation was induced in left and right ears of the mice that received topical ATLa2 (left ear only in the mice receiving i.v. delivery of ATLa2) by topical application of either LTB4 (1 μg) or PMA (100 ng) in acetone (10 μl). After 24 hr, 6-mm-diameter tissue punch biopsies were taken (Acu-Punch) from the ears and assayed by the method of Bradley et al. (25) for myeloperoxidase activity as an index of PMN number. Isolated murine PMNs were enumerated by light microscopy and processed in the same manner to obtain a calibration curve.
Plasma Clearance.
The time course for the clearance of ATLa2 from plasma after tail vein injection was determined over 50 min. Male BALB/c mice (6- to 8-wk old, 20 g) were anesthetized with pentobarbital (70 mg/kg) and received bolus tail vein injections of 27 μM ATLa2 (0.1 mg/kg) in 100 μl of sterile 0.9% saline. Blood was taken from the mice by cardiac puncture at 2, 5, 10, 15, and 50 min postinjection. The plasma was obtained and extracted as above, with the methyl formate fractions from the solid phase extraction dried down for LC/MS/MS analysis. Values for ATLa2 quantified in plasma are expressed in units of ng/ml plasma taken from the mouse, with n = 3 for each time point.
RESULTS
Biostability.
After 3-hr incubations of LXA4 in mouse whole blood ex vivo, the predominant metabolite peak observed in the LC/MS chromatogram of the extracted sample had a retention time and MS/MS spectrum matching that of 15-oxo-LXA4, as generated by recombinant 15-hydroxy-prostaglandin dehydrogenase from synthetic LXA4 (Fig. 1A). To determine whether addition of bulky substituents to the native LX structure enhances biostability, two aspirin-triggered lipoxin stable analogs, ATLa1 and ATLa2, were incubated in mouse whole blood and compared with LXA4. A methyl group at carbon-15 was placed as a racemate to protect both LXA4 and 15-epi-LXA4 in ATLa1, and a fluoride was placed at the para position of the phenoxy ring of 15-epi-16-phenoxy-LXA4 in ATLa2 (Fig. 1B). LC/MS/MS analysis of whole blood incubations showed that ≈40% of LXA4 was lost, whereas both ATLa1 and ATLa2 exhibited greater stability with ≈90% and ≈100% remaining, respectively (Fig. 1B). In human whole blood, quantitatively similar results were obtained with ATLa1 (data not shown). Because ATLa1 and ATLa2 appear to give essentially equivalent activity, to spare animals we performed in vivo experiments with ATLa2.
Intravenous and Local Delivery of ATLa2 Inhibits TNF-α-Induced PMN Infiltration in the Dorsal Air Pouch.
The 6-day murine dorsal air pouch is characterized by the presence of a nascent lining that encloses the air cavity and is composed of both fibroblast-like cells, which are indistinguishable from type B cells of murine knee synovium, and macrophage-like cells, which share morphology with synovial type A cells (26). The air pouch therefore serves as an in vivo model of the rheumatoid synovium (24, 26) and was used here to evaluate the impact of intravenous and local delivery of ATLa2 in the inhibition of cytokine-mediated inflammation and for direct comparison to the actions of ASA and dexamethasone. TNF-α induces leukocyte infiltration, predominantly neutrophils (>75%), into the pouch with maximal cell accumulation occurring between 2–4 hr postinjection (27). ATLa2, dexamethasone, and ASA were each injected locally into the air pouch of individual mice and immediately before the administration of murine TNF-α. For systemic delivery of ATLa2, injections were given via the mouse tail vein before local air pouch injection of murine TNF-α. Here, local delivery of TNF-α alone (20 ng/mouse) induced the recruitment of 4.8 ± 1.1 × 106 PMN into the air pouch at 4 hr (Fig. 2). When ATLa2 was delivered locally into the air pouch (10 μg/mouse), only 1.1 ± 0.3 × 106 PMN were present in the pouch exudate, representing ≈77% inhibition of the TNF-α-induced PMN infiltration. Delivery of ATLa2 (10 μg/mouse) by i.v. injection proved to be an even more potent method of inhibiting TNF-α-driven PMN infiltration. The PMN recruitment values dropped to an average of 7.9 ± 2.9 × 105 PMN/air pouch, representing an inhibition of ≈85%. Moreover, no apparent toxicity of ATLa2 to the mice was observed. Local administration of either ASA or dexamethasone also inhibited PMN recruitment. An equivalent dose of dexamethasone (10 μg/mouse) led to 61% inhibition of PMN recruitment (infiltration of 1.7 ± 0.5 × 106 PMN), whereas a 100-fold greater dose of ASA (1.0 mg/mouse) was required to inhibit PMN infiltration to a similar degree as ATLa2. The presence of 1.5 ± 0.6 × 106 cells with 1.0 mg ASA represents 69% inhibition compared with TNF-α administration alone given to mice in parallel. The mean values of PMN in the air pouch exudates indicate a trend toward increased potency with local or i.v. delivery of ATLa2 compared with dexamethasone. The mean number of PMN after i.v. ATLa2 treatment was significantly different from that after local administration of dexamethasone at 90% confidence (P = 0.085; Student’s two-tailed t test), but not within the 95% confidence interval (P > 0.05).
Figure 2.

ATLa2 inhibits TNF-α-induced PMN infiltration by both local air pouch and i.v. delivery. When injected locally into the air pouch, after injection of vehicle (900 μl PBS), murine TNF-α (20 ng/100 μl PBS) induced the infiltration of 4.8 ± 1.1 × 106 PMN by 4 hr. Dexamethasone (10 μg/air pouch; 0.5 mg/kg), ASA (1 mg/air pouch; 50 mg/kg), and ATLa2 (10 μg/air pouch; 0.5 mg/kg) were locally administered in 900 μl PBS and before TNF-α. Systemic delivery of ATLa2 was by i.v. injection into the mouse tail vein (10 μg/mouse; 0.5 mg/kg). 2.1 ± 0.7 × 105 PMN was found in the air pouch 4 hr after injection of vehicle (1 ml sterile PBS) alone. Values represent mean ± SEM (n = 3–5). ∗, P < 0.05; †, P < 0.15 Student’s two-tailed t test.
Histological analysis of the tissue lining surrounding the air pouch cavity showed that the addition of TNF-α resulted in a markedly increased number of neutrophils (Fig. 3A), which was reduced when ATLa2 was delivered either by intrapouch injection (Fig. 3B) or i.v. via the tail vein (Fig. 3C) before TNF-α administration. Moreover, microscopic analyses of dermal tissue from mice that received ATLa2 treatment were indistinguishable from those exposed only to vehicle (Fig. 3D), which also showed a mild neutrophil infiltrate accompanying this wound model.
Figure 3.

Tissue biopsies of air pouch linings: inhibition of TNF-α-induced PMN accumulation. (A) Lining section taken 4 hr after exposure to TNF-α (20 ng/mouse) showing increased neutrophil number, low-power field inset. (B) Section taken 4 hr after exposure to TNF-α (20 ng/mouse), with prior local delivery of ATLa2 (10 μg/mouse). (C) Section taken 4 hr after exposure to TNF-α (20 ng/mouse), with prior i.v. delivery of ATLa2 (10 μg/mouse). (D) Section of 6-day air pouch lower lining taken from a mouse 4 hr after exposure to vehicle alone. Arrows denote neutrophils. Sections were prepared as in Materials and Methods and were stained with hematoxylin-eosin.
ATLa2 Does Not Inhibit PMN Recruitment by Regulating Vasoactivity.
LXA4 exhibits both concentration- and vascular bed-dependent vasoactive properties. For example, topical administration of LXA4 (1 μM) induces arteriolar dilation in the hamster cheek pouch with no change in venular diameters, whereas systemic delivery into rats produces a vasoconstrictor response in the mesenteric bed (28). In addition, 20 min infusion of 1 or 2 μg/kg LXA4 induces renal vasorelaxation in rats without changing mean arterial pressure (29). To determine whether the increased stability of ATLa2 enhances potential vasoreactivity at the therapeutic dose found to inhibit PMN infiltration in Figs. 2 and 5, vascular changes in response to ATLa2 were compared directly to those of Iloprost, a prostacyclin stable analog that rapidly stimulates arterial vasodilation (30). Added to organ baths, ATLa2 relaxed precontracted isolated rat aorta to ≈40% of the level of relaxation caused by equimolar treatment (1 μM) with Iloprost (results are available as supplemental material on the PNAS web site, www.pnas.org). However, when 10 μg, or ≈24 nmol/mouse, of ATLa2 was injected into the tail vein as in Fig. 2, no apparent changes in mean arterial pressure were observed (Fig. 4). In sharp contrast, injection of equimolar quantities of Iloprost elicited a maximum mean decrease of ≈28 mmHg (1 Hg = 133 Pa) ≈50 s postinjection, with pressure returning to baseline after ≈8 min.
Figure 5.

ATLa2 inhibits both PMA- and LTB4-induced PMN infiltration by topical application. ATLa2 was applied topically (20 μg in 10 μl acetone) to the left mouse ear or delivered intravenously (10 μg in 100 μl of 0.9% sterile saline) through the tail vein. Inflammation was induced in left and right ears by topical application of either LTB4 (1 μg) or PMA (100 ng) in acetone (10 μl). Punch biopsies were obtained after 24 hr and myeloperoxidase activity was measured as an index of PMN number in the ear. Values represent mean ± SEM (n = 3). ∗, P < 0.05 Student’s two-tailed t test.
Figure 4.

ATLa2 does not inhibit PMN recruitment to a site of inflammation by regulating vasodilatation. Mouse arterial pressure was monitored with a pressure transducer via the cannulated carotid artery. Tail vein injection of vehicle (100 μl; 0.9% saline) showed no changes in arterial pressure, whereas 10 μg Iloprost elicited a maximum mean decrease of ≈28 mmHg ≈50 s postinjection, with pressure returning to baseline after ≈500 s. ATLa2 (10 μg) was injected into three mice with no change in mean arterial pressure. Values represent mean ± SEM (n = 3). See supplemental material on the PNAS web site (www.pnas.org) for additional results.
ATLa2 Inhibits PMN Infiltration in Murine Ear Skin to Both Exogenous and Endogenous Chemoattractants.
Topical application of ATLa1, a carbon-15 racemic analog with properties of both 15-epi-LXA4 and native LXA4, and 16-phenoxy-LXA4 (an analog of LXA4) to mouse ear epidermis inhibits LTB4-induced PMN influx as well as vascular permeability changes (22). Here, this ear skin model of inflammation was used to determine whether i.v. or topical delivery of the whole blood stable ATLa2 could also inhibit PMN influx, which is maximal at 24 hr after topical application of either LTB4 or PMA to skin. Topical application of ATLa2 inhibited both LTB4-and PMA-induced inflammation, by ≈78% and ≈49% respectively (Fig. 5). In contrast to i.v. and dorsal administration in the air pouch, a single bolus i.v. injection of ATLa2 (10 μg) did not inhibit PMN influx at 24 hr with either agonist applied topically to ear skin (Fig. 5). But, when a second i.v. injection of this analog was repeated at 20 hr (4 hr before PMN determinations), LTB4-induced PMN recruitment was inhibited by ≈22% (not shown).
ATLa2 Is Rapidly Cleared from Plasma After I. V. Injection.
Because i.v. tail vein delivery of ATLa2 elicited a potent antiinflammatory response blocking PMN infiltration within a 4-hr period in the dorsal air pouch (Fig. 2) but not at 24 hr in the ear skin (Fig. 5), the question arose as to what extent ATLa2 possessed enhanced biostability in circulation after bolus tail vein injections. To address this question, ATLa2 was extracted from mouse plasma collected at several time intervals after tail vein injections, and the recovered materials were quantitated by LC/MS/MS. At 2 min postinjection, ≈34 ng/ml plasma was detected. The levels of the analog decreased with time and were not detected after 15 min. These results indicate rapid clearance from blood and therefore rapid distribution and/or elimination (Fig. 6).
Figure 6.

ATLa2 bolus tail vein injection: time course in plasma. BALB/c mice (6- to 8-wk old) received i.v. tail vein injections of ATLa2 (2 μg/mouse) in 100 μl sterile 0.9% saline. Blood was obtained by cardiac puncture, and ATLa2 was extracted from the plasma by solid phase extraction. The amounts of ATLa2 remaining were quantitated by LC/MS/MS. Values represent mean ± SEM (n = 3). See supplemental material on the PNAS web site (www.pnas.org) for conversion kinetics in mouse whole blood.
DISCUSSION
We report here that a fluorinated analog of 15-epi-LXA4, ATLa2, is a stable analog inhibitor of both direct- (LTB4) and indirect- (TNF-α, PMA) acting chemoattractants. These in vivo observations further support the role of the aspirin-triggered lipoxin circuit as an additional mechanism underlying aspirin’s antiinflammatory therapeutic impact and provide evidence for endogenous antiinflammatory signaling pathways.
Our results indicate that specific design modifications of the native LXA4 structure, such as the addition of a C-15 methyl group (ATLa1) or a bulky ω-chain (para-fluoro)-phenoxy group (ATLa2), prolong the lifetime in blood of these compounds and therefore, potentially, their bioavailabilities as well. Such modifications sterically hinder conversion of the analogs, relative to rapid bioinactivation of the native structure, by recombinant 15-hydroxy-prostaglandin dehydrogenase in vitro (6). As evidenced by LC/MS/MS analyses, the major product of this human dehydrogenase incubated with LXA4 is 15-oxo-LXA4. LC/MS/MS analyses showed that 15-oxo-LXA4 also was produced from LXA4 in mouse whole blood (Fig. 1A), suggesting that the mouse shares with the human a common pathway for LXA4 inactivation.
ATLa2 proved to be a potent inhibitor of TNF-α-induced PMN infiltration into the air pouch cavity, as doses as low as 24 nmol per mouse delivered locally into the air pouch or by systemic i.v. injection via the tail vein resulted in ≈77% and ≈85% inhibition, respectively. Histologically, this wound model is thought to resemble rheumatoid synovium (26), and TNF-α injection initiates PMN recruitment to the cavity (Fig. 2). Injection of TNF-α into the air pouch increases, within the surrounding tissue, C-C chemokine (murine monocyte chemotactic peptide-1 and macrophage inflammatory protein-1α) and C-X-C chemokine (macrophage inflammatory protein-2) production and increases messenger RNA levels for the aforementioned chemokines as well as murine growth-related oncogene protein-α, all of which are collectively required for neutrophil recruitment (27). Because ATLa2 blocked TNF-α-induced PMN infiltration (Fig. 2), ATL disrupts this chemokine network in vivo. This finding may have therapeutic implications, as a variety of pathological conditions, including rheumatoid arthritis, psoriasis, and Crohn’s disease, have associated with them an overproduction of TNF-α and therefore control of this cytokine’s actions is highly sought (31).
We also found that ATLa2 was more potent than ASA because a 100-fold greater dose of ASA, delivered locally to the air pouch, resulted in a level of inhibition of TNF-α-driven PMN recruitment that was less than that of ATLa2. Furthermore, local administration of an equivalent dose of dexamethasone inhibited PMN infiltration at a lower mean value than inhibition by ATLa2 (Fig. 2). Given the unwanted side effects associated with the structures of both ASA (acidity that can lead to ulceration) and dexamethasone (steroid structure that can also impact physiologic steroidal functions), structurally distinct compounds such as ATL analogs designed on the basis of endogenous regulators of leukocyte function may prove to be advantageous therapeutic alternatives.
Applied topically to the ear, ATLa2 also inhibited both LTB4- and PMA-induced PMN recruitment by ≈78% and ≈49%, respectively (Fig. 5). LXA4 and ATLa1 exhibit similar IC50s in vitro in the inhibition of PMN transmigration across polarized epithelial monolayers or PMN adherence to vascular endothelial cells (21). Topical delivery of ATLa1 inhibits LTB4-induced PMN recruitment, but interestingly the level of inhibition afforded by the native LXA4 applied topically was less than 25% compared with that of either ATLa1 or ATLa2 (22). These observations regarding in vitro vs. in vivo potencies between the analogs and the native structure indicate that the ATL analogs possess enhanced bioavailability in vivo. Thus, in addition to protection from enzymatic inactivation, the structural modifications to the native LXA4 structure incorporated in ATLa1 and ATLa2 also improved their topical delivery and contributed to rapid distribution to tissue (Fig. 2).
Results obtained from the air pouch model 4 hr after administration of the analog indicate that i.v. delivery of ATLa2 to a remote site of inflammation was surprisingly even more effective than topical application. In sharp contrast are the findings with ear skin, where topical application of ATLa2 elicited substantial inhibition of topically applied proinflammatory mediators; i.v. delivery of the analog showed no apparent inhibition of LTB4-induced PMN recruitment. We also found that the ATL analog was stable ex vivo in whole blood suspensions, with essentially complete quantitative recovery at 3 hr, and was rapidly cleared from plasma after i.v. injection into the tail vein (between 15 and 50 min). Taken together, these results suggest that ATLa2 is rapidly distributed to tissues from i.v. injections, rather than eliminated, and could remain in an active form for several hours, e.g., during the time course of the TNF-α-driven PMN recruitment to the wounded dorsal pouch (Fig. 2). Furthermore, the absence of PMN inhibition through systemic delivery in the mouse ear model suggests that ATLa2 displays site-selective bioaction from circulation, such as to the dorsal pouch rather than to ear skin. An alternate explanation for the differences in efficacy in ear skin vs. the air pouch after i.v. administration may result from differential distribution within the tissues. Hence, after injection of ATLa2, a rapid distribution to the air pouch might serve as a reservoir permitting the compound’s potent bioactivity in this tissue. Along these lines, ear skin may lack such storage of ATLa2 and thus, coupled with rapid plasma clearance, systemic impact is muted at 24 hr within ear skin.
In summary, these results indicate that the inhibitory actions of aspirin-triggered lipoxins are both tissue- and delivery site-dependent and are the first to show that stable analogs of ATL inhibit acute inflammation at sites distant from the point of delivery. Because ATL stable analogs were designed as mimetics to incorporate the native aspirin-triggered structural features, the present findings, taken together, provide new tools to examine endogenous antiinflammatory pathways as well as avenues to approach the development of both topical and intravenous anti-PMN therapies.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grants GM-38765 and DK-50305 (to C.N.S.) and a research grant from Schering Berlex AG (to C.N.S.). K.G. is the recipient of a postdoctoral fellowship from the National Arthritis Foundation. We thank Mary Halm Small for expert assistance in the preparation of this manuscript and Dr. B. Schmidt (Department of Pathology, Brigham and Women’s Hospital) for microscopic analyses of mouse air pouches.
ABBREVIATIONS
- ASA
aspirin (acetylsalicylic acid)
- ATL
aspirin-triggered lipoxins
- ATLa1
15(R/S)-methyl-lipoxin A4
- ATLa2
15-epi-16-(para-fluoro)-phenoxy-lipoxin A4
- COX I and II
cyclooxygenases I and II
- 15-epi-LXA4
5S,6R,15R-trihydroxyeicosa-7E,9E,11Z,13E-tetraenoic acid
- 15-epi-LXB4
5S,6R,15R-trihydroxyeicosa-6E,8Z,10E,12E-tetraenoic acid
- LC/MS/MS
liquid chromatography tandem mass spectrometry
- LTB4
leukotriene B4, 5S,12R-dihydroxyeicosa-6E,8Z,10Z,14E-tetraenoic acid
- LX
lipoxins
- LXA4
lipoxin A4, 5S,6R,15S-trihydroxyeicosa-7E,9E,11Z,13E-tetraenoic acid
- LXB4
lipoxin B4, 5S,14R,15S-trihydroxyeicosa-6E,8Z,10E,12E-tetraenoic acid
- PMA
phorbol 12-myristate 13-acetate
- PMN
polymorphonuclear leukocyte
- TNF-α
tumor necrosis factor α
- MS/MS
product ion mass spectra
References
- 1.Weissmann G. Sci Am. 1991;264:84–90. doi: 10.1038/scientificamerican0191-84. [DOI] [PubMed] [Google Scholar]
- 2.Ridker P M, Cushman M, Stampfer M J, Tracy R P, Hennekens C H. N Engl J Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 3.Giovannucci E, Egan K M, Hunter D J, Stampfer M J, Colditz G A, Willett W C, Speizer F E. N Engl J Med. 1995;333:609–614. doi: 10.1056/NEJM199509073331001. [DOI] [PubMed] [Google Scholar]
- 4.Marcus A J. N Engl J Med. 1995;333:656–658. doi: 10.1056/NEJM199509073331011. [DOI] [PubMed] [Google Scholar]
- 5.Herschman H R. Trends Cardiovasc Med. 1998;8:145–150. doi: 10.1016/S1050-1738(98)00004-8. [DOI] [PubMed] [Google Scholar]
- 6.Serhan C N. Prostaglandins. 1997;53:107–137. doi: 10.1016/s0090-6980(97)00001-4. [DOI] [PubMed] [Google Scholar]
- 7.Chiang N, Takano T, Clish C B, Petasis N A, Tai H-H, Serhan C N. J Pharmacol Exp Ther. 1998;287:779–790. [PubMed] [Google Scholar]
- 8.Lee T H, Crea A E, Gant V, Spur B W, Marron B E, Nicolaou K C, Reardon E, Brezinski M, Serhan C N. Am Rev Respir Dis. 1990;141:1453–1458. doi: 10.1164/ajrccm/141.6.1453. [DOI] [PubMed] [Google Scholar]
- 9.Chavis C, Chanez P, Vachier I, Bousquet J, Michel F B, Godard P. Biochem Biophys Res Commun. 1995;207:273–279. doi: 10.1006/bbrc.1995.1183. [DOI] [PubMed] [Google Scholar]
- 10.Chavis C, Vachier I, Chanez P, Bousquet J, Godard P. J Exp Med. 1996;183:1633–1643. doi: 10.1084/jem.183.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas E, Leroux J L, Blotman F, Chavis C. Inflamm Res. 1995;44:121–124. doi: 10.1007/BF01782022. [DOI] [PubMed] [Google Scholar]
- 12.Gewirtz A T, McCormick B, Neish A S, Petasis N A, Gronert K, Serhan C N, Madara J L. J Clin Invest. 1998;101:1860–1869. doi: 10.1172/JCI1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pillinger M H, Abramson S B. Rheum Dis Clin N Am. 1995;21:691–714. [PubMed] [Google Scholar]
- 14.Hagihara H, Nomoto A, Mutoh S, Yamaguchi I, Ono T. Atherosclerosis (Dallas) 1991;91:107–116. doi: 10.1016/0021-9150(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 15.McLaughlan J M, Seth R, Vautier G, Robins R A, Scott B B, Hawkey C J, Jenkins D. J Pathol. 1997;181:87–92. doi: 10.1002/(SICI)1096-9896(199701)181:1<87::AID-PATH736>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 16.Anezaki K, Asakura H, Honma T, Ishizuka K, Funakoshi K, Tsukada Y, Narisawa R. Intern Med. 1998;37:253–258. doi: 10.2169/internalmedicine.37.253. [DOI] [PubMed] [Google Scholar]
- 17.Iverson L, Kragballe K. In: Skin Immune System (SIS) Bos J D, editor. Boca Raton, FL: CRC; 1997. pp. 227–237. [Google Scholar]
- 18.Ensor C M, Tai H-H. In: Prostaglandins, Leukotrienes, Lipoxins, and PAF. Bailey J M, editor. New York: Plenum; 1991. pp. 39–52. [Google Scholar]
- 19.Serhan C N, Fiore S, Brezinski D A, Lynch S. Biochemistry. 1993;32:6313–6319. doi: 10.1021/bi00076a002. [DOI] [PubMed] [Google Scholar]
- 20.Maddox J F, Colgan S P, Clish C B, Petasis N A, Fokin V V, Serhan C N. FASEB J. 1998;12:487–494. doi: 10.1096/fasebj.12.6.487. [DOI] [PubMed] [Google Scholar]
- 21.Serhan C N, Maddox J F, Petasis N A, Akritopoulou-Zanze I, Papayianni A, Brady H R, Colgan S P, Madara J L. Biochemistry. 1995;34:14609–14615. doi: 10.1021/bi00044a041. [DOI] [PubMed] [Google Scholar]
- 22.Takano T, Clish C B, Gronert K, Petasis N, Serhan C N. J Clin Invest. 1998;101:819–826. doi: 10.1172/JCI1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takano T, Fiore S, Maddox J F, Brady H R, Petasis N A, Serhan C N. J Exp Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sin Y M, Sedgwick A D, Chea E P, Willoughby D A. Ann Rheum Dis. 1986;45:873–877. doi: 10.1136/ard.45.10.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradley P P, Priebat D A, Christensen R D, Rothstein G. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 26.Edwards J C W, Sedgwick A D, Willoughby D A. J Pathol. 1981;134:147–156. doi: 10.1002/path.1711340205. [DOI] [PubMed] [Google Scholar]
- 27.Tessier P A, Naccache P H, Clark-Lewis I, Gladue R P, Neote K S, McColl S R. J Immunol. 1997;159:3595–3602. [PubMed] [Google Scholar]
- 28.Dahlén S E, Serhan C N. In: Lipoxygenases and Their Products. Crooke S T, Wong A, editors. San Diego, CA: Academic; 1991. pp. 235–276. [Google Scholar]
- 29.Katoh T, Takahashi K, DeBoer D K, Serhan C N, Badr K F. Am J Physiol. 1992;263:F436–442. doi: 10.1152/ajprenal.1992.263.3.F436. [DOI] [PubMed] [Google Scholar]
- 30.Grant S M, Goa K L. Drugs. 1992;43:899–924. doi: 10.2165/00003495-199243060-00008. [DOI] [PubMed] [Google Scholar]
- 31.Marriott J B, Westby M, Dalgleish A G. Drug Discov Today. 1997;2:273–282. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}