

Abstract
The protein stabilizing effects of the small molecule osmolyte, trimethylamine N-oxide, against chemical denaturant was investigated by NMR spin-relaxation measurements and model-free analysis. In the presence of 0.7 M guanidine hydrochloride increased picosecond-nanosecond dynamics are observed in the protein ribonuclease A. These increased fluctuations occur throughout the protein, but the most significant increases in flexibility occur at positions believed to be the first to unfold. Addition of 0.35 M trimethylamine N-oxide to this destabilized form of ribonuclease results in significant rigidification of the protein backbone as assessed by 1H-15N order parameters. Statistically, these order parameters are the same as those measured in native ribonuclease indicating that TMAO reduces the amplitude of backbone fluctuations in a destabilized protein. These data suggest that TMAO restricts the bond vector motions on the protein energy landscape to resemble those motions that occur in the native protein and points to a relation between stability and dynamics in this enzyme.
Keywords: guanidine, protein dynamics, protein stability, NMR spin-relaxation, TMAO
Many organisms produce and accumulate small organic solutes to counteract the effects of environmental stresses. Such inhospitable living conditions can include high temperature, pressure, salt, and dehydration. One such solute, trimethylamine N-oxide, (TMAO; Scheme 1) provides protection to proteins against the effects of chemical denaturant, high pressures, and high temperatures (Yancey et al. 2002; Somero 2003; Yancey 2005).
Scheme 1.

Chemical structure of trimethylamine N-oxide
In the enzymes ribonuclease A (RNase A) (Yancey and Somero 1979) and lysozyme (Arakawa and Timasheff 1985), TMAO was shown to increase the midpoint of the thermal unfolding profile. Other protective properties of TMAO include the ability to counteract the deleterious effects of urea as probed by enzyme assays (Yancey and Somero 1979; Palmer et al. 2000) and protein folding studies (Yancey and Somero 1979; Lin and Timasheff 1994). Alexandrescu and coworkers have shown that TMAO increases the protection of amide protons from exchange with solvent by solution NMR hydrogen exchange (HX) experiments (Jaravine et al. 2000). HX experiments on RNase A in urea solutions (Qu and Bolen 2003) suggest that TMAO suppresses the native-state protein fluctuations, thereby dampening the increased motional amplitude caused by denaturant (Wang and Bolen 1997; Qu and Bolen 2003). These results suggest that TMAO directly impacts protein dynamics. In contrast, phosphorescent lifetime and quenching experiments indicate that 1.8 M TMAO does not affect the dynamics of the native protein fold (Gonnelli and Strambini 2001). Thus, there remains some uncertainty as to the role of TMAO on protein stability and dynamics. The stability of proteins depends on the difference in free energy between the native and denatured states, and it is thought that the action of TMAO results in differential stabilizations of the native and denatured states (Lin and Timasheff 1994).
Computational and physical methods have also been used to investigate the mode of action of TMAO on protein stability. Densimetry studies showed that the chemical potential changes of a protein in denaturant alone or in denaturant + TMAO are the same, indicating that TMAO acts independently of denaturant and does not affect the binding of denaturant to proteins (Lin and Timasheff 1994). Other work has demonstrated that TMAO exerts its effect primarily through changes in solvent hydrogen bonding and by unfavorable interactions between TMAO and the amide moiety of proteins (Wang and Bolen 1997; Zou et al. 2002; Bennion and Daggett 2004; Athawale et al. 2005; Esposito and Daggett 2005). However, a somewhat different picture emerged from recent solution NMR experiments that suggest that a hydrated form of TMAO may interact with the peptide backbone (Hovagimyan and Gerig 2005). Thus, there is not complete consensus on the mechanism of TMAO action. Because organisms produce TMAO and chemically similar molecules as a general stress response, their effects would be expected to be general and not specific to any particular protein or nucleic acid.
Here, we use a well-characterized system to probe the effects of TMAO on protein dynamics using solution NMR techniques. NMR spin-relaxation experiments can provide a unique glimpse at protein motions. In particular, analysis of the NH order parameter (S 2) can reveal important information about the distribution of conformational states including the amplitude and timescale of backbone fluctuations. The backbone dynamics of native RNase A in buffer, in the presence of guanidine (Gdn) hydrochloride (RNase A/Gdn) and in the presence of Gdn and TMAO (RNase A/Gdn/TMAO), were investigated by solution NMR spin-relaxation experiments.
Results
Circular dichroism spectroscopy
The effects of an increase in temperature on the secondary structure of RNase A were detected from 29°C–80°C by monitoring the molar ellipticity at 222 nm (Fig. 1). In phosphate buffer (pH 6.4) the RNase A melting temperature (T m) is 62.9°C. In the presence of 700 mM Gdn, the T m decreases to 48°C. Addition of 350 mM TMAO to this solution raises the Tm by >2°C to 50.2°C. The magnitude and trend of these effects are similar to those observed with RNase A and with other proteins (Yancey and Somero 1979; DeKoster and Robertson 1997; Poklar et al. 1999).
Figure 1.

Thermal denaturation of RNase A. The molar ellipticity at 222 nm was monitored by circular dichroism spectroscopy for 20 μM native RNase A (•), 15 μM RNase A + 700 mM guanidine hydrochloride (▾), and 20 μM RNase A + 700 mM guanidine hydrochloride + 350 mM TMAO (♦).
NMR spin-relaxation
In this study, TMAO and Gdn have little effect on the RNase A resonance positions similar to observations with cold-shock protein A (Jaravine et al. 2000). Therefore, the assignment of all resonances in RNase A could be determined in the presence of solutes by monitoring the peak positions during titration of Gdn or TMAO into the RNase A solution.
Motions of the N-H bond vector at discrete frequencies, which modulate the 15N chemical-shift anisotropy (CSA) and 1H-15N dipolar interaction, will result in decay (relaxation) of nonequilibrium nuclear magnetization (Abragam 1961). Therefore, there is a direct linkage between protein dynamics and NMR spin-relaxation. Protein motion at these frequencies is commonly described by the spectral density function J(ω), which relates the frequency of these motions to the measured NMR relaxation rates (Abragam 1961). The amplitudes and timescales of these protein motions are described by the model-free formalism, which models the spectral density function as (Lipari and Szabo 1982a,b; Clore et al. 1990)
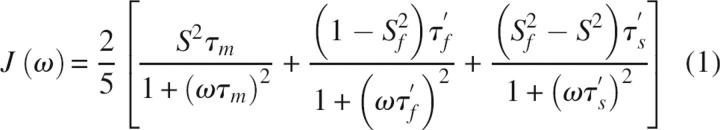 |
in which τf′ = τeτm/(τe+τm), τs′ = τsτm/(τs+τm), τm is the isotropic rotational correlation time of the macromolecule, τe is the effective correlation time for fast (<200 psec) internal motions, and τs the correlation time for slow (∼nsec) motions (τe < τs < τm). Corrections for nonisotropic macromolecular tumbling, in most cases, are readily implemented (Woessner 1962; Lee et al. 1997). S 2 = S f 2 S s 2 is the square of the generalized order parameter that characterizes the amplitude of internal motions, and S f 2 and S s 2 are the order parameters for internal dynamics on the fast and slow time scales, respectively. The order parameter describes the amplitude of bond vector motions in the time range from picoseconds to nanoseconds and has values from 0 for unrestricted motions to 1 indicating a static bond vector in the molecular reference frame. Consequently, measurement of 15N longitudinal and transverse relaxation rates in combination with the 1H-15N heteronuclear NOE allows sampling of the spectral density function and thus amplitudes and time scales of protein motion can be determined.
15N-longitudinal and transverse relaxation rates and the 1H-15N heteronuclear NOE were determined for RNase A, RNase A + 700 mM Gdn (RNase A/Gdn), and RNase A + 700 mM Gdn + 350 mM TMAO (RNase A/Gdn/TMAO) at 600 MHz and 300 K. For the RNase A backbone, the average relaxation rates for nonproline residues with good resolution and signal-to-noise values are R 1 = 1.64 ± 0.08 sec−1 (n = 99); R 2 = 9.77 ± 2.39 sec−1 (n = 98), and NOE = 0.782 ± 0.044 (n = 94). In the presence of 700 mM Gdn, R 1 = 1.57 ± 0.07 sec−1 (n = 97); R 2 = 9.27 ± 1.29 sec−1 (n = 93) and NOE = 0.724 ± 0.068 (n = 92). When both 700 mM Gdn and 350 mM TMAO are added to the RNase A solution, the spin-relaxation values are R 1 = 1.35 ± 0.10 sec−1 (n = 97), R 2 = 11.38 ± 2.39 sec−1 (n = 96), and NOE = 0.758 ± 0.075 (n = 96). The residue specific values are shown in Figure 2.
Figure 2.
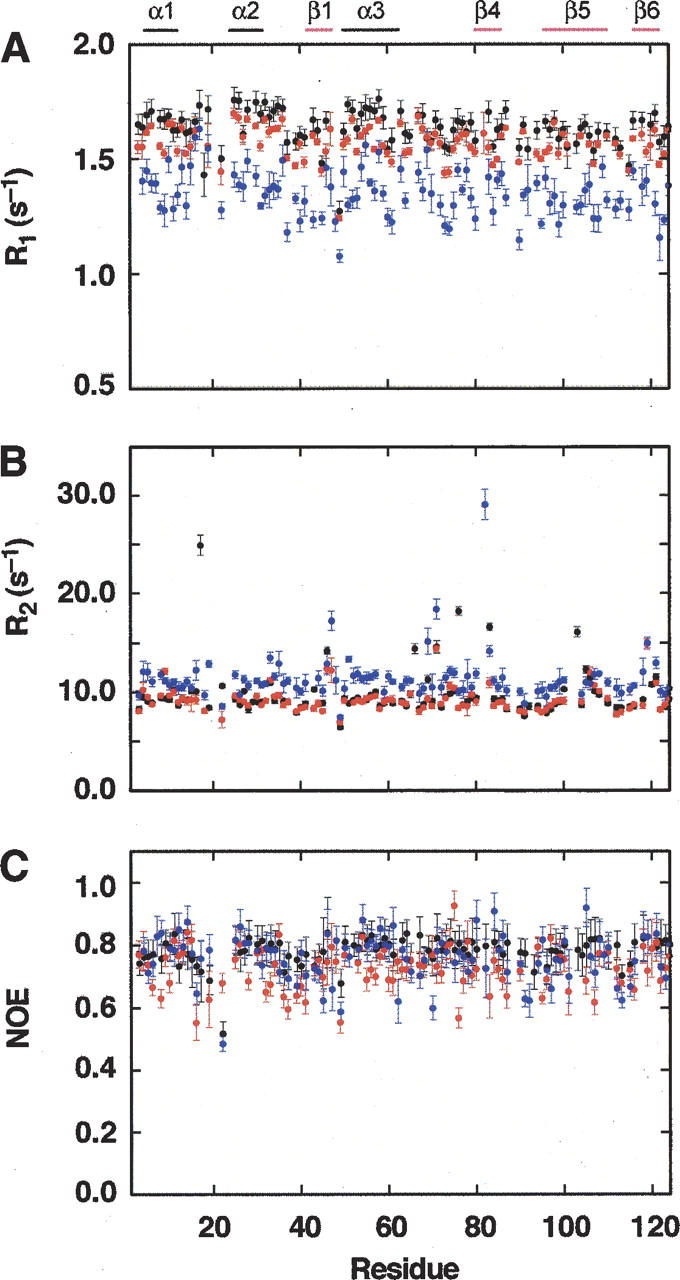
Residue-specific NMR spin-relaxation rates. Longitudinal (R 1) (A) and transverse (R 2) (B) relaxation rates and the steady-state heteronuclear NOE (C) are shown as a function of the RNase A primary sequence for native RNase A (black circles), RNase A + 700 mM guanidine hydrochloride (red circles), and RNase A + 700 mM guanidine hydrochloride + 350 mM TMAO (blue circles). The location of secondary structure elements is shown at the top of the figure.
Model-free protein dynamics
The model-free analysis of the NMR spin-relaxation data provides information on the overall rotational properties of RNase A as well as information on the internal backbone dynamics within the molecular reference frame. For RNase A, RNase A/Gdn, and RNase A/Gdn/TMAO the average rotational correlation times, τm, determined from the model free analysis of the NMR relaxation data are 6.65 ± 0.01, 6.83 ± 0.01, and 8.49 ± 0.02 nsec. The value of 6.65 nsec for native RNase A is in good agreement with values previously determined in this lab of 6.61 nsec (Kovrigin et al. 2003). The increase in τm with added Gdn and TMAO is consistent with the increase in solution viscosity of these samples caused by the addition of cosolute. At the RNase A concentrations used in these studies, no evidence of protein aggregation is observed (data not shown). In all cases, the spin-relaxation was best fit in the context of an axially symmetric rotational diffusion tensor with values of D‖/D⊥ = 0.84 ± 0.01, 0.87 ± 0.01, and 0.89 ± 0.01 for RNase A, RNase A/Gdn, and RNase A/Gdn/TMAO, respectively, and are similar to values previously determined (Cole and Loria 2002, 2003; Kovrigin et al. 2003).
The results of the model-free analysis for RNase A are shown in Figure 3. The overall average order parameter (±SD) for RNase A in each cosolute is S 2 = 0.868 ± 0.042 (n = 91), 0.831 ± 0.047, (n = 91), and 0.858 ± 0.061 (n = 86) for RNase A, RNase A/Gdn, and RNase A/Gdn/TMAO, respectively. The residue-specific values are provided in the Supplemental Material. Amino acid residues with the same motional model, under the different solution conditions, have significant correlations in their corresponding S 2 values. For RNase A and RNase A/Gdn, the correlation coefficient is 0.88 (n = 40); for RNase A and RNase A/Gdn/TMAO, r = 0.76 (n = 51), and for RNase A/Gdn/TMAO and RNase A/Gdn, the correlation coefficient is 0.77 (n = 43).
Figure 3.
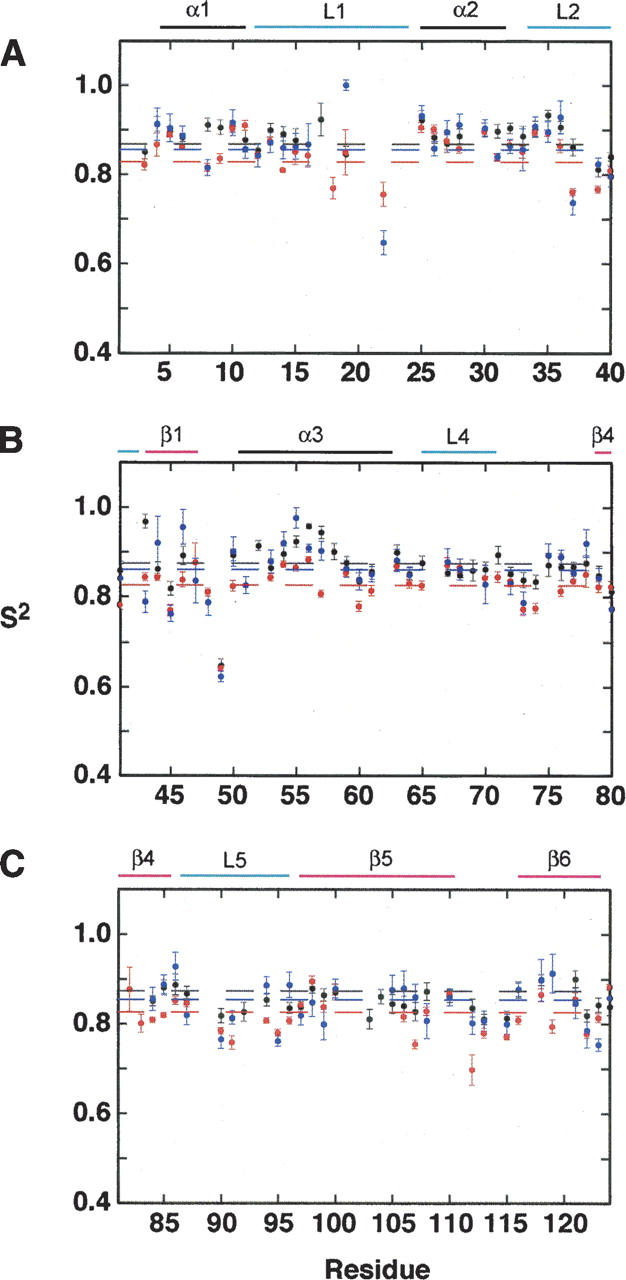
Residue specific order parameters (S 2). (A–C) S 2 values as a function of RNase A primary sequence for native RNase A (black circles), RNase A + 700 mM guanidine hydrochloride (red circles), and RNase A + 700 mM guanidine hydrochloride + 350 mM TMAO (blue circles). The location of secondary structure elements is shown at the top of the figure. Black, blue, and red horizontal dashed lines show the protein-wide average S 2 value for native, RNaseA/Gdn/TMAO, and RNase A/Gdn samples, respectively.
For all residues in which model-free analysis could be performed, a t-test comparison of S 2 values for RNase A and RNase A/Gdn (t = 9.40, dof = 81, p < 0.0001) indicate that the difference in S 2 values is significant from zero. Comparison of S 2 for RNase A and RNase A/Gdn/TMAO gives t = 1.97, dof = 78, p = 0.053, and therefore these differences are not different from zero at the strict 95% level. Further comparison between RNase A/Gdn and RNase A/Gdn/TMAO (t = 4.02, dof = 82, p = 0.0001) indicates statistically significant differences in the internal dynamics parameters. Thus, native RNase A and RNase A/Gdn/TMAO have similar dynamics parameters and both are significantly different from RNase A in the presence of 700 mM guanidine hydrochloride alone.
Further dissection of S 2 values by RNase A secondary structure shows a similar trend as that observed for the S 2 values for the enzyme as a whole (Table 1), that is, the enzyme experiences increased backbone flexibility in the presence of guanidine-HCl, whereas TMAO partially reverses this trend, resulting in dynamics more similar to native RNase A. A t-test of these 2° structure order parameters shows that there are significant differences (p ≤ 0.05) for loop2, α3, loop5, and β6 (Table 2) between RNase A and RNase A/Gdn. At p ≤ 0.10 an additional three 2° structure elements (α1, β1, and β4) can be considered to be statistically different from each other for these two solution conditions. In contrast, comparison of RNase A and RNase A/Gdn/TMAO at the 2° structure level shows no significant differences from zero for these order parameters (Table 2).
Table 1.
Average S 2 values for secondary structure elements

Table 2.

In addition to the increase in flexibility of RNase A in guanidinium hydrochloride, a significant number of residues (n = 41) require the model-free models 2, 4, or 5 for proper fitting. A common feature of models 2, 4, and 5 is the inclusion of the τe term to account for additional picosecond time-scale motions (Lipari and Szabo 1982a, b). These residues include A6, F8, S16, S22, Q28, S32, R33, T36, K37, R39, C40, K41, V43, N44, T45, E49, Q55, A56, V57, Q60, V63, C65, C72, Y76, S77, M79, S80, D83, E86, T87, S90, C95, A96, I106, I107, G112, N113, V116, H119, A122, and S123 (Fig. 4), whereas in native RNase A, only A19, E49, and N113 require a τe term in the model-free analysis. In RNase A/Gdn, all of these residues have τe values ranging between 22 psec for C65 and 130 psec for S16 with the exception of G112 (τe value of 1700 ± 700 psec). The addition of TMAO results in a significant decrease in the number of residues (n = 19) requiring a τe term (Fig. 4C) relative to the RNase A/Gdn sample. These residues are A5, S16, S22, K37, R39, K41, T45, V47, E49, N67, T70, T78, T87, K91, Y97, G112, N113, Y115, and S123. Again, these data are consistent with TMAO acting to reverse the non-native protein dynamics caused by the denaturant, guanidine.
Figure 4.

Structural context of RNase A backbone dynamics. Residues in RNase A in which an internal correlation time (τe) is part of the model-free dynamics analysis are colored red on the illustration of RNase A. (A) Data for native RNase A, (B) RNase A + 700 mM guanidine hydrochloride + 350 mM TMAO, and (C) RNase A + 700 mM guanidine hydrochloride. The two panels in C are related by 180° rotation. For aid in viewing, selected residues are labeled with the one-letter amino acid code; in A, 2° structure elements are identified to orient the reader. Residues in gray are those that were not analyzed, including proline, overlapped residues, or those with low signal-to-noise values. The figure was prepared with the program MacPyMOL (DeLano 2005).
The extent of microsecond–millisecond (μs–ms) protein conformational exchange motion manifests, in the model-free analysis as an exchange term, R ex (models 3 and 4). There are no significant differences in the protein sites experiencing conformational exchange in native, Gdn, and Gdn/TMAO samples of RNase A. Twenty-one residues in native RNase A experience conformational exchange, consistent with previous observations (Cole and Loria 2002, 2003; Beach et al. 2005; Kovrigin and Loria 2006a,b). In RNase A/Gdn and RNase A/Gdn/TMAO there are 17 and 10 residues, respectively, that undergo conformational exchange motion at similar amino acid sites as native RNase A. Many of these seemingly unaccounted for differences in sites in conformational exchange are the result of residues in RNase A/Gdn and RNase A/Gdn/TMAO that are broad and have low signal-to-noise, which is characteristic of conformational exchange motion, and were therefore not analyzed further. Consequently, conformational exchange motion appears to not be affected by the presence of these cosolutes.
Discussion
Protein dynamics and stability
There is great interest in the physicochemical factors that determine protein stability and those that modulate protein dynamics. Forces (stabilizing) that protect proteins from denaturation may or may not be distinct from those forces that rigidify the protein. For example, some crystallographic studies have identified thermophilic proteins as possessing smaller Debye-Waller factors than their mesophilic counterparts, suggesting a link and inverse correlation between thermal stability and protein dynamics (Vihinen 1987). In addition, there have been a number of studies, both experimental and computational, that provide a correlation for (Wagner and Wuthrich 1979; Lazaridis et al. 1997; Tang and Dill 1998; Zavodszky et al. 1998; Svingor et al. 2001; Tsai et al. 2001) or show no evidence for (Colombo and Merz 1999; Fitter and Heberle 2000; Hernandez et al. 2000; Fitter et al. 2001; Grottesi et al. 2002) a link between dynamics and stability. In general, the results of this work supports an inverse correlation between protein dynamics and stability, though the increase in protein rigidity with TMAO in the presence of guanidine must be considered in light of the slight (2°C increase) in T m that is measured by CD spectroscopy.
CD spectroscopy
As expected, submolar amounts of guanidinium hydrochloride have a small but detectable effect on the melting transition of RNase A, and in molar ratios of 2:1 denaturant to TMAO this effect is partially reversed. However, it should be noted that at the temperature of these NMR experiments (300 K), RNase A exists in essentially the fully folded, native conformation under all conditions used in this study (Udgaonkar and Baldwin 1990; Qu and Bolen 2003). The native state is populated to >99.5% under all conditions investigated here (Qu and Bolen 2003). Thus, measured changes in protein dynamics can be considered to occur within the native-state ensemble. The native conformation is also supported by the nearly identical 1H-15N HSQC experiments under the various solution conditions (data not shown).
RNase A/Gdn
The addition of 700 mM Gdn to RNase A resulted in a decrease in average order parameters for the N-H bond vector from S 2 = 0.868 for native RNase A to S 2 = 0.831 for RNase A in denaturant. These effects occur throughout the RNase A structure, thereby indicating a global effect of the denaturant on protein dynamics. Because the N-H order parameter is directly related to the backbone conformational entropy (Akke et al. 1993; Li et al. 1996; Yang and Kay 1996), these data indicate that at nondenaturing concentrations Gdn acts to increase the backbone entropy relative to the native state of RNase A. If this bond vector motion is modeled assuming diffusion in a cone, then the relation between S 2 and the semi-angle, θ for motion in this cone is given by (Lipari and Szabo 1982a)
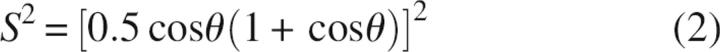 |
Applying this relation to the RNase A order parameters indicates a modest increase in average motional amplitude from 17.5° for native RNase A to 20° for RNase A/Gdn. The increase in S 2 values to 0.858 upon TMAO addition to the RNase A/Gdn solution gives an average bond vector amplitude of 18°, nearly that of native RNase A. These data indicate that the amplitude of bond vector motions in native RNase A and RNase A/Gdn/TMAO are nearly identical.
Though the effects of Gdn on RNase dynamics are global, the most statistically significant increases in psec-nsec dynamics occur in loop2, loop5, α3, and β6 (Fig. 5). Moreover, three amino acid residues show large (ΔS 2 = S 2 apo – S 2 Gdn > 0.1) increases in dynamics upon the addition of Gdn: V43, ΔS 2 = 0.125; V57, ΔS 2 = 0.138; and G112, ΔS 2 = 0.138. Valine 57 is in the middle of α helix 3 and has its amide nitrogen hydrogen bonded to the carbonyl oxygen of V54. V43 is positioned at the N terminus of β1 and G112 is located in the loop connecting β5 and β6, with both of these residues having solvent-exposed amide nitrogen atoms. Interestingly, G112 is located in the loop that facilitates formation of the C-domain swapped dimer in RNase A (Liu et al. 2001). These three regions of enhanced dynamics (loop2, loop5, α3, and β6) are solvent exposed, thereby indicating that the primary effects of low concentrations of denaturant occur on the surface of the protein, suggesting that backbone fluctuations that lead to local unfolding may be the first step in unfolding of the entire enzyme. Several residues in these regions have been shown by hydrogen exchange experiments to be some of the first regions to undergo unfolding including N34, V43, S59, and K91 (Juneja and Udgaonkar 2002). In somewhat of a contrast, other experiments indicate that N34, V43, and S59 are weakly protected from exchange in an early folding intermediate (Udgaonkar and Baldwin 1990). In addition, the N-terminal, α1 region is shown to be formed last during folding and is one of the first regions to become destabilized in the unfolding process (Udgaonkar and Baldwin 1990; Houry and Scheraga 1996; Juneja and Udgaonkar 2002). The studies described in this work, at low denaturant concentration, indicate small differences (p = 0.01) in psec-nsec dynamics in α1 between native and RNase A/Gdn and, therefore, may report on motional processes different from those sampled by hydrogen exchange measurements.
Figure 5.

Guanidine-induced changes in protein backbone dynamics. Regions of RNase A in which backbone dynamics show the largest statistical difference from native RNase A (by t-test,Table 2) are colored black. Statistically significant differences are those in which p < 0.05 (Devore 2000). The figure was prepared with the program MacPyMOL (DeLano 2005).
An interesting feature that emerged from these dynamics studies is the increased number of amino acid sites that experience additional picosecond fluctuations in the presence of Gdn (Fig. 4). These picosecond motions, characterized by a τe term, cluster together in the RNase A structure, suggesting that the model-free analysis is capturing real changes in protein dynamics and that these fluctuations are structurally correlated. For example, residues comprising two of the four disulfide bonds (C95-C40 and C65-C72) in RNase A possess additional picosecond dynamics. Residues I106, I107, A122, S123, C72, and C65 are located near each other on adjacent β-sheets (Fig. 4C). Additionally, β5 (97–111) and β6 (116–123) pack onto α3 (51–63), and residues in each of these secondary structure elements experience additional picosecond dynamics. This is also the case of adjacent β strands 1 and 4 and loops 2 and 5. Finally, whereas the S 2 values in α1 did not indicate that Gdn had a significant effect on the amplitude of motion relative to native RNase A (vide supra), several residues in α1 for the RNase A/Gdn sample also require τe in the model-free fitting. These Gdn-induced effects observed here may indicate a correlation between instability and increased dynamics. In fact, previous studies suggest that this N-terminal region is a site of instability and potentially one of the first regions of RNase A to unfold (Udgaonkar and Baldwin 1990; Houry and Scheraga 1996; Juneja and Udgaonkar 2002). These additional picosecond motions observed in the presence of Gdn suggest a hierarchical mechanism in which very fast native-state bond vector motions increase in amplitude and begin to sample additional conformational substates.
RNase A/Gdn/TMAO
As noted, osmolytes such as TMAO should exert their stabilizing effects in a general way because their role is to stabilize many distinct cellular proteins (Yancey 2001). In organisms that produce TMAO to counteract the effects of chemical denaturant, the ratio of denaturant to TMAO is typically 2:1 and the absolute concentrations are found to be <1 M (Yancey 1994, 2001; Yancey et al. 2001). Therefore, in this NMR work similar conditions were used. As shown in Tables 1 and 2 and Figure 3, TMAO effects changes in protein dynamics caused by Gdn to render them statistically indistinguishable from native RNase A. This is somewhat at odds with the work of Gonnelli and Strambini, in which, based on phosphorescence lifetime measurements with apoazurin, alcohol dehydrogenase, alkaline phosphatase, and glyceraldehyde-3-phosphate dehydrogenase, they observe no effect of TMAO on the internal dynamics of these proteins (Gonnelli and Strambini 2001). This contrasting view of the effects of TMAO may be due to the much different time scale that is probed by the phosphorescence and NMR techniques or by the limited site resolution of phosphorescence measurements. Moreover, our results clearly show significant effects on protein dynamics caused by TMAO and are in agreement with other experimental and theoretical studies, indicating that TMAO reverses the effects of chemical denaturants by decreasing the fluctuations of the native state (Jaravine et al. 2000; Qu and Bolen 2003; Bennion and Daggett 2004; Gahl et al. 2004). However, the NMR spin-relaxation experiments described here provide two additional important pieces of information that complement and add to the experimental studies noted above. First, these NMR spin-relaxation experiments typically allow analysis of significantly more residues than other methods and thus result in a more complete dynamics picture. Second, motions over a large time window (psec-msec) can be examined by these experiments, thereby providing additional insight on the types of protein motions involved.
For example, as demonstrated in Figure 4C, in the presence of guanidine the dynamics description of 41 residues requires the inclusion of an internal correlation time τe, whereas in native RNase A there are only three τe requiring residues and in the RNase A/Gdn/TMAO sample this number is 19. τe in the model-free formalism suggests the presence of additional internal motion occurring on a timescale from 20 to 200 psec. Of these 41 sites in both the native and Gdn/TMAO RNase A samples, all but nine (K37, R39, K41, T45, E49, T87, G112, N113, and S123) are best described by models not containing τe, indicating that for these 32 residues the internal correlation time is very fast (<20 psec) in the native and Gdn/TMAO RNase A forms. TMAO reverses the effects of guanidine by reducing the amplitude of fluctuations as measured by the NMR order parameters and causes a reduction in the number of residues experiencing internal motions on the τe time scale (20 psec < τe < τm).
These studies demonstrate that TMAO restricts the increase in conformational space sampled by the N-H bond vectors in the presence of guanidine hydrochloride alone. Thus, TMAO causes more restricted, native-like protein fluctuations, possibly limiting access to higher energy conformational substates that would ultimately lead to protein denaturation. Several of the protein sites experiencing this TMAO-induced reversal of dynamics reflect those identified by hydrogen exchange experiments (Idiyatullin et al. 2003), which occurs on a much slower time scale, suggesting that NMR spin-relaxation experiments are a useful and complementary tool for studying protein stability. The nearly uniform nature of the effects on protein dynamics and NMR chemical shifts caused by TMAO argues against specific protein binding (Lin and Timasheff 1994; Zou et al. 2002). Finally, these NMR experiments are independent of the mechanism of amide hydrogen exchange with solvent and, therefore, their interpretation is often more straightforward.
Materials and methods
Protein preparation
The plasmid (pBXR) encoding RNase A was a gift from Professor Ronald T. Raines (University of Wisconsin-Madison). 15N-labeled RNase A was expressed and purified from Escherichia coli strain BL21 (DE3) as described previously (Cole and Loria 2002). The 15N-labeled RNase A sample used here was provided by Dr. Evgenii Kovrigin (Yale University). RNase A was judged pure by SDS-PAGE and MALDI-TOF mass spectrometry. The concentration of RNase A was determined using a molar extinction coefficient, ɛ278 = 9800 M−1 cm−1 (Anderson et al. 1968). Trimethylamine N-oxide (TMAO) and guanidine hydrochloride (Gdn) were purchased from Sigma-Aldrich and were of the highest purity available. Concentrations of these cosolutes were determined gravimetrically. All reference to the RNase A three-dimensional structures are based on the X-ray crystal structure 7RSA (Wlodawer et al. 1988).
Circular dichroism spectroscopy
For CD experiments, [RNase A] = 20 or 15 μM in 10 mM potassium phosphate buffer (pH 6.4). In the temperature-dependent studies, guanidine hydrochloride (Gdn) and/or trimethylamine N-oxide (TMAO) were included in this buffer at the noted concentrations. In all cases, blanks were run with identical buffer components excluding RNase A. Melting profiles were determined by monitoring the change in molar ellipticity at 222 nm between 302 and 353 K (29°C–81°C). The temperature was increased at a rate of 1°C/min with data acquisition for 5 sec at each temperature on an Aviv CD spectrometer. All melting curves were performed in triplicate and the values were averaged.
NMR spectroscopy
For NMR experiments, [RNase A] = 0.65 mM in 5 mM MES buffer (pH 6.4) and 10% D2O. NMR experiments were performed on either a Varian Inova 600 or Varian Unity+ 600 MHz instruments. The temperature was calibrated prior to each experiment at 300 K using 100% methanol as a standard. The 1H carrier frequency was set to the water resonance and the 15N frequency was set to 120 ppm. All experiments were collected using gradient-selected sensitivity-enhanced pulse sequences (Kay et al. 1992; Kördel et al. 1992; Palmer et al. 1992; Skelton et al. 1993) with 128 t 1 increments and 2k points in the direct dimension with spectral widths of 1800 and 9000 Hz, respectively. The data were zero filled prior to Fourier transformation. A Kaiser window (Cavanagh et al. 1996) was applied to the t 1 dimension with Lorentz-to-Gauss resolution enhancement in t 2. All NMR were processed using NMRPipe (Delaglio et al. 1995) and visualized with Sparky (T.D. Goddard and D.G. Kneller, University of California, San Francisco). Peak heights were determined from 3 × 3 grids centered on the peak maximum.
For the R 1 and R 2 relaxation rate measurements, each experiment was preceded by a 2.7-sec recycle delay. Relaxation delays for the R 1 experiments were 2 (×2), 102, 222, 357, 512, 692, 912 (×2), and 1202 msec. The R 2 experiments were acquired with relaxation delays of 0 (×2), 10, 22, 38 (×2), 70, 92, 122, and 162 msec. All relaxation data was fit with a single exponential decay function to obtain the relaxation rates using Curvefit (http://cpmcnet.columbia.edu/dept/gsas/biochem/labs/palmer/software.html). 1H(15N) 90° pulse lengths (microseconds) for the RNase A, RNase A/700 mM Gdn-HCl, and RNase A/700mM Gdn-HCl/350 mM TMAO samples were 9.5 (37.5), 14.0 (37.3), and 12.5 (38.0), respectively. The heteronuclear NOE effect was measured using interleaved experiments with and without proton saturation pulses with 10-sec recycle delays.
Model-free dynamics analysis
Amide backbone dynamics were characterized by fitting the experimentally determined R 1, R 2, and NOE to each of five spectral density models with the corresponding free parameters (Lipari and Szabo 1982a,b; Clore et al. 1990).
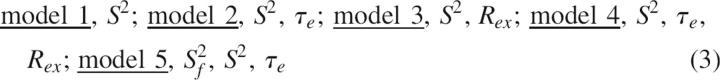 |
in which S 2 is the generalized order parameter, S f 2 is the order parameter for motion with a correlation time faster than 10–20 psec, R ex is the contribution due to conformational exchange motion (τc ∼ μsec-msec), and τe is the internal correlation time (20 psec < τe < 200 psec). Motional parameters were determined using the program FAST-model free (Cole and Loria 2003) interfaced with Model-Free 4.1 (http://cpmcnet.columbia.edu/dept/gsas/biochem/labs/palmer/software.html). The statistical criteria used for model selection is as described by Mandel et al. (1995). Prior to model fitting, the RNase A rotational diffusion tensor was estimated from R 2/R 1 ratios and the program pdbinertia (http://cpmcnet.columbia.edu/dept/gsas/biochem/labs/palmer/software.html) using the crystal structure of RNase A (7RSA), in which hydrogens had been added and the molecule minimized to <0.1 kcal energy difference using the minimize routine in the program TINKER v4.2. For the model-free analysis, the N–H bond length was assumed to be 1.02 Å and the 15N chemical-shift anisotropy was assumed to be axially symmetric with a value of –160 ppm. Other aspects of the model-free fitting are as described previously (Cole and Loria 2003; Kovrigin et al. 2003).
Electronic supplemental material
Supplemental material contains the NMR spin-relaxation rates and the results of model free analysis.
Acknowledgments
We thank Professor Andrew Hamilton (Yale University) for the use of his CD spectrometer and Dr. Evgenii Kovrigin (Yale University) for the RNase A sample and for critical reading of this manuscript. J.P.L. acknowledges funding from an NSF-CAREER award and a fellowship from the Alfred P. Sloan Foundation. This work, in part, fulfilled the undergraduate chemistry degree requirements for V.D.N. V.D.N. was part of the STARS undergraduate research program at Yale that was partially funded by the Howard Hughes Medical Institute and Boehringer-Ingelheim Pharmaceuticals.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: J. Patrick Loria, Department of Chemistry Yale University, P.O. Box 208107, New Haven, CT 06520, USA; e-mail: Patrick.loria@yale.edu; fax: (203) 432-6144.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062393707.
Abbreviations: CD, circular dichroism spectroscopy; Gdn, guanidine hydrochloride; MALDI-TOF, matrix assisted laser desorption ionization time of flight; RNase A, bovine pancreatic ribonuclease A; TMAO, trimethylamine N-oxide.
References
- Abragam, A. 1961. Principles of nuclear magnetism. Clarendon Press, Oxford, UK.
- Akke, M., Brüschweiler, R., and Palmer, A.G. 1993. NMR order parameters and free energy: An analytic approach and application to cooperative Ca2+ binding by calbindin D9k . J. Am. Chem. Soc. 115: 9832–9833. [Google Scholar]
- Anderson, D.G., Hammes, G.G., and Walz Jr., F.G. 1968. Binding of phosphate ligands to ribonuclease A. Biochemistry 7: 1637–1645. [DOI] [PubMed] [Google Scholar]
- Arakawa, T. and Timasheff, S.N. 1985. The stabilization of proteins by osmolytes. Biophys. J. 47: 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athawale, M.V., Dordick, J.S., and Garde, S. 2005. Osmolyte trimethylamine-N-oxide does not affect the strength of hydrophobic interactions: Origin of osmolyte compatibility. Biophys. J. 89: 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach, H., Cole, R., Gill, M., and Loria, J.P. 2005. Conservation of μs-ms enzyme motions in the apo- and substrate-mimicked state. J. Am. Chem. Soc. 127: 9167–9176. [DOI] [PubMed] [Google Scholar]
- Bennion, B.J. and Daggett, V. 2004. Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: A chemical chaperone at atomic resolution. Proc. Natl. Acad. Sci. 101: 6433–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh, J., Fairbrother, W.J., Palmer, A.G., and Skelton, N.J. 1996. Protein NMR spectroscopy: Principles and practice. Academic Press, San Diego, CA.
- Clore, G.M., Szabo, A., Bax, A., Kay, L.E., Driscoll, P.C., and Gronenborn, A.M. 1990. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 112: 4989–4991. [Google Scholar]
- Cole, R. and Loria, J.P. 2002. Evidence for flexibility in the function of ribonuclease A. Biochemistry 41: 6072–6081. [DOI] [PubMed] [Google Scholar]
- Cole, R. and Loria, J.P. 2003. FAST-Modelfree: A program for rapid automated analysis of solution NMR spin-relaxation data. J. Biomol. NMR 26: 203–213. [DOI] [PubMed] [Google Scholar]
- Colombo, G. and Merz, K.M. 1999. Stability and activity of mesophilic subtilisin E and its thermophilic homologue: Insights from molecular dynamics simulations. J. Am. Chem. Soc. 121: 6895–6903. [Google Scholar]
- DeKoster, G.T. and Robertson, A.D. 1997. Calorimetrically-derived parameters for protein interactions with urea and guanidine-HCl are not consistent with denaturant m values. Biophys. Chem. 64: 59–68. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- DeLano, W.L. 2005. MacPyMOL: A PyMOL-based molecular graphics application for MacOS X. DeLano Scientific LLC, South San Francisco, CA.
- Devore, J. 2000. Probability and statistics for engineering and the sciences. Brooks/Cole Publishing Company, Monterey, CA.
- Esposito, L. and Daggett, V. 2005. Insight into ribonuclease A domain swapping by molecular dynamics unfolding simulations. Biochemistry 44: 3358–3368. [DOI] [PubMed] [Google Scholar]
- Fitter, J. and Heberle, J. 2000. Structural equilibrium fluctuations in mesophilic and thermophilic α-amylase. Biophys. J. 79: 1629–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitter, J., Herrmann, R., Dencher, N.A., Blume, A., and Hauss, T. 2001. Activity and stability of a thermostable α-amylase compared to its mesophilic homologue: Mechanisms of thermal adaptation. Biochemistry 40: 10723–10731. [DOI] [PubMed] [Google Scholar]
- Gahl, R.F., Narayan, M., Xu, G., and Scheraga, H.A. 2004. Trimethylamine-N-oxide modulates the reductive unfolding of onconase. Biochem. Biophys. Res. Commun. 325: 707–710. [DOI] [PubMed] [Google Scholar]
- Gonnelli, M. and Strambini, G.B. 2001. No effect of trimethylamine N-oxide on the internal dynamics of the protein native fold. Biophys. Chem. 89: 77–85. [DOI] [PubMed] [Google Scholar]
- Grottesi, A., Ceruso, M.A., Colosimo, A., and Di Nola, A. 2002. Molecular dynamics study of a hyperthermophilic and a mesophilic rubredoxin. Proteins 46: 287–294. [DOI] [PubMed] [Google Scholar]
- Hernandez, G., Jenney Jr., F.E., Adams, M.W., and LeMaster, D.M. 2000. Millisecond time scale conformational flexibility in a hyperthermophile protein at ambient temperature. Proc. Natl. Acad. Sci. 97: 3166–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houry, W.A. and Scheraga, H.A. 1996. Structure of a hydrophobically collapsed intermediate on the conformational folding pathway of ribonuclease A probed by hydrogen- deuterium exchange. Biochemistry 35: 11734–11746. [DOI] [PubMed] [Google Scholar]
- Hovagimyan, K.G. and Gerig, J.T. 2005. Interactions of trimethylamine N-oxide and water with cyclo-alanylglycine. J. Phys. Chem. B. 109: 24142–24151. [DOI] [PubMed] [Google Scholar]
- Idiyatullin, D., Nesmelova, I., Daragan, V.A., and Mayo, K.H. 2003. Heat capacities and a snapshot of the energy landscape in protein GB1 from the pre-denaturation temperature dependence of backbone NH nanosecond fluctuations. J. Mol. Biol. 325: 149–162. [DOI] [PubMed] [Google Scholar]
- Jaravine, V.A., Rathgeb-Szabo, K., and Alexandrescu, A.T. 2000. Microscopic stability of cold shock protein A examined by NMR native state hydrogen exchange as a function of urea and trimethylamine N-oxide. Protein Sci. 9: 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja, J. and Udgaonkar, J.B. 2002. Characterization of the unfolding of ribonuclease a by a pulsed hydrogen exchange study: Evidence for competing pathways for unfolding. Biochemistry 41: 2641–2654. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Keifer, P., and Saarinen, T. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114: 10663–10665. [Google Scholar]
- Kördel, J., Skelton, N.J., Akke, M., Palmer, A.G., and Chazin, W.J. 1992. Backbone dynamics of calcium-loaded calbindin D9k studied by two-dimensional proton-detected NMR spectroscopy. Biochemistry 31: 4856–4866. [DOI] [PubMed] [Google Scholar]
- Kovrigin, E.L. and Loria, J.P. 2006a. Characterization of the transition state of functional enzyme dynamics. J. Am. Chem. Soc. 128: 7724–7725. [DOI] [PubMed] [Google Scholar]
- Kovrigin, E.L. and Loria, J.P. 2006b. Enzyme dynamics along the reaction coordinate: Critical role of a conserved residue. Biochemistry 45: 2636–2647. [DOI] [PubMed] [Google Scholar]
- Kovrigin, E.L., Cole, R., and Loria, J.P. 2003. Temperature dependence of the backbone dynamics of Ribonuclease A in the ground state and bound to the inhibitor 5′-phosphothymidine (3′-5′) pyrophosphate adenosine 3′-phosphate. Biochemistry 42: 5279–5291. [DOI] [PubMed] [Google Scholar]
- Lazaridis, T., Lee, I., and Karplus, M. 1997. Dynamics and unfolding pathways of a hyperthermophilic and a mesophilic rubredoxin. Protein Sci. 6: 2589–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, L.K., Rance, M., Chazin, W.J., and Palmer, A.G. 1997. Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13Cα nuclear spin relaxation. J. Biomol. NMR 9: 287–298. [DOI] [PubMed] [Google Scholar]
- Li, Z., Raychaudhuri, S., and Wand, A.J. 1996. Insights into the local residual entropy of proteins provided by NMR relaxation. Protein Sci. 5: 2647–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T.Y. and Timasheff, S.N. 1994. Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine N-oxide interactions with protein. Biochemistry 33: 12695–12701. [DOI] [PubMed] [Google Scholar]
- Lipari, G. and Szabo, A. 1982a. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104: 4546–4559. [Google Scholar]
- Lipari, G. and Szabo, A. 1982b. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104: 4559–4570. [Google Scholar]
- Liu, Y., Gotte, G., Libonati, M., and Eisenberg, D. 2001. A domain-swapped RNase A dimer with implications for amyloid formation. Nat. Struct. Biol. 8: 211–214. [DOI] [PubMed] [Google Scholar]
- Mandel, A.M., Akke, M., and Palmer, A.G. 1995. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 246: 144–163. [DOI] [PubMed] [Google Scholar]
- Palmer, A.G., Skelton, N.J., Chazin, W.J., Wright, P.E., and Rance, M. 1992. Suppression of the effects of cross-correlation between dipolar and anisotropic chemical shift relaxation mechanisms in the measurement of spin-spin relaxation rates. Mol. Phys. 75: 699–711. [Google Scholar]
- Palmer, H.R., Bedford, J.J., Leader, J.P., and Smith, R.A. 2000. 31P and 1H NMR studies of the effect of the counteracting osmolyte trimethylamine-N-oxide on interactions of urea with ribonuclease A. J. Biol. Chem. 275: 27708–27711. [DOI] [PubMed] [Google Scholar]
- Poklar, N., Petrovcic, N., Oblak, M., and Vesnaver, G. 1999. Thermodynamic stability of ribonuclease A in alkylurea solutions and preferential solvation changes accompanying its thermal denaturation: A calorimetric and spectroscopic study. Protein Sci. 8: 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, Y. and Bolen, D.W. 2003. Hydrogen exchange kinetics of RNase A and the urea:TMAO paradigm. Biochemistry 42: 5837–5849. [DOI] [PubMed] [Google Scholar]
- Skelton, N.J., Palmer, A.G., Akke, M., Kördel, J., Rance, M., and Chazin, W.J. 1993. Practical aspects of two-dimensional proton-detected 15N spin relaxation measurements. J. Magn. Reson. B 102: 253–264. [Google Scholar]
- Somero, G.N. 2003. Protein adaptations to temperature and pressure: Complementary roles of adaptive changes in amino acid sequence and internal milieu. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 136: 577–591. [DOI] [PubMed] [Google Scholar]
- Svingor, A., Kardos, J., Hajdu, I., Nemeth, A., and Zavodszky, P. 2001. A better enzyme to cope with cold. Comparative flexibility studies on psychrotrophic, mesophilic, and thermophilic IPMDHs. J. Biol. Chem. 276: 28121–28125. [DOI] [PubMed] [Google Scholar]
- Tang, K.E. and Dill, K.A. 1998. Native protein fluctuations: The conformational-motion temperature and the inverse correlation of protein flexibility with protein stability. J. Biomol. Struct. Dyn 16: 397–411. [DOI] [PubMed] [Google Scholar]
- Tsai, A.M., Udovic, T.J., and Neumann, D.A. 2001. The inverse relationship between protein dynamics and thermal stability. Biophys. J. 81: 2339–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udgaonkar, J.B. and Baldwin, R.L. 1990. Early folding intermediate of ribonuclease A. Proc. Natl. Acad. Sci. 87: 8197–8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen, M. 1987. Relationship of protein flexibility to thermostability. Protein Eng. 1: 477–480. [DOI] [PubMed] [Google Scholar]
- Wagner, G. and Wuthrich, K. 1979. Correlation between the amide proton exchange rates and the denaturation temperatures in globular proteins related to the basic pancreatic trypsin inhibitor. J. Mol. Biol. 130: 31–37. [DOI] [PubMed] [Google Scholar]
- Wang, A. and Bolen, D.W. 1997. A naturally occurring protective system in urea-rich cells: Mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry 36: 9101–9108. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Svensson, L.A., Sjolin, L., and Gilliland, G.L. 1988. Structure of phosphate-free ribonuclease A refined at 1.26 Å. Biochemistry 27: 2705–2717. [DOI] [PubMed] [Google Scholar]
- Woessner, D.E. 1962. Spin relaxation processes in a two-proton system undergoing anisotropic reorientation. J. Chem. Phys. 36: 1–4. [Google Scholar]
- Yancey, P.H. 1994. Compatible and counteracting solutes. In Cellular and molecular physiology of cell volume regulation (ed. K. Strange), pp. 81–109. CRC Press, Boca Raton, FL.
- Yancey, P.H. 2001. Protein, osmolytes and water stress. Am. Zool. 41: 699–709. [Google Scholar]
- Yancey, P.H. 2005. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 208: 2819–2830. [DOI] [PubMed] [Google Scholar]
- Yancey, P.H. and Somero, G.N. 1979. Counteraction of urea destabilization of protein structure by methylamine osmoregulatory compounds of elasmobranch fishes. Biochem. J. 183: 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey, P.H., Fyfe-Johnson, A.L., Kelly, R.H., Walker, V.P., and Aunon, M.T. 2001. Trimethylamine oxide counteracts effects of hydrostatic pressure on proteins of deep-sea teleosts. J. Exp. Zool. 289: 172–176. [DOI] [PubMed] [Google Scholar]
- Yancey, P.H., Blake, W.R., and Conley, J. 2002. Unusual organic osmolytes in deep-sea animals: Adaptations to hydrostatic pressure and other perturbants. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 133: 667–676. [DOI] [PubMed] [Google Scholar]
- Yang, D. and Kay, L.E. 1996. Contributions to conformational entropy arising from bond vector fluctuations measured from NMR-derived order parameters: Application to protein folding. J. Mol. Biol. 263: 369–382. [DOI] [PubMed] [Google Scholar]
- Zavodszky, P., Kardos, J., Svingor, A., and Petsko, G.A. 1998. Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proc. Natl. Acad. Sci. 95: 7406–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, Q., Bennion, B.J., Daggett, V., and Murphy, K.P. 2002. The molecular mechanism of stabilization of proteins by TMAO and its ability to counteract the effects of urea. J. Am. Chem. Soc. 124: 1192–1202. [DOI] [PubMed] [Google Scholar]