

Abstract
Myocardin (MC) family proteins are transcriptional coactivators for serum response factor (SRF). Each family member possesses a conserved N-terminal region containing three RPEL motifs (the “RPEL domain”). MAL/MKL1/myocardin-related transcription factor A is cytoplasmic, accumulating in the nucleus upon activation of Rho GTPase signaling, which alters interactions between G-actin and the RPEL domain. We demonstrate that MC, which is nuclear, does not shuttle through the cytoplasm and that the contrasting nucleocytoplasmic shuttling properties of MAL and MC are defined by their RPEL domains. We show that the MAL RPEL domain binds actin more avidly than that of MC and that the RPEL motif itself is an actin-binding element. RPEL1 and RPEL2 of MC bind actin weakly compared with those of MAL, while RPEL3 is of comparable and low affinity in the two proteins. Actin binding by all three motifs is required for MAL regulation. The differing behaviors of MAL and MC are specified by the RPEL1-RPEL2 unit, while RPEL3 can be exchanged between them. We propose that differential actin occupancy of multiple RPEL motifs regulates nucleocytoplasmic transport and activity of MAL.
The myocardin (MC) family of transcriptional coactivators regulates the activity of the transcription factor serum response factor (SRF) through association with its DNA-binding domain (2, 14, 17, 21, 24, 27). Two of the proteins, MAL/MKL1/myocardin-related transcription factor A (MRTF-A) and MAL16/MKL2/MRTF-B, are ubiquitously expressed, while the expression of MC, the founding family member, is restricted to smooth and cardiac muscle. In contrast to MC, which appears constitutively nuclear (24), the other MC family members redistribute from the cytoplasm to the nucleus upon activation of Rho signaling in many other cell lines (5, 14).
In fibroblasts, the regulation of MAL localization and activity is controlled largely by Rho-dependent changes in the dynamics of actin turnover between its monomeric (G-actin) and filamentous (F-actin) states, and blockade of Rho-induced actin polymerization prevents MAL-mediated activation of SRF target genes (11, 13, 14, 23). MAL constantly circulates between nucleus and cytoplasm in serum-starved cells. Its cytoplasmic steady-state localization is maintained by very efficient CRM1-dependent nuclear export, which also requires its interaction with actin in the nucleus (23). MAL senses the cellular G-actin concentration by direct interaction (Fig. 1A), and reduction of this interaction, whether it results from Rho-induced depletion of the G-actin pool or from direct disruption by actin-binding drugs, such as cytochalasin D (CD), leads to MAL nuclear accumulation (Fig. 1A) (14, 23).
FIG. 1.

MAL and MC are differentially regulated through their N-terminal RPEL domains. (A) Schematic representation of Rho-actin signaling to SRF. Depletion of the G-actin pool is sensed by the actin-binding SRF cofactor MAL. C3 transferase blocks Rho-mediated changes in actin dynamics; CD disrupts the MAL-actin complex; LatB increases the G-actin pool by blocking actin polymerization. (B) Domain organization of MAL and MC. B1, basic region 1; Q, Q-rich region; SAP, SAF-AIB, Acinus, Pias domain, LZ, leucine zipper motif; TAD, transcription activation domain. B2 is in yellow. (C) Localization of transiently expressed MAL, MC, and chimeras generated by the reciprocal crossover of the RPEL domains, as shown in panel B, in serum-starved NIH 3T3 fibroblasts detected by immunofluorescence microscopy. See Fig. 6B for quantitation. (D) Activation of an SRF luciferase reporter by expression of the indicated MAL and MC derivatives without (−C3) and with (+C3) coexpression of C3 transferase in serum-starved NIH 3T3 fibroblasts. Reporter activation is normalized to that conferred by SRF-VP16 or SRF-VP16 plus C3 transferase (100%). Three independent experiments were performed. Error bars, SEM. (E) MC does not shuttle through the cytoplasm. Nuclear export rates of MAL-GFP, MC-GFP, and chimeras measured by FLIP under the indicated conditions. The cytoplasm is bleached repeatedly, and nuclear fluorescence is monitored. Left, bleaching kinetics of nuclear fluorescence; right, initial bleach rates (>10 cells per condition). Error bars, standard deviations (SD).
MC family proteins possess a conserved N-terminal region containing three RPEL motifs (Pfam no. 02755) (6), termed the RPEL domain, and form one of two families of RPEL-containing proteins in metazoans (Fig. 1B). The MAL RPEL domain forms a stable complex with three molecules of actin in solution (18, 23). Alanine substitution at the conserved R or P residues of all three MAL RPEL motifs effectively reduces the interaction of the MAL RPEL domain with actin, abolishing nuclear export and resulting in nuclear accumulation; in addition, such mutants strongly activate SRF-dependent transcription independently of Rho signaling (14, 18, 23). Curiously, although MC possesses RPEL motifs, it is constitutively nuclear and insensitive to Rho signaling (11, 24). It has remained unclear whether these differences reflect sequence divergence within and between the individual RPEL motifs or other regulatory domains in the protein (9).
Here, we investigate the role of the RPEL domain in regulation in detail. We show that the MAL RPEL domain is necessary and sufficient to confer Rho-regulated nuclear accumulation upon a heterologous protein, while that of MC confers constitutive nuclear localization. We show that the RPEL motif defines an actin-binding element. MAL RPEL1 and RPEL2 bind actin relatively strongly, while RPEL3 binds more weakly. In contrast, whereas MC RPEL3 is of an affinity similar to that of MAL, MC RPEL1 and RPEL2 have a much lower affinity for actin. We show that the three RPEL motifs functionally cooperate in MAL to control nucleocytoplasmic shuttling. Using point mutants and protein chimeras, we show that RPEL1 and RPEL2 together control whether the protein is constitutively nuclear or linked to Rho signaling, while RPEL3 is interchangeable between MAL and MC. These results establish a central role for actin binding and the RPEL domain in control of MAL subcellular localization. We propose that MAL regulation is achieved by regulation of actin binding to multiple RPEL motifs.
MATERIALS AND METHODS
MC, mutant MAL derivatives, and MAL-MC chimeras.
A cDNA encoding MC (transcript variant A; GenBank accession number NM_145136) was obtained by reverse transcription-PCR from mouse cardiac RNA. The following point mutations were introduced into MAL(full length): R81A, RR81/82DD, R125A, and R169A (see Fig. 5A). Crossover points for reciprocal exchanges of portions of the N termini between MAL and MC were placed in the center of the RPEL-RPEL linker sequences. The reciprocal RPEL domain exchange included six amino acids C-terminal of the Pfam-defined RPEL motif 3. The C-terminal residues of N extensions were six amino acids N-terminal of Pfam-defined RPEL1. RPEL2 as defined by Pfam was reciprocally exchanged. For Pfam definitions, see Fig. 4C.
FIG. 5.

RPEL motifs cooperate in MAL regulation. (A) Schematic representation of generated mutants. x23, R81A; 1x3, R125A; 12x, R169A and combinations thereof; MAL-1DD, RR81/82D. (B) Left, quantitation of immunofluorescence microscopy: localization of the indicated C-terminally HA-tagged MAL derivatives was scored in 200 serum-starved cells. N, nuclear; N/C, comparable intensity in nucleus and cytoplasm; C, cytoplasmic. Data from three independent experiments are shown. Error bars, SEM. Right, illustration of the localization scoring system. MAL derivatives, greyscale; F-actin, red; DAPI, blue. (C and E) Activation of an SRF luciferase reporter by expression of the indicated MAL derivatives, analyzed as for Fig. 1D. (D) Quantitation of immunofluorescence microscopy as in panel B. See Fig. S4 in the supplemental data for expression levels, and see Fig. S2 in the supplemental data for representative micrographs.
FIG. 4.

RPEL motifs have different affinities for actin. (A) GST pulldown assay. The indicated GST fusion proteins were used to immobilize endogenous actin from a total NIH 3T3 cell lysate in the absence or presence of 0.5% TX-100. WB, Western blot with detection of β-actin (two exposures shown). Ponceau stain of membrane to show GST fusion proteins. (B) FRET analysis of the interaction of MAL-GFP or MC-GFP (donors) with Cy3-immunolabeled MYC-β-actin (acceptor). Left, micrographs: top, donor intensities; middle, lifetime maps of donor fluorescence with higher lifetimes in blue and lower lifetimes in red; median FRET efficiencies are indicated; bottom, acceptor intensities. 0.3%, serum-starved cells. Right, FRET efficiencies for MC in serum-starved cells in a box-and-whisker plot showing the median (18 cells). (C) Top, multiple sequence alignment of RPEL motifs of mouse MAL and MC generated by ClustalX (version 1.81 (22). The Pfam definition of the RPEL motif is indicated by brackets. “x” denotes the first and most conserved R residue of the RPEL motif targeted by the R→A mutation. Bottom, corresponding phylogram also containing MAL16, generated by ClustalW (3). The phylogram is based on the RPEL motifs as defined by Pfam. Distances to the closest nodes are: MAL RPEL1, 0.06250; MAL16 RPEL1, 0.07468; MC RPEL1, 0.19805; MAL RPEL2, 0.08807; MAL16 RPEL2, 0.13920; MC RPEL2, 0.24148; MAL RPEL3, 0.0; MAL16 RPEL3, 0.0; MC RPEL3, 0.05114. (D) GST pulldown assay as in panel A, but only in the absence of TX-100. Peptides fused to GST comprise 32 amino acids as shown in panel C. R→A mutations refer to R81 in RPEL1, R125 in RPEL2, and R169 in RPEL3 (see panel C). RR→DD refers to RR81/82 in RPEL1. (E) Fluorescence anisotropy assay. Anisotropy of FITC-conjugated 32-amino acid RPEL peptides (as shown in panel C) at 0.5 μM was measured over a range of LatB-actin concentrations. Anisotropy values were multiplied by 1,000. KD values for RPEL-actin interactions were determined by nonlinear regression using GraFit (bottom right). Data from three parallel experiments are shown. Error bars, SD.
Immunofluorescence microscopy.
Immunofluorescence microscopy was performed as described previously (23). In a six-well dish, 150,000 cells per well were transfected with 100 ng of C-terminally hemagglutinin (HA)-tagged MAL, MC, or derivatives; 50 ng of FLAG-MAL; 10 ng of FLAG-MAL(2-204)-pyruvate kinase (PK); or FLAG-MC(2-150)-PK and 50 ng MYC-β-actin R62D. Primary antibodies were as follows: anti-FLAG (F7425; Sigma), anti-HA (12CA5; Roche) and anti-MYC (gE10; CR-UK). Before stimulation, cells were maintained in medium containing 0.5% serum for 20 h after transfection. Unless stated otherwise, cells were treated with 15% fetal calf serum, 2 μM CD, 0.1 μM jasplakinolide, 0.3 μM latrunculin B (LatB), or 20 nM leptomycin B (LMB) for 30 min. MAL localization was scored as predominantly nuclear, pancellular, or predominantly cytoplasmic in 150 to 200 cells. Data from three independent experiments are shown in the figures with standard errors of the means (SEM).
Luciferase reporter assay.
Cells in a 24-well dish (30,000 cells/well) were transfected with SRF reporter p3DA.luc (8 ng), reference reporter ptk-RL (20 ng) with or without SRF-VP16 (40 ng), MAL and MAL mutant derivative constructs (10 ng), or C3 transferase (2 ng). Cells were maintained in 0.5% fetal calf serum for 22 h, after which luciferase activities were measured. Firefly luciferase activity was normalized to Renilla luciferase activity. Data from three independent experiments with SEM are expressed relative to reporter activation by coexpressed SRF-VP16. Data with C3 transferase coexpression were normalized to reporter activation by SRF-VP16 and C3 transferase coexpression.
GST pulldown assays.
Glutathione-agarose (Sigma) was saturated with glutathione S-transferase (GST) or GST fusion proteins and peptides from Escherichia coli lysates, washed, and used as affinity resin in a binding reaction with total NIH 3T3 cell extract, generated by lysis in binding buffer (50 mM Tris-HCl [pH 8], 100 mM NaCl, 3 mM MgCl2, 0.2 mM EGTA, 0.2 mM ATP, 1 mM dithiothreitol [DTT], and protease inhibitors) through syringing and removal of insoluble material by centrifugation. An equivalent of a confluent 150-mm dish of NIH 3T3 cells was used for two binding reactions. Binding was for 2 h in binding buffer, supplemented with 0.5% TX-100 where indicated. The resin was washed three times in the respective binding buffer without protease inhibitors and subjected to 4 to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting with detection of endogenous β-actin (AC-15; Sigma). The blot was Ponceau stained to reveal bait input.
Fluorescence anisotropy.
Fluorescein isothiocyanate (FITC)-conjugated peptides were synthesized by the Cancer Research UK peptide synthesis facility and quantified using the equation ɛ492 = 83,000 m−1 cm−1. Actin was purified from rabbit skeletal muscle, rendered nonpolymerizable by incubation in G buffer (2 mM Tris-HCl [pH 8.0], 0.3 mM MgCl2, 0.2 mM EGTA, 0.2 mM ATP, 0.5 mM DTT) with a 10-fold molar excess of LatB (Calbiochem) for 15 h. Uncomplexed actin was polymerized by the addition of 20× initiation buffer (2 M NaCl, 60 mM MgCl2, 10 mM ATP) and removed by ultracentrifugation. Fluorescence anisotropy was measured in a total volume of 50 μl in Mg2+-F buffer (2 mM Tris-HCl, pH 8.0; 100 mM NaCl; 3 mM MgCl2; 0.2 mM EGTA; 0.7 mM ATP; 0.5 mM DTT). FITC-conjugated peptides were used at 0.5 μM while LatB-actin was added from 1 nM up to 59 μM. Plates were read after a minimal coincubation period of 5 h at room temperature to ensure the establishment of binding equilibrium using a Safire2 microplate reader (Tecan) in fluorescence polarization mode (excitation, 470 ± 20 nm; emission, 525 ± 20 nm; 10 reads; 40-μs integration time) and its Magellan software (version 5.03). Anisotropy (A) was calculated by the Magellan software using the formula A = (Iparallel − Iperpendicular)/(Iparallel + 2Iperpendicular), where Iparallel and Iperpendicular denote the fluorescence intensities parallel and perpendicular to the excitation plane, respectively, and a G factor of 1.2041. Nonlinear regression to determine equilibrium dissociation constant (KD) values was done with GraFit version 5.0.11 (Erithacus Software) using the following equation (7):
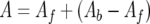 |
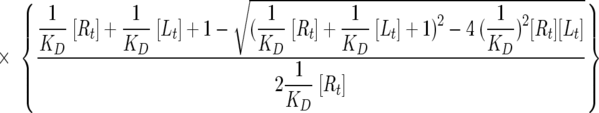 |
where A is the measured value of anisotropy; Af and Ab are the anisotropy values corresponding to free and bound peptide, respectively; and [Rt] and [Lt] are the total peptide (“receptor”) and total LatB-actin (“ligand”) concentrations, respectively.
Live-cell imaging and photobleaching.
Live-cell imaging was performed essentially as described in reference 23. Briefly, for fluorescence loss in photobleaching (FLIP), cells were plated on MatTek dishes (MatTek Corporation), transfected with 50 ng of MAL-green fluorescent protein (GFP) and chimera-GFP plasmids or 20 ng of MAL(1-204)-2GFP or its derivatives, and maintained in phenol red-free medium containing 0.3% serum for 18 h prior to imaging. Cells were stimulated 30 to 60 min prior to imaging, and an area in the cytoplasm was repeatedly bleached for 80 s. For analysis, the background was subtracted, and nuclear fluorescence prior to bleaching was set to 1. At least 10 cells from at least two independent experiments were analyzed per condition. For analysis of nuclear accumulation of MAL(1-204)-2GFP upon stimulation, a stable cell line expressing MAL(1-204)-2GFP was used. The nuclear fluorescence prior to stimulation was set to 0, and the nuclear fluorescence after complete accumulation was set to 1. At least 12 cells from three independent movies were analyzed per condition. For details, see reference 23.
FRET by FLIM.
Fluorescence resonance energy transfer (FRET) monitored by fluorescence lifetime imaging microscopy (FLIM) has been described in detail elsewhere (12) and was performed essentially as described in reference 23. NIH 3T3 cells were transfected with 100 ng of MAL-GFP or MC-GFP (donors), with or without 100 ng of MYC-β-actin (acceptor). MYC-β-actin was subsequently detected with Cy3-9E10 monoclonal antibody. The image sequences of the donor were processed by using IP Lab software with automated analysis to provide nonbiased analysis of the data (P. Leboucher and B. Larijani, unpublished data). For details, see reference 23.
RESULTS
MC does not shuttle between nucleus and cytoplasm.
We compared the localization of transiently expressed MAL and MC in transfected NIH 3T3 cells. MC was nuclear in both serum-starved and -stimulated cells and strongly activated an SRF reporter gene, in agreement with earlier reports (Fig. 1C and D; data not shown) (11, 24). Reporter activation by MC was unaffected by the coexpression of C3 transferase, which inactivates Rho (8), and was therefore independent of functional Rho, unlike activation by MAL (Fig. 1D) (14). We considered the possibility that, like MAL, MC also shuttles through the nucleus, its nuclear localization reflecting enhanced import or decreased export rates compared to MAL. To investigate this, we performed FLIP experiments, in which decay of nuclear fluorescence of a test protein fused to GFP is monitored during photobleaching of the cytoplasm (Fig. 1E). A MC-GFP fusion protein was localized to the nucleus; its bleaching rate was slower than that of MAL-GFP in serum-stimulated or CD-treated cells and very similar to that of MAL-GFP in the presence of LMB, which blocks CRM1-dependent nuclear export (Fig. 1E) (see reference 23). Thus, MC is a constitutively nuclear SRF coactivator that does not shuttle between nucleus and cytoplasm.
Since actin binding to the RPEL domain regulates nucleocytoplasmic shuttling of MAL, we next investigated whether differential regulation of MAL and MC result from differences between the proteins’ RPEL domains. Reciprocal exchange of the entire RPEL domain between MAL and MC resulted in exchange of their regulatory properties. MC-N123-MAL, like MC, was predominantly nuclear (Fig. 1C). The two proteins potently activated the SRF reporter to very similar extents, and the activation was not dependent on functional Rho (Fig. 1D). Analysis by FLIP indicated that, like MC itself, MC-N123-MAL was not appreciably exported from the nucleus (Fig. 1E). Conversely, MAL-N123-MC was predominantly cytoplasmic in serum-starved cells (Fig. 1C); it moderately activated the SRF reporter in a Rho-dependent manner, as did MAL (Fig. 1D). Treatment with serum or CD resulted in MAL-N123-MC nuclear accumulation, under which conditions its nuclear export rate was comparable to that of MAL (Fig. 1E).
Together, these results show that the RPEL domains of MAL and MC determine their differential regulation, at the levels of both subcellular localization and activation of SRF-mediated transcription.
The RPEL domain controls dynamic nucleocytoplasmic shuttling of MAL.
We next tested whether the RPEL domain is sufficient to mediate Rho-actin-regulated nucleocytoplasmic shuttling in the absence of other MAL or MC sequences. The MAL and MC RPEL domains were fused to PK, a cytoplasmic tetramer of 60-kDa subunits (MAL(2-204)-PK and MC(2-150)-PK) (Fig. 2A) (10). We assessed the behavior of the fusions or point mutant derivatives under a number of conditions previously shown to affect MAL subcellular localization (Fig. 2B) (14, 23).
FIG. 2.

The RPEL domain of MAL is sufficient to confer nucleocytoplasmic shuttling. (A) Schematic representation of PK fusion proteins. (B) Immunofluorescence microscopy of PK fusion proteins. Transiently transfected cells were serum starved, starved and stimulated, or treated as indicated (PK derivative, green; F-actin, red). Bottom, quantitation of immunofluorescence microscopy: localization of MAL, MAL(2-204)-PK, MC, and MC(2-150)-PK was scored in 150 to 200 cells as follows: N, nuclear; N/C, comparable intensity in nucleus and cytoplasm; C, cytoplasmic (for an example, see also Fig. 5B). Three independent experiments were performed. Error bars, SEM; FCS, fetal calf serum; Jasp, jasplakinolide; R62D, coexpression of β-actin R62D. xxx refers to mutation of all three RPEL motifs by R→A exchange (Fig. 5A). See Fig. S4 in the supplemental material for the expression of MAL(2-204)-PK.
MAL(2-204)-PK behaved very similarly to full-length MAL. It was predominantly cytoplasmic in serum-starved cells, and it accumulated in the nucleus following treatments that induce MAL nuclear accumulation (Fig. 2B, compare left and right bars). Serum-induced MAL(2-204)-PK nuclear accumulation was rapidly reversed by serum washout or LatB treatment and was prevented by Rho inactivation or overexpression of the nonpolymerizable β-actin mutant R62D. As seen with intact MAL, alanine substitutions at the conserved arginine in all three RPEL motifs (subsequently referred to as R→A mutations) resulted in constitutive nuclear localization of MAL(2-204)-PK. Nuclear accumulation, whether induced by serum stimulation or mutation of the RPEL motifs, was dependent on the RPEL domain B2 region (Fig. 2B, compare left and right bars) (14, 23). This result supports the notion that the B2 region is a nuclear localization signal that is regulated by actin binding (23). Like MC, MC(2-150)-PK was nuclear in unstimulated cells (Fig. 2B). Serum stimulation and LatB treatment did not affect nuclear localization of either MC or MC(2-150)-PK, although the coexpression of β-actin R62D (19) resulted in a moderate relocalization of both MC and MC(2-150)-PK to the cytoplasm, suggesting that the MC RPEL domain may, in principle, bind to actin. Deletion of the B2 region in MC and MC(2-150)-PK resulted in their substantial or complete cytoplasmic localization, respectively (data not shown). Taken together, these observations show that the RPEL domain is sufficient to respond to Rho-actin signaling and that the different behaviors of MAL and MC reflect different properties of their RPEL domains.
To assess the dynamics of RPEL fusion proteins, we linked the MAL and MC RPEL domains to a double-GFP tag [MAL(1-204)-2GFP and MC(1-150)-2GFP] (Fig. 3A). In serum-starved cells, MAL(1-204)-2GFP was cytoplasmic; its basal rate of nuclear import, revealed by the rate of its nuclear accumulation upon LMB treatment, was comparable to that following serum or CD stimulation, as previously observed for MAL-GFP (Fig. 3B) (23). To monitor the nuclear export of MAL(2-204)-2GFP, we again performed FLIP experiments. Under the experimental conditions, the nuclear fluorescence of GFP rapidly decayed during cytoplasmic bleaching (Fig. 3C). The nuclear fluorescence of MC(1-150)-2GFP, like that of MAL-GFP in the presence of LMB, was largely insensitive to cytoplasmic bleaching. Thus, the RPEL domain of MC cannot facilitate nuclear export. Bleaching of nuclear MAL(1-204)-2GFP fluorescence following CD treatment or serum stimulation was somewhat more efficient and was comparable to the that of the actin-binding-deficient xxx derivative of MAL(1-204)-2GFP. Similar results were previously observed with MAL-GFP (23). In summary, the MAL and MC RPEL domains suffice for conferring actin-controlled and CRM1-dependent nucleocytoplasmic shuttling or constitutively nuclear localization on MRTFs.
FIG. 3.

The MAL RPEL domain is sufficient to mediate dynamic nucleocytoplasmic shuttling of MAL. (A) Schematic representation of 2GFP fusion proteins. (B) Nuclear accumulation kinetics of MAL(1-204)-2GFP upon indicated stimulus; top, representative micrographs; bottom, quantitation of nuclear fluorescence (at least 12 cells per condition; error bars, SD). t[s], time in seconds. (C) The MAL RPEL domin confers serum- and CD-induced nuclear accumulation via a block of nuclear export. FLIP analysis was performed as for Fig. 1D. See Fig. S1 in the supplemental material for the localization of MAL(1-204)-2GFP and MC(1-150)-2GFP.
MAL and MC RPEL domains bind actin differentially.
Since actin binding is essential for coupling MAL localization to Rho signaling, we tested whether the link of MAL but not MC to Rho signaling is a consequence of differential actin binding to their RPEL domains. In GST pulldown assays, GST-MAL(2-261) recruited endogenous β-actin from a total cell lysate; actin recruitment by GST-MC(2-209) was substantially less efficient and was further weakened in the presence of detergent (Fig. 4A). To test whether differential actin binding to MAL and MC occurs in a cellular environment, we used FRET detected by FLIM (Fig. 4B). In this assay, FRET between MAL-GFP and Cy3-immunolabeled MYC-β-actin could be readily detected (FRET efficiencies, 4.55 and 4.91 in serum-starved and LMB-treated cells, respectively) (23). In contrast, we did not detect FRET for MC-GFP and actin under identical conditions (FRET efficiency, 0.5). Thus, the differential regulatory properties of the two RPEL domains are associated with differences in their actin-binding properties in vitro and in vivo.
The isolated RPEL motif defines an actin-binding element.
To investigate the basis for the differential actin-binding properties of the MAL and MC RPEL domains, we analyzed the properties of the individual RPEL motifs. Phylogenetic analysis shows that each of the three MAL RPEL motifs is more closely related to the corresponding motifs in the other family members than to the other motifs in MAL. RPEL1 and RPEL2 are most divergent between MAL and MC, while RPEL3 is relatively conserved (Fig. 4C).
We first performed GST pulldown assays using individual RPEL motifs as baits. GST fusions of MAL RPEL1 or RPEL2 efficiently recruited β-actin from cell lysates, while MAL RPEL3 recovered β-actin inefficiently (Fig. 4D, left). In this assay, the MAL RPEL1 R→A substitution greatly reduced but did not abolish interaction with actin, whereas the analogous substitutions in RPEL2 and RPEL3 had a much greater effect. A more severe, charge-reversal mutation in MAL RPEL1, RR81/82DD, blocked its interaction with actin in this assay. In contrast to MAL, MC RPEL1 and RPEL2 did not recover detectable amounts of β-actin in this assay, although MC RPEL3 appeared to bind actin similarly to MAL RPEL3 (Fig. 4D, right).
To measure RPEL-actin binding affinities quantitatively under conditions of solution-binding equilibrium, we employed fluorescence anisotropy assays. Increasing amounts of nonpolymerizable LatB-actin were titrated into binding reactions containing a constant amount of 32-residue RPEL peptides, N-terminally modified with fluorescein. The fluorescence anisotropy at 525 nm was measured, and the equilibrium dissociation constants were derived as described in Materials and Methods. MAL RPEL1 and RPEL2 bound actin with comparable affinities of 5.4 ± 0.5 μM and 2.3 ± 0.2 μM, respectively (Fig. 4E). MAL RPEL3 bound three to seven times weaker, with a KD of only 18.8 ± 1.0 μM. The R→A mutation decreased the affinities of MAL RPEL1 and RPEL2 for actin to 16.4 ± 2.1 μM and 15.0 ± 3.3 μM, respectively, and reduced the affinity of MAL RPEL3 to below the detection limit, as did the RPEL1 charge reversal mutation. Actin bound to MC RPEL1 with a KD of 15.4 ± 0.8 μM, threefold lower than to MAL RPEL1, but strikingly, binding of actin to MC RPEL2 was undetectable under our assay conditions. The affinity of MC RPEL3 was very similar to that of MAL RPEL3, with a KD of 16.6 ± 0.2 μM. Taken together, these data demonstrate that the RPEL motif defines an actin-binding element with a wide range of affinities for actin and suggest that the different properties of the MAL and MC RPEL domains may reflect differences in the affinities of their RPEL motifs for actin.
The three MAL RPEL motifs functionally cooperate in regulation.
We next used the insights from the actin-binding analysis to address the functional significance of RPEL motifs for MAL regulation. To this end, we introduced the R→A mutation into each of the RPEL motifs, generating single (x23; 1x3; 12x), double (1xx; x2x; xx3), and triple (xxx) MAL RPEL domain mutants (Fig. 5A). The proteins were expressed in NIH 3T3 cells and analyzed for subcellular localization and their potential to activate an SRF reporter.
Under serum-starved conditions, wild-type MAL was found detectably nuclear (nuclear or with a comparable intensity in the nucleus and cytoplasm) in less than 10% of the cells (Fig. 5B) (14). An alanine substitution at RPEL1, RPEL2, and RPEL3 increased this to approximately 25%, 40%, and 70% of the cells, respectively. Compared to single RPEL mutants, all double RPEL mutants displayed a much stronger tendency to accumulate in the nucleus, and MAL 1xx and xxx were virtually indistinguishable, with 90% of cells displaying predominantly nuclear localization (Fig. 5B). Following serum stimulation, all proteins were predominantly nuclear in >80% of the cells (data not shown). The ability of these mutants to activate the SRF reporter correlated well with the proportion of cells displaying appreciable nuclear accumulation (Fig. 5C). Single-alanine substitutions reduced reporter dependence on Rho, with mutation of RPEL3 showing the greatest effect, while double substitutions further decreased Rho dependence (Fig. 5C). The integrity of all three RPEL motifs is thus required for MAL regulation.
The alanine-substituted MAL RPEL1 and RPEL2 peptides both retain an affinity for actin similar to that of MAL RPEL3 (Fig. 4E). To analyze the role of actin binding to RPEL1 more thoroughly, we introduced the RPEL1 charge reversal mutation (RR81/82DD), which effectively abolishes actin binding in vitro, into MAL (MAL-1DD) (Fig. 5A). MAL-1DD was more severely deregulated than MAL x23: it was cytoplasmic in only 30% of the cells, pancellular in about 60%, and nuclear in approximately 10% (Fig. 5D), and it activated the SRF reporter more strongly (Fig. 5E). We used a similar approach with MAL RPEL2, in this case precisely exchanging it for MC RPEL2, which does not bind actin detectably in vitro (MAL-2MC) (Fig. 5A). This mutant also exhibited striking deregulation, with nuclear localization in 20% of the cells and with most of the remainder displaying pancellular localization (Fig. 5D); the expression of MAL-2MC strongly activated the SRF reporter substantially independently of Rho (Fig. 5E).
These results demonstrate that the three MAL RPEL motifs functionally cooperate, with even the weakest actin-binding motif, RPEL3, playing an important role in MAL regulation. Mutations in any single MAL RPEL motif are sufficient to cause deregulation of MAL subcellular localization, provided such mutations effectively abolish RPEL-actin interaction in vitro. Taken together, the results suggest that MAL regulation involves assembly of a higher-order actin-MAL complex(es) (see Discussion).
The RPEL1-RPEL2 unit specifies differential regulation of MAL and MC.
The different actin-binding affinities of MAL and MC RPEL1 and RPEL2 suggest that the reason for the differential regulation of MAL and MC might reside in the unit containing these two RPEL motifs. Indeed, the observation above that substitution of MAL RPEL2 by that of MC results in substantial deregulation of MAL subcellular localization and activity is consistent with this idea. To address the role of the RPEL motifs in MC localization more systematically, we generated a series of MAL-MC chimeras, in which increasing extents of N-terminal sequences were reciprocally exchanged, with crossover points in the center of the RPEL-RPEL linker sequence (see Fig. 6A for nomenclature). The chimeras were analyzed for their subcellular localization and their ability to activate an SRF reporter in serum-starved cells, and their nuclear export rates were analyzed using the FLIP assay.
FIG. 6.

An intact unit of MAL RPEL motifs 1 and 2 is required for MAL regulation. (A) Schematic representation of amino termini of MAL-MC chimeras. Nomenclature: N, N extension; numbers refer to position of RPEL motifs. The B2 region is in yellow. (B, E, and G) Quantitation of immunofluorescence microscopy: localization of the indicated C-terminally HA-tagged MAL and MC derivatives was scored in 150 to 200 serum-starved cells as in Fig. 5B. See Fig. S4 in the supplemental material for expression levels, and see Fig. S3 in the supplemental material for representative micrographs. (C, F, and H) Activation of the SRF luciferase reporter by expression of the indicated MAL and MC derivatives, analyzed as for Fig. 1D. (D) Nuclear export of MAL-MC chimeras measured by FLIP, as in Fig. 1E.
We first used the chimeras to map which RPEL domain sequences are necessary to specify the nuclear localization of MC. Replacement of the MAL N extension (those sequences N-terminal to RPEL1) by that of MC had a slight effect on MAL regulation, with an increased number of cells showing pancellular localization. However, replacement of the MAL N extension and RPEL1 by the corresponding MC sequences resulted in its profound deregulation: MC-N1-MAL was nuclear in about 70% of the cells and pancellular in almost all of the remaining cells (Fig. 6B), and it strongly activated the SRF reporter independently of functional Rho (Fig. 6C). In the FLIP assay, MC-N1-MAL exhibited a slightly increased bleach rate compared to MC and MC-N123-MAL, indicating that it was weakly susceptible to export (Fig. 6D; compare Fig. 1E). Further substitution of the MAL RPEL domain with MC sequences increased the proportion of cells with predominantly nuclear protein, and allowed strong activation of the SRF reporter in a Rho-independent manner. Taken together with the properties of the MAL-2MC mutant presented in Fig. 5, these results suggest that MC RPEL1 and RPEL2 sequences play an important role in determining its nuclear localization.
To determine which parts of the RPEL domain specify MAL-like regulation, we replaced MC RPEL domain sequences with the corresponding sequences from MAL (Fig. 6A). Replacement of the MC N extension with that of MAL had little effect on MC subcellular localization or its ability to activate the SRF reporter. Substitution of the MC N extension and RPEL1 sequences with those of MAL had a greater effect: most cells expressed MAL-N1-MC throughout the nucleus and cytoplasm, and as with MAL, SRF reporter activation was now substantially Rho dependent (Fig. 6B and C). Replacement of the MC N extension, RPEL1, and RPEL2 by the equivalent sequences from MAL conferred authentic MAL-like regulation on MC, at the levels of both steady-state subcellular localization and of SRF reporter activation (Fig. 6B and C). MAL-N12-MC accumulated in the nucleus upon LMB treatment, indicating that it shuttles continuously through the nucleus, and analysis of its nuclear export rates by FLIP indicated that it behaves very similarly to intact MAL or MAL-N123-MC upon serum or CD stimulation (Fig. 6D; compare Fig. 1E). Finally, we tested whether substitution of MC RPEL2 by that of MAL is sufficient to confer MAL-like regulation on MC. The resulting mutant, MC-2MAL, behaved effectively identically to MC itself (Fig. 6E and F). These results show that the regulatory properties of MAL require the integrity of its RPEL1-RPEL2 unit, consistent with the analysis presented in Fig. 5.
Taken together, the behaviors of MAL-MC chimeras demonstrate that the different regulatory behaviors of MAL and MC are both specified by the identity of their RPEL1-RPEL2 unit.
The functional significance of RPEL3 depends on its context.
The results presented above show that the different regulatory behaviors of MAL and MC are specified by the identity of their RPEL1-RPEL2 unit. The data also suggest that although both MAL RPEL3 and MC RPEL3 bind actin with a similarly low affinity, actin binding by RPEL3 is functionally relevant only in the context of MAL. To test this idea, we examined the functional significance of RPEL3 R→A mutations, which abolish actin binding in vitro (Fig. 4). The R→A mutation of MAL RPEL3 induced strong deregulation of MAL at the levels of both localization and reporter activation, as described above, while the analogous mutation of MC RPEL3 affected neither MC nuclear localization nor its ability to activate the SRF reporter (Fig. 6G and H). In contrast, the MC RPEL3 mutation caused pronounced deregulation in the context of MAL-N12-MC (MAL-N12-MC 3x): while the intact protein behaved very similarly to MAL, its RPEL3 R→A derivative was nuclear in about 40% of the cells, pancellular in approximately 50%, and cytoplasmic in about only 10% and strongly activated the SRF reporter. These results show that RPEL3 is functionally interchangeable between MAL and MC and suggest that its ability to confer regulation depends on the identity of the RPEL1-RPEL2 unit N-terminal to it. Mutation of RPEL motifs 1 and 2 (MAL xx3) (Fig. 5A) results in a degree of deregulation comparable to that of MAL 12x which suggests that it is the ability of the RPEL1-RPEL2 unit to bind actin that determines whether actin binding to RPEL3 is significant for MAL regulation.
DISCUSSION
In this paper, we studied the role of actin binding in the regulation of members of the MC family of SRF coactivators. We found that differences between their N-terminal RPEL domains define the distinct regulatory behaviors of MAL, which exhibits Rho-dependent nucleocytoplasmic shuttling, and MC, which is confined to the nucleus. The MAL and MC RPEL domains are sufficient to confer their characteristic regulatory properties on heterologous proteins. Chimera and point-mutant studies show that the identity of the RPEL1-RPEL2 unit defines the regulatory properties of MAL and MC, while RPEL3 is exchangeable between them, and functions in a context-dependent manner. The MAL RPEL domain exhibits greater affinity for actin than that of MC both in vivo and in vitro, and we found that individual RPEL motifs represent actin-binding elements with various affinities. Our observations show that the MAL RPEL domain acts as a G-actin sensor that links nucleocytoplasmic shuttling to Rho signaling and that it is the loss of actin binding that accounts for Rho-independent activity of MC. The reciprocal switch of regulatory behaviors of MAL and MC by mutual substitution of their RPEL domains provides yet another example of how the exchange of protein domains can rewire signaling pathways (1, 16).
A working model of MC family RPEL domain function is shown in Fig. 7. We propose that changes in actin-MAL interaction control the relative abundance of complexes which differ in their competence for nuclear export and import. Our fluorescence anisotropy studies show that two MAL RPEL motifs represent high-affinity actin-binding sites, while the third is weak; nevertheless, all three sites in MAL must be competent to bind actin for proper regulation to occur, indicating that they functionally cooperate in vivo. The higher affinity of RPEL1 and RPEL2 requires that more severe mutations must be introduced into these sites to significantly affect their activity. The B2 sequence within the RPEL domain is required for MAL nuclear import, which can be inhibited by actin overexpression; actin binding is also required for CRM1-dependent nuclear export (14, 23). One simple interpretation of our data is thus that the B2 sequence is a target for as-yet-unidentified nuclear import factors and that it is occluded by actin interacting with the neighboring RPEL motifs, particularly the adjacent RPEL3 (Fig. 7, top). According to this view, MAL does not necessarily need to be devoid of actin to enter the nucleus, but resolution of this issue awaits further studies. Conversely, export factors would bind only to the actin-bound RPEL domain. While Fig. 7 shows interaction predominantly with actin bound to all three RPEL motifs, our data do not rule out other modes of interaction.
FIG. 7.

Model for regulation of MAL nucleocytoplasmic shuttling and transcriptional activity by interaction of actin with RPEL motifs. Actin is shown in dark red and RPEL motifs in red (MAL) or green (MC), with relative affinities indicated. In resting cells, actin concentrations are sufficiently high to ensure actin-dependent recruitment of a putative export factor. Actin binding may also occlude the MAL B2 nuclear import signal (yellow) at high G-actin concentrations (23). The interaction of actin with RPEL3 of MAL confers regulation in a way that is dependent on the interaction of actin with the RPEL1-RPEL2 unit. It is therefore likely that regulation involves a higher-order actin-MAL complex. For simplicity, the figure shows a complex in which all three motifs are occupied; however, other binding possibilities cannot be excluded. In contrast, MC, or actin-binding-defective MAL RPEL mutants, do not bind actin, and are substrates for import but not export. In stimulated cells, depletion of the G-actin pool results in a block of nuclear export, while import is not impaired. It is possible that actin remains bound to a subset of the RPEL motifs in such complexes.
We propose that in resting cells, the prevailing G-actin concentration is such that MAL can readily access both export-competent and import-competent states, allowing its rapid shuttling through the nucleus (Fig. 7). Upon depletion of the G-actin pool following Rho activation, it is the binding of actin to RPEL3, the weakest binding motif, that is affected first; this reduces MAL's access to the export-competent state and thus inhibits its nuclear export (Fig. 7). We have confirmed by FRET that actin-MAL interactions indeed change during signaling in vivo (23). Our chimera experiments show that the regulatory relevance of an actin-MAL RPEL3 interaction is critically dependent on engagement of actin with the RPEL1-RPEL2 unit. Biochemical studies have identified a 3:1 actin-MAL RPEL domain complex (23), but while it is tempting to speculate that this contains an actin bound to each of the three motifs, our data cannot rule out the possibility of other actin-actin or actin-RPEL domain interactions. Structural studies are in progress to resolve these issues.
Even though MC and MAL RPEL domain mutants may have residual affinity for actin, our data show that this must be below the threshold for effective interaction with actin at the G-actin concentrations prevailing in vivo. These proteins are therefore refractory to actin-mediated export, although actin overexpression does cause some redistribution of MC to the cytoplasm. The conservation of RPEL3 in MC is puzzling, since MC does not appear to be regulated by actin, at least in our system. It remains possible that other unknown MC family regulatory factors interact with RPEL3, and we are currently seeking to identify such proteins.
It is likely that actin recruitment to MAL RPEL motifs involves positive cooperativity, either through direct actin-actin interactions or allosteric mechanisms. We speculate that such cooperativity might confer resistance of the Rho-actin-MAL-SRF system to subtle fluctuations in G-actin concentration while allowing “switch-like” activation of MAL once the G-actin concentration drops below a specific threshold. Cooperativity in actin binding may explain the discrepancy between our fluorescence anisotropy results, which show that individual RPEL motifs recruit actin with micromolar affinity and the nanomolar affinity of the intact MAL RPEL domain previously estimated from actin polymerization inhibition studies (18). Although functional cooperativity between multiple actin-binding motifs or domains has previously been observed for many F-actin regulatory proteins (see, e.g., references 4, 20, and 26), the actin-MAL interaction appears unique in that functional cooperation occurs between G-actin binding sites, and the actin-binding partner is the target, not the regulator.
While this work was in progress, a study of MC subcellular localization in 10T1/2 cells was presented by others (9). That study proposed that in addition to MAL RPEL2, the basic region B2, together with RPEL3 and/or basic region B1, determine MAL cytoplasmic localization. Our data demonstrate that the RPEL domain is necessary and sufficient to mediate MC- or MAL-like subcellular localization properties, and therefore do not support this interpretation. We also note that chimeras used for the study by Hinson et al. used the MAL(met) isoform of MAL, which does not contain MAL RPEL1 (14). Nevertheless, the possibility that the discrepancy reflects cell line-specific effects cannot be excluded.
The cytoplasmic steady-state localization of MAL(2-204)-PK and MAL(1-204)-2GFP is more sensitive to their expression level than that of intact MAL (S.G. and M.K.V., unpublished observations). It is possible that this reflects a lack of transcription-mediated cytoskeletal homeostasis upon the expression of RPEL domain fusions, although we cannot rule out the possibility that it is a consequence of the fusion.
What is the functional advantage of uncoupling SRF activation from Rho-actin signaling in muscle cells via MC? The specialized role of actin in differentiated muscle cells might impinge on the actin polymerization cycle in a way that might interfere with growth factor signaling via the actin cytoskeleton. Indeed, actin filaments are stabilized in muscle cells, as documented by their relative resistance to CD or LatB (for references, see reference 25), which rely on actin filament turnover. Filament stabilization in muscle cells is, for example, conferred by the actin-filament binder tropomyosin, which inhibits ADF/cofilin activity (15). Maintenance of smooth and cardiac muscle identity may require MC as an actin-independent SRF coactivator.
In conclusion, our data show that regulation of MAL does not merely require its interaction with actin per se but is dependent on specific signal-mediated changes in actin's interaction with the different RPEL motifs, and possibly other sequences, within the RPEL domain. The elucidation of the molecular mechanism by which binding of multiple actin molecules controls MAL activity will form an interesting subject for future work.
Supplementary Material
Acknowledgments
We thank Nicola O'Reilly and the peptide synthesis lab for outstanding peptide synthesis, Maureen Biggerstaff and Rachel Coulthard for assistance with fluorescence anisotropy measurements and data analysis, Stéphane Mouilleron for helpful discussions, and Rob Nicolas for continuous support. We thank colleagues of the lab and Caroline Hill, Michael Way, Neil McDonald, and laboratory members for technical help, discussions, and comments on the manuscript. We thank the London Research Institute Light Microscopy Facility for assistance with life cell imaging.
This work was funded by Cancer Research UK. M.K.V. is supported by an EMBO long-term fellowship, and S.G., a fellow of the Studienstiftung des deutschen Volkes, by a Boehringer Ingelheim Fonds predoctoral scholarship.
The authors have no conflicting financial interests.
Footnotes
Published ahead of print on 19 November 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Bhattacharyya, R. P., A. Remenyi, B. J. Yeh, and W. A. Lim. 2006. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75655-680. [DOI] [PubMed] [Google Scholar]
- 2.Cen, B., A. Selvaraj, R. C. Burgess, J. K. Hitzler, Z. Ma, S. W. Morris, and R. Prywes. 2003. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol. Cell. Biol. 236597-6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chenna, R., H. Sugawara, T. Koike, R. Lopez, T. J. Gibson, D. G. Higgins, and J. D. Thompson. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 313497-3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conibear, P. B., and M. A. Geeves. 1998. Cooperativity between the two heads of rabbit skeletal muscle heavy meromyosin in binding to actin. Biophys. J. 75926-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du, K. L., M. Chen, J. Li, J. J. Lepore, P. Mericko, and M. S. Parmacek. 2004. Megakaryoblastic leukemia factor-1 transduces cytoskeletal signals and induces smooth muscle cell differentiation from undifferentiated embryonic stem cells. J. Biol. Chem. 27917578-17586. [DOI] [PubMed] [Google Scholar]
- 6.Finn, R. D., J. Mistry, B. Schuster-Bockler, S. Griffiths-Jones, V. Hollich, T. Lassmann, S. Moxon, M. Marshall, A. Khanna, R. Durbin, S. R. Eddy, E. L. Sonnhammer, and A. Bateman. 2006. Pfam: clans, web tools and services. Nucleic Acids Res. 34D247-D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heyduk, T., and J. C. Lee. 1990. Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc. Natl. Acad. Sci. USA 871744-1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hill, C. S., J. Wynne, and R. Treisman. 1995. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 811159-1170. [DOI] [PubMed] [Google Scholar]
- 9.Hinson, J. S., M. D. Medlin, K. Lockman, J. M. Taylor, and C. P. Mack. 2007. Smooth muscle cell-specific transcription is regulated by nuclear localization of the myocardin-related transcription factors. Am. J. Physiol. Heart Circ. Physiol. 292H1170-H1180. [DOI] [PubMed] [Google Scholar]
- 10.Kalderon, D., B. L. Roberts, W. D. Richardson, and A. E. Smith. 1984. A short amino acid sequence able to specify nuclear location. Cell 39499-509. [DOI] [PubMed] [Google Scholar]
- 11.Kuwahara, K., T. Barrientos, G. C. Pipes, S. Li, and E. N. Olson. 2005. Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol. Cell. Biol. 253173-3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larijani, B., V. Allen-Baume, C. P. Morgan, M. Li, and S. Cockcroft. 2003. EGF regulation of PITP dynamics is blocked by inhibitors of phospholipase C and of the Ras-MAP kinase pathway. Curr. Biol. 1378-84. [DOI] [PubMed] [Google Scholar]
- 13.Lockman, K., J. S. Hinson, M. D. Medlin, D. Morris, J. M. Taylor, and C. P. Mack. 2004. Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J. Biol. Chem. 27942422-42430. [DOI] [PubMed] [Google Scholar]
- 14.Miralles, F., G. Posern, A. I. Zaromytidou, and R. Treisman. 2003. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113329-342. [DOI] [PubMed] [Google Scholar]
- 15.Ono, S., and K. Ono. 2002. Tropomyosin inhibits ADF/cofilin-dependent actin filament dynamics. J. Cell Biol. 1561065-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pawson, T., and P. Nash. 2003. Assembly of cell regulatory systems through protein interaction domains. Science 300445-452. [DOI] [PubMed] [Google Scholar]
- 17.Pipes, G. C., E. E. Creemers, and E. N. Olson. 2006. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 201545-1556. [DOI] [PubMed] [Google Scholar]
- 18.Posern, G., F. Miralles, S. Guettler, and R. Treisman. 2004. Mutant actins that stabilise F-actin use distinct mechanisms to activate the SRF coactivator MAL. EMBO J. 233973-3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Posern, G., A. Sotiropoulos, and R. Treisman. 2002. Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol. Biol. Cell 134167-4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selden, L. A., H. J. Kinosian, J. Newman, B. Lincoln, C. Hurwitz, L. C. Gershman, and J. E. Estes. 1998. Severing of F-actin by the amino-terminal half of gelsolin suggests internal cooperativity in gelsolin. Biophys. J. 753092-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selvaraj, A., and R. Prywes. 2003. Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum response factor, is required for skeletal myogenic differentiation. J. Biol. Chem. 27841977-41987. [DOI] [PubMed] [Google Scholar]
- 22.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 254876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vartiainen, M. K., S. Guettler, B. Larijani, and R. Treisman. 2007. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 3161749-1752. [DOI] [PubMed] [Google Scholar]
- 24.Wang, D., P. S. Chang, Z. Wang, L. Sutherland, J. A. Richardson, E. Small, P. A. Krieg, and E. N. Olson. 2001. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105851-862. [DOI] [PubMed] [Google Scholar]
- 25.Wang, J., J. M. Sanger, and J. W. Sanger. 2005. Differential effects of Latrunculin-A on myofibrils in cultures of skeletal muscle cells: insights into mechanisms of myofibrillogenesis. Cell Motil. Cytoskeleton 6235-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang, Y., A. L. Miller, M. S. Mooseker, and A. J. Koleske. 2001. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc. Natl. Acad. Sci. USA 9814865-14870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaromytidou, A. I., F. Miralles, and R. Treisman. 2006. MAL and ternary complex factor use different mechanisms to contact a common surface on the serum response factor DNA-binding domain. Mol. Cell. Biol. 264134-4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.