

Abstract
The retroviral Gag polyprotein directs virus particle assembly, resulting in the release of virions from the plasma membranes of infected cells. The earliest steps in assembly, those immediately following Gag synthesis, are very poorly understood. For Rous sarcoma virus (RSV), Gag proteins are synthesized in the cytoplasm and then undergo transient nuclear trafficking before returning to the cytoplasm for transport to the plasma membrane. Thus, RSV provides a useful model to study the initial steps in assembly because the early and later stages are spatially separated by the nuclear envelope. We previously described mutants of RSV Gag that are defective in nuclear export, thereby isolating these “trapped” Gag proteins at an early assembly step. Using the nuclear export mutants, we asked whether Gag protein-protein interactions occur within the nucleus. Complementation experiments revealed that the wild-type Gag protein could partially rescue export-defective Gag mutants into virus-like particles (VLPs). Additionally, the export mutants had a trans-dominant negative effect on wild-type Gag, interfering with its release into VLPs. Confocal imaging of wild-type and mutant Gag proteins bearing different fluorescent tags suggested that complementation between Gag proteins occurred in the nucleus. Additional evidence for nuclear Gag-Gag interactions was obtained using fluorescence resonance energy transfer, and we found that the formation of intranuclear Gag complexes was dependent on the NC domain. Bimolecular fluorescence complementation allowed the direct visualization of intranuclear Gag-Gag dimers. Together, these experimental results strongly suggest that RSV Gag proteins are capable of interacting within the nucleus.
The Gag polyprotein directs the assembly and budding of retrovirus particles from the plasma membranes of infected cells. The expression of Gag alone in cells is sufficient to induce the formation and release of virus-like particles (VLPs) that resemble authentic virus particles (18, 61, 62). Shortly after budding, the Gag polyprotein is cleaved into the MA (matrix), CA (capsid), and NC (nucleocapsid) proteins, and for Rous sarcoma virus (RSV), additional cleavage products include p2a, p2b, p10, SP (spacer), and PR (protease) (Fig. 1). Analysis of Gag-mediated budding led to the identification of three independent assembly motifs common to all retroviruses: the membrane binding domain (M) in MA, which targets Gag to the plasma membrane; the interaction (I) domains in the NC domain that mediate Gag-Gag and Gag-RNA interactions; and the late (L) domain, which facilitates the final steps of VLP release (37, 64).
FIG. 1.

Schematic of Gag expression constructs. The wild-type RSV Gag polyprotein is illustrated at the top, with MA, p2, p10, CA, NC, and PR domains indicated. The wild-type (WT) p10 NES sequence is shown above the schematic, and mutant sequences are indicated below the schematic. Critical hydrophobic residues are indicated in bold. GagΔPR lacks the entire PR domain. For each Gag fusion protein, the PR sequence was replaced with spectral variants of GFP: YFP, CFP, mCherry, and the N- or C-terminal domain of Venus (VN and VC). Gag.ΔNC has a deletion of amino acid residues 495 to 577 in the NC domain. Gag.Zip has the leucine zipper domain of the human CREB binding protein (Zip) substituted for the NC domain. The MLV Gag polyprotein, consisting of MA, p12, CA, and NC, is shown fused to the N- or C-terminal domain of Venus.
The alpharetrovirus RSV follows the C-type morphogenetic pathway, meaning that virus particles appear to form and to be released directly from the plasma membrane (reviewed in reference 10). However, several independent lines of evidence indicate that Gag-Gag interactions occur in the cytoplasm prior to reaching the plasma membrane. Complementation experiments reveal that budding can be restored when Gag mutants defective in membrane binding are coexpressed with wild-type Gag (2, 4, 5, 35). Moreover, cytoplasmic extracts from cells expressing Gag contain higher-order Gag assembly intermediates that increase in size in a stepwise fashion, suggesting an ordered process of dimerization, oligomerization, and multimerization (29). Similarly, oligomeric and multimeric complexes of Gag proteins can be isolated from the cytoplasm of intact cells (56). Furthermore, sequences downstream of MA, primarily those within the I domains, promote membrane binding, suggesting that Gag multimerization occurs prior to plasma membrane localization (34, 42, 66). Recently, analysis of fluorescently tagged Gag fusion proteins using confocal microscopy and fluorescence resonance energy transfer (FRET) provided direct evidence that Gag proteins interact in the cytoplasm (13, 27).
Previously, it was thought that Gag proteins were targeted directly from their sites of synthesis on free cytosolic ribosomes to the plasma membrane. However, subsequent studies revealed that Gag proteins appear to be transiently present in the nucleus for RSV, murine leukemia virus (MLV), human immunodeficiency virus type 1 (HIV-1), and foamy viruses (2, 14, 44, 47). For RSV Gag, a CRM1-dependent nuclear export signal (NES) was identified in the p10 sequence, and treatment of RSV-infected cells with leptomycin B (LMB), an inhibitor of CRM1 export, results in the accumulation of Gag in the nucleus (44). Mutation of any or all of the critical hydrophobic residues (L219, W222, V225, or L229) in the p10 NES interferes with the nuclear egress of Gag (46). NES mutant Gag proteins are sequestered in the nucleus, and the rate of virus budding is severely impaired. These results imply that nuclear trafficking of RSV Gag is a rate-limiting, early step in the assembly pathway. Thus, the nuclear envelope separates early and late stages of virus particle formation for RSV.
For retrovirus particles, the earliest “assembly unit” consists of a dimer of Gag proteins bound to an RNA molecule (3, 17, 33, 40, 60). Gag dimers and multimers spontaneously form in vitro when recombinant Gag proteins are mixed with nucleic acids, which promote Gag-Gag multimerization (8, 9, 12, 16, 31, 32). However, an RNA-independent protein-protein interaction domain can substitute for I domain activity in NC to mediate dimer formation (22). Taken together, these results indicate that both protein-protein and protein-RNA interactions are important for Gag-mediated particle assembly.
Higher-order Gag complexes isolated from the cytoplasm represent dimers and multimeric structures that are larger than simple dimers (29, 31, 56). However, it is not known where Gag-Gag dimers are initially formed within the cell. The RSV Gag NES mutants provide a unique set of tools to investigate the location of Gag dimer formation. To address whether Gag-Gag interactions occur very early in assembly, either prior to nuclear entry or within the nucleus, we coexpressed wild-type and NES mutant Gag proteins. As described in this report, we found compelling evidence for intranuclear Gag-Gag dimer formation.
MATERIALS AND METHODS
Expression vectors, plasmids, and cells.
RSV Gag expression plasmid pGagΔPR was described previously (46). The Gag p10 NES mutations encoding L219A, LWVL-A, and NES-A in pGag-GFP (6, 45) were transferred into pGag.CFP and pGag.YFP expression vectors (a gift from V. Vogt, Cornell University) (27) using SstI-SdaI fragment exchange. pΔNC.CFP and pΔNC.YFP were created using SacI-ApaI fragment exchange between pΔNC.GFP (a gift from J. Wills, Penn State College of Medicine) (7) and pGag.CFP or pGag.YFP. pGag.mCherry was made by PCR amplification of the fluorophore sequence from pRSET8-mCherry (a gift from R. Tsien, University of California at San Diego) (49) and transfer into pGag-GFP using ApaI-NotI. pGFP.NLS.PK was created using QuikChange mutagenesis (Stratagene) to insert the simian virus 40 T-antigen nuclear localization signal (NLS) into pGFP-PK (a gift from Warner Greene, University of California at San Francisco) (51). pRSVGag.VN173 and pRSVGag.VC155 were constructed by using PCR amplification of bJunVN173 or bFosVC155 (a gift from C. Hu, Purdue University) (53) and insertion into the pRSVGag.GFP vector in place of green fluorescent protein (GFP) using ApaI-NotI. pMLV.Gag.VN173 and pMLV.Gag.VC155 were made by PCR and NheI-SmaI fragment exchange into pMLVGag.YFP (50) (a gift from W. Mothes, New Haven, CT). Mutants were screened using restriction endonuclease digestion and confirmed with automated DNA sequencing. All experiments were performed using either the chemically transformed QT6 quail fibroblast cell line or immortalized chicken embryo fibroblast DF-1 cells, maintained as previously described (19, 38). Transfections were performed by using the calcium phosphate method or Fugene 6 transfection reagent (Roche Applied Science).
Radioimmunoprecipitation assays.
Budding assays were performed as previously described (39, 59). Immunoprecipitated RSV Gag proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed using a PhosphorImager (Molecular Dynamics). Budding efficiency was calculated as the ratio of Gag proteins in the media divided by the sum of the Gag proteins expressed in the cell lysates and media.
Confocal imaging and subcellular quantification.
Live cells were plated onto 35-mm glass-bottomed dishes (MatTek Corporation) and imaged using a Leica AOBS SP2 confocal microscope with sequential scan settings at 17 to 24 h posttransfection. Quantification of the total fluorescent intensity of the fluorescently tagged Gag proteins was performed using a single optical slice through the nuclear plane, and image analysis was performed using Leica Microsystems software. The percent nuclear fluorescence was calculated as the fluorescence intensity of the nucleus divided by the fluorescence intensity of the entire cell.
FRET measurements.
Acceptor photobleaching FRET was performed on transfected cells fixed with 4% paraformaldehyde. Prebleach images of both cyan fluorescent protein (CFP) (excitation at 458 nm, emission at 460 to 500 nm, and 20% laser power) and yellow fluorescent protein (YFP) (excitation at 514 nm, emission at 550 to 600 nm, and 10% laser power) channels were acquired. YFP was specifically photobleached using the 514-nm laser at 100% laser power until the fluorescence intensity was decreased to 10% of the prebleach level. Postbleach images were acquired at the prebleach settings. FRET efficiency was calculated using the formula 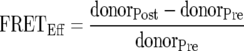 , when donorPost is greater than donorPre. FRET analysis was performed on at least two separate days using a minimum of 10 different cells per day. Nuclear FRET was performed by bleaching the entire nucleus through a single optical section of the nuclear plane.
, when donorPost is greater than donorPre. FRET analysis was performed on at least two separate days using a minimum of 10 different cells per day. Nuclear FRET was performed by bleaching the entire nucleus through a single optical section of the nuclear plane.
BiFC analysis.
QT6 cells were transfected in duplicate with 100 ng of each plasmid DNA using Fugene 6. At 4.5 h posttransfection, cells were fixed in 4% paraformaldehyde, stained with 4′,6′-diamidino-2-phenylindole (DAPI) (Calbiochem) at a 1:10,000 dilution, washed in phosphate-buffered saline, mounted using a Slowfade antifade kit (Invitrogen), and imaged using a Leica SP2 confocal microscope. Overall intensity was increased equally for each bimolecular fluorescence complementation (BiFC) image after acquisition using CorelDRAW X3 (Corel Corporation). Duplicate plates were lysed in Laemmli sample buffer for analysis of intracellular protein expression levels using Western blotting with polyclonal anti-GFP (ab290) antibody (Abcam).
RESULTS
The subcellular trafficking pathways of type C retroviruses remain poorly delineated. In particular, the intracellular sites of initial Gag-Gag intermolecular interactions were previously thought to be in the cytoplasm or at the plasma membrane (55). However, because RSV Gag transiently trafficks through the nucleus, it is possible that protein-protein dimers form within the nucleus prior to cytoplasmic relocalization and plasma membrane targeting. To address this question, we coexpressed nucleus-restricted Gag mutants with wild-type Gag proteins and used several complementary approaches to monitor protein-protein interactions within cell nuclei.
Subcellular localization of p10 NES mutant Gag proteins coexpressed with wild-type Gag.
To determine whether the expression of NES mutant Gag proteins would alter the subcellular localization of the wild-type Gag protein, each Gag variant was fused to either YFP or CFP at the C terminus (Fig. 1). Because the wild-type Gag protein shuttles through the nucleus, we tested the possibility that it might dimerize with the nucleus-restricted NES mutant. Upon coexpression of the differentially tagged proteins, we envisioned three possible outcomes. First, if the NES mutant and wild-type Gag proteins did not associate either prior to nuclear entry or within the nucleus, the intracellular distribution of each protein population would be unchanged. Second, if the NES mutant exerted a trans-dominant negative effect on trafficking of the wild-type protein, there would be an increase in the amount of wild-type Gag in the nucleus. Third, the wild-type Gag protein might associate with the NES mutant within the nucleus, restoring cytoplasmic relocalization to the mutant that is normally “trapped” within the nucleus.
Using sequential scanning to eliminate spectral overlap between the YFP and CFP channels, optical slices through the nuclear plane of each cell were obtained using confocal microscopy. Coexpression of wild-type Gag-YFP and wild-type Gag-CFP revealed primarily cytoplasmic fluorescence with punctate foci within the cytoplasm and along the plasma membrane (Fig. 2a and b, where pseudocolored images show YFP as red and CFP as green). Localization appeared to be similar to data from previous reports of singly and coexpressed RSV Gag proteins in avian cells (6, 27, 44). Of note, the nuclei of these cells did not display substantial fluorescence, reflecting the transient nature of Gag nuclear trafficking and the greater efficiency of nuclear export compared to import. In contrast, when a mutant NES Gag-CFP protein was expressed with the wild-type Gag-YFP protein, a distinct change in the localization of the wild-type Gag protein was observed (Fig. 2c to h). In cells marked with white arrows, the wild-type Gag protein was sequestered in the nucleus, as indicated by an increase in the nuclear fluorescence. The accumulation of wild-type Gag within the nucleus was most striking in those cells expressing higher levels of the NES Gag mutant, as shown clearly in the case of the LWVL-A.Gag-CFP protein (Fig. 2d). These results support the hypothesis that the NES mutant Gag proteins act in a trans-dominant negative fashion to alter the nuclear export of wild-type Gag through protein-protein interactions within the nucleus.
FIG. 2.

Confocal microscopic images of cells transfected with Gag-YFP and NES mutant Gag-CFP. Live cells expressing wild-type Gag-YFP (left) (pseudocolored red) and NES mutants LWVL-A.Gag-CFP, NES-A.Gag-CFP, and L219A.Gag-CFP (right) (pseudocolored green) were analyzed using sequential scanning laser confocal microscopy at wavelengths of 458 nm (CFP) and 514 nm (YFP). A single optical section through the nuclear plane is shown. Arrows indicate the nuclear accumulation of wild-type Gag-YFP with the coexpression of NES mutant Gag-CFP proteins.
To quantify the amount of nuclear accumulation of the wild-type Gag protein, confocal images of living cells were analyzed for nuclear fluorescence compared to total cellular fluorescence (Fig. 3). This method was preferred over subcellular fractionation because we used a transient transfection system, and therefore, it was not possible to ensure that every cell in the population expressed both the NES mutant and wild-type Gag proteins. The confocal imaging allowed us to choose to analyze only those cells coexpressing the mutant and wild-type proteins. To avoid complications resulting from potential weak interactions between the YFP and CFP domains (65), we substituted a monomeric far-red fluorescent tag (mCherry) for the fluorophore on the wild-type Gag protein, and CFP was used to tag the NES mutant Gag proteins (49). Western blot analysis of each fusion protein used for analysis revealed little or no free mCherry or CFP (data not shown).
FIG. 3.

Quantification of the nuclear localization of wild-type and NES mutant Gag proteins. QT6 cells transfected with the indicated plasmid DNA were imaged with laser confocal microscopy using an excitation wavelength of 543 nm for mCherry. Total cellular and nuclear fluorescence intensities were measured, and Gag-mCherry nuclear fluorescence was calculated as the nuclear intensity divided by the whole-cell intensity. Each bar represents the average ratio of the nuclear to total cellular fluorescence. At least 100 cells were measured from three separate transfections, except for GFP.NLS.PK, for which 46 cells from two transfections were analyzed.
The wild-type Gag-mCherry fusion protein produced a subcellular distribution that was indistinguishable from that of Gag-GFP, Gag-YFP, or Gag-CFP (data not shown), and 7.3% of the wild-type protein was detected in the nucleus under steady-state conditions (Fig. 3). The coexpression of Gag-mCherry with LWVL-A.Gag-CFP, L219A.Gag-CFP, or NES-A.Gag-CFP resulted in an increase in the nuclear fluorescence of Gag-mCherry to 14.7%, 15.5%, or 16.4%, respectively, of the total cellular fluorescence. As a control, we expressed Gag-CFP with GFP.NLS.PK, which contains the simian virus 40 large-T-antigen NLS fused to chicken pyruvate kinase (51). The coexpression of the nucleus-localized GFP.NLS.PK protein with wild-type Gag-mCherry led to a slight increase in the amount of wild-type Gag in the nucleus (9.8%). However, the nuclear accumulation of wild-type Gag was significantly increased in the presence of each of the NES mutants compared to intranuclear levels of wild-type Gag expressed alone or with GFP.NLS.PK (P value of <0.0001). This finding indicated that the interaction between the wild-type and NES mutant Gag proteins was specific. Together, these results suggest that the NES mutant forms dimers or oligomers with wild-type Gag in the nucleus, retarding the nuclear egress of the wild-type protein.
Because Gag nuclear sequestration was not complete under conditions of coexpression with the NES mutants, we tested the possibility that the intranuclear interactions between Gag and the NES mutant facilitated the trafficking of the mutant into the cytoplasm. The cytoplasmic localization of each NES mutant was measured using confocal microscopy in cells coexpressing wild-type and NES mutant Gag proteins (data not shown). There was very little change in the cytoplasmic relocalization of the NES mutant proteins. However, we considered it likely that interactions between the wild-type protein and the NES Gag mutant would result in a loss of the mutant protein from the cytoplasm due to its release into the medium during budding. If the coexpression of wild-type Gag partially reversed the budding defect of the NES mutants, the cytoplasmic fluorescence of the NES mutants might be reduced.
Rescue of budding by complementation between wild-type and mutant Gag proteins.
To determine whether the wild-type Gag protein could enhance the incorporation of NES mutant Gag proteins into VLPs, we performed budding assays on cells coexpressing wild-type Gag-GFP and NES mutants LWVL-A.Gag-CFP, NES-A.Gag-CFP, and L219A.Gag-CFP (Fig. 4). Cells were metabolically labeled for 2.5 h, cell lysates and medium samples were immunoprecipitated with anti-RSV serum, and proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and quantified by PhosphorImager analysis. Budding efficiency was calculated as the amount of Gag protein released into the medium compared to the total amount of Gag protein detected in the cell lysate and medium. The expression of each p10 NES mutant Gag-CFP protein with wild-type Gag-GFP resulted in an increase in budding for each mutant, although LWVL-A.Gag-CFP (11.4% increased to 50.0%) and L219A.Gag-CFP (10.5% increased to 34.3%) demonstrated the greatest degree of rescue (Fig. 4A and B). Of note, the magnitude of the increase in budding might be underestimated in these experiments because particle release for wild-type Gag-CFP was reduced in comparison to the budding efficiency of wild-type Gag-GFP.
FIG. 4.

Rescue of budding for NES mutant Gag proteins coexpressed with wild-type Gag. (A) Autoradiogram of a typical budding rescue assay (see Materials and Methods). Gag-GFP can be differentiated from Gag-CFP by its faster migration during electrophoresis. Note the increase in the amount of Gag in the medium for L219A.Gag-CFP when coexpressed with wild-type Gag.GFP. (B) PhosphorImager quantification of budding efficiency. Particle release for wild-type Gag-GFP was arbitrarily set to 100%, and each coexpressed CFP protein was compared to Gag-GFP. Error bars represent standard errors of the means. (C) Analysis of budding efficiency for untagged NES GagΔPR mutants expressed with wild-type Gag-GFP resulted in an increase in NES mutant budding.
To eliminate any potential influence of the C-terminal CFP/GFP tags with Gag-Gag interactions, we performed budding complementation experiments using untagged NES mutants in which the PR domains were deleted (Fig. 4C). For LWVL-A.GagΔPR and NES-A.Gag ΔPR, budding efficiency was rescued from 9.3% to 41.6% and 45.5% of wild-type levels, respectively. The restoration of budding for the NES mutants suggests that the wild-type Gag protein interacts with the NES mutants in the nucleus. While it is possible that the Gag-mutant interaction occurs prior to nuclear entry, this conclusion is less likely given the confocal microscopy results shown in Fig. 2. However, to test rigorously whether true intermolecular Gag-Gag interactions were established in the nucleus, a biophysical method was utilized to detect protein-protein associations.
Analysis of intranuclear Gag-Gag interactions.
Although Gag-Gag interactions in the cytoplasm and at the plasma membrane have been reported using FRET analysis (27), there are no studies of intranuclear Gag-Gag binding. For FRET to occur, two proteins must be between 10 and 100 Å from one another, a distance consistent with a biologically relevant, direct, and physical interaction (21, 54, 57). The magnitude of the energy transfer between the CFP and YFP fluorophores depends on proximity, so more closely associated proteins demonstrate higher FRET efficiencies (48).
For the FRET experiments, cells were transfected with Gag-CFP/Gag-YFP pairs, and representative images obtained for the YFP channel are shown in Fig. 5. To assess intranuclear interactions between wild-type Gag proteins, cells were treated with LMB to concentrate Gag within the nucleus. Two patterns of intranuclear distribution were observed: either Gag proteins were diffuse throughout the nucleoplasm, excluding nucleoli (Fig. 5a), or there were distinct punctate foci within the nucleoplasm, again excluding nucleoli (Fig. 5a′). The discrete puncta likely represent large aggregates of Gag proteins that form more frequently with higher levels of intracellular Gag expression.
FIG. 5.

Intranuclear distribution patterns of Gag proteins and FRET analysis. Fixed QT6 cells were imaged through the nuclear plane using confocal microscopy. Diffuse intranuclear patterns are shown on the left, punctate distributions are on the right, and cells having different subnuclear distribution patterns were derived from a single transfection. Cells treated with LMB are indicated as +LMB. Punctate foci were not obtained for ΔNCGag-YFP/CFP or Gag.Zip-YFP/CFP.
To determine whether LMB was affecting the pattern of intranuclear localization, the NES-A.Gag-YFP/CFP proteins were examined without LMB treatment; however, the same diffuse and punctate distributions were observed (Fig. 5b and b′). Interestingly, the deletion of the Gag NC domain, which mediates Gag-RNA and Gag-Gag interactions, abrogated the formation of punctate foci (Fig. 5c), indicating that the puncta were NC dependent. To test whether the punctate pattern was mediated through protein-protein or protein-RNA interactions, we examined LMB-treated cells expressing the Gag.Zip-YFP/CFP proteins, which have the human CREB binding domain leucine zipper substituted for NC (22). The Gag.Zip proteins interact via a nucleic acid-independent mechanism and failed to form punctate foci, suggesting that the Gag aggregates are mediated through NC-RNA interactions in the nucleus.
Acceptor photobleaching FRET analysis (23) was performed using single confocal z sections in which the maximal nuclear radius was evident in cells expressing wild-type or mutant Gag-CFP/YFP pairs. Representative images from a FRET experiment for NES-A.Gag-CFP coexpressed with NES-A.Gag-YFP are shown in Fig. 6A. A region of interest encompassing the entire nucleus was specifically bleached using the 514-nm laser (excitation wavelength for YFP) at 100% laser intensity. A marked decrease in YFP fluorescence was evident in the postbleach YFP panel compared to the prebleach image, as expected (Fig. 6A, right). A concomitant increase in the intensity of the postbleach CFP fluorescence compared to the prebleach level was observed, indicating that FRET had occurred (Fig. 6A, left).
FIG. 6.

Intranuclear Gag-Gag interactions assessed by acceptor photobleaching FRET analysis. (A) FRET analysis was performed on cells coexpressing NES-A.Gag-CFP and NES-A.Gag-YFP. Confocal images through the nuclear plane were obtained prior to photobleaching (top). The nuclear region of interest, indicated by the white circle, was subjected to bleaching of the YFP (acceptor) fluorophore using a high-intensity laser at a wavelength of 514 nm, resulting in a significant loss of the signal in the postbleach NES-A.Gag-YFP image (lower right). The increased intensity of the NES-A.Gag-CFP fluorescence in the postbleach image (lower left) resulted from the decreased transfer of energy from the CFP fluorophore (donor) to YFP. (B) QT6 cells expressing the indicated proteins demonstrating punctate (P) or diffuse (D) phenotypes (as shown in Fig. 5) were analyzed using FRET. The FRET efficiency (percent) for each condition was calculated according to the equation shown in Materials and Methods. FRET experiments were performed a minimum of 10 times from at least two separate transfections. The mean FRET efficiency was calculated and graphed, with error bars representing standard errors of the means.
Average FRET efficiency values were obtained from at least 10 cells derived from two separate transfections in which Gag was distributed in diffuse or punctate patterns (Fig. 6B). As a negative control, cells expressing free CFP and YFP produced very low FRET efficiencies (0.6%) in the nucleus. Cells coexpressing wild-type Gag-CFP and Gag-YFP fusion proteins were treated with LMB, causing the relocalization of Gag to the nucleus, and FRET efficiencies of 2.0% were observed in nuclei with a diffuse pattern and 9.4% in cells containing punctate foci. For the NES mutants, the range of nuclear FRET values was 1.4% to 4.9% for nuclei with a diffuse distribution and 8.5% to 12.8% for nuclei with punctate fluorescence. Deletion of the NC domain did not eliminate Gag-Gag associations, as the FRET efficiency was 4.9%, although there were no large multimeric intranuclear complexes of Gag seen in these cells. For the Gag.Zip-CFP/YFP pair, the FRET efficiency was 11.1%, indicating that the proteins were interacting in the nucleus. However, even though the Gag.Zip proteins were in close proximity based on the FRET results, they were unable to form punctate fluorescent complexes in the nucleus.
Detection of Gag-Gag interactions using BiFC.
Accurate results with FRET required that Gag proteins be expressed at high levels. Although both FRET and BiFC detect protein-protein interactions within distinct subcellular compartments, BiFC offers an advantage because it identifies transitory protein complexes (20). Furthermore, the specificity of interactions detected using BiFC requires low expression levels of the test proteins (53), avoiding potential problems arising from protein overexpression.
To determine whether Gag-Gag interactions could be detected at lower intracellular levels of Gag, we utilized BiFC analyses. For these experiments, wild-type and mutant Gag proteins were fused to either the N-terminal 173 residues of the YFP variant Venus (VN173) or the C-terminal 155 amino acids (VC155) (Fig. 1 and 7). A low level of protein expression was needed to preserve the specificity of the interactions, so a small amount of plasmid DNA (100 ng) was used for transfection, and cells were examined by confocal microscopy a short time (4.5 h) after transfection (20, 53). If Gag proteins were closely juxtaposed, the N- and C-terminal halves of Venus would be brought together and fold into a functional fluorophore (63). The expression of each fusion protein containing VN or VC in the absence of the complementing fluorophores revealed no fluorescence (data not shown). However, the coexpression of RSV Gag-VN with RSV Gag-VC led to perinuclear, cytoplasmic, and plasma membrane epifluorescence (Fig. 7a and a′). The NES-A.Gag-VN/VC proteins appeared predominantly within the nucleus and faintly along the plasma membrane, indicating that these sites were the major sites of interaction. Removal of the NC domain resulted in cytoplasmic interactions without plasma membrane epifluorescence. As a negative control, RSV Gag-VN was expressed with MLV Gag-VC, and no fluorescence was detected, as expected, since these heterologous Gag proteins are not copackaged into VLPs (1, 4). The lack of fluorescence was not due to defective MLV Gag proteins, as complementation between MLV Gag-VN/VC resulted in Venus expression within discrete cytoplasmic foci. Thus, interactions between intranuclear RSV Gag proteins occurred under conditions that allowed the discrimination of specific intermolecular interactions.
FIG. 7.

BiFC analysis of Gag-Gag complex formation. Cells were transfected with 100 ng of each plasmid construct bearing N- or C-terminal halves of the Venus fluorophore fused to the indicated Gag protein. Cells were fixed 4.5 h posttransfection and imaged using confocal microscopy. Excitation with the 514-nm laser produced a fluorescent signal when Gag dimers formed. The YFP (Venus) images are shown on the left, and Venus overlaid with DAPI shows the location of nuclei on the right.
DISCUSSION
The early events in C-type retrovirus assembly are poorly characterized due to limitations in distinguishing distinct steps in Gag subcellular trafficking (55). However, because RSV Gag undergoes nuclear trafficking at a critical, rate-limiting stage of the assembly process (44, 46), the phases of assembly can be separated into early (prenuclear and intranuclear localization), mid (nuclear egress and cytoplasmic trafficking), and late (plasma membrane binding and budding). The results presented in this report suggest that early Gag-Gag interactions, defined as the formation of dimers and oligomers, occur either prior to nuclear import or within the nuclear compartment. At a high concentration of intranuclear Gag proteins, larger aggregates of Gag complexes form in an NC- and nucleic acid-dependent fashion.
Using confocal microscopy, FRET, and BiFC analysis, the data demonstrated that Gag-Gag intermolecular contacts occur in the nucleus, although our experiments could not differentiate whether the initial site of complex formation occurred in the cytoplasm preceding nuclear import or within the nucleus. If dimeric or oligomeric Gag complexes form in the cytoplasm, then they must be capable of trafficking through the nuclear pore using an active nuclear targeting mechanism since Gag is detected in the nucleus during 1.5 to 2 h of incubation with LMB (44). The nuclear pore can accommodate large protein complexes and macromolecules that are 39 nm in diameter (36), so the import of Gag multiprotein complexes, depending on their quaternary structures, is theoretically possible. If Gag-Gag dimerization depends on RNA binding, as generally accepted (32, 52, 55, 58), then Gag-RNA interactions would promote Gag protein-protein contacts. Therefore, a logical conclusion is that RSV Gag-RNA binding occurs either prior to Gag nuclear entry or within the nucleus. Future experiments will test this hypothesis directly.
Support for the idea that intranuclear Gag-Gag interactions depend on RNA binding was provided by the behavior of the ΔNC.Gag and Gag.Zip proteins. The formation of large aggregates of Gag proteins in the nucleus depends on the presence of the NC domain, suggesting that the punctate foci (Fig. 5) represent organized higher-order complexes mediated through NC-NC and NC-RNA interactions. The replacement of NC with a nucleic acid-independent protein-protein interaction “Zip” domain resulted in strong protein-protein contacts, but Gag aggregates were not observed in the nucleus, suggesting that the formation of intranuclear foci was augmented by nucleic acid binding. Furthermore, the magnitude of FRET efficiency values for punctate nuclear foci (8.5% to 12.8%) of NC-bearing Gag variants was similar to the levels of FRET for Gag.Zip proteins (11.1%), suggesting that in both cases, the Gag proteins are interacting at close proximity. The simplest interpretation of these data is that NC-RNA binding in the nucleus promotes Gag-Gag dimerization and possibly oligomerization; higher-order multimerization appears to occur only when nuclear export is inhibited, resulting in very high intranuclear Gag concentrations.
Interestingly, we have not been successful in visualizing intranuclear particle formation when we examined LMB-treated, RSV-infected cells by electron microscopy, suggesting that the formation of intranuclear virus particles is regulated (our unpublished results). The mechanism of regulation during virus infection could be through constraining the conformation of Gag oligomers to prevent the formation of spherical arrays or through the activities of viral or host factors that interfere with particle formation. In the experiments that we reported here, no additional viral factors were present, and the overexpression of Gag might have saturated cellular factors that normally control particle assembly. We will attempt to obtain electron microscopic images of the punctate nuclear foci observed by confocal microscopy to determine whether the Gag aggregates are in the form of VLPs.
The FRET and BiFC analyses confirm the idea that the interaction of Gag proteins is not mediated solely by the NC domain. However, the images generated from the ΔNC.Gag BiFC experiments demonstrate that the deletion of NC resulted in a more diffuse cytoplasmic localization and reduced plasma membrane association compared to those of the wild-type Gag protein. Our results are consistent with previous conclusions that the CA domain plays a major role in promoting Gag-Gag interactions that dictate virion size, morphology, and copackaging of Gag (1, 26, 28). It should be noted that the ΔNC.Gag constructs used in our experiments contain the 8 amino acids at the N-terminal sequence of NC, and these remaining residues might provide the minimal requirement for Gag-Gag interactions, as reported previously for the HIV-1 Gag protein (41).
Dimerization is a common mechanism for regulating the subcellular localizations and biological activities of cellular and viral proteins. For some proteins, including the mitogen-activated protein kinase extracellular signal-regulated kinase 2, the formation of dimers promotes nuclear import (24). For others, such as the antiapoptotic factor survivin, dimerization masks the NES and prevents cytoplasmic relocalization (15). For the androgen receptor, the formation of dimers occurs in the nucleus, and self-association is necessary for activity in transcriptional regulation (43). Similarly, the HIV-1 Rev protein multimerizes in the nucleus, although weaker interactions also occur in the cytoplasm, as we observed with RSV Gag (11, 30). Oligomers of Rev bind the HIV-1 Rev-responsive element to facilitate viral mRNA nuclear export (reviewed in reference 25).
Our working model, supported by data presented here and previously, is that RSV Gag binds to viral unspliced RNA in the nucleus, promoting Gag dimerization (44, 45). We hypothesize that RNA binding and dimer formation induce a conformational change to unmask the p10 NES, resulting in the nuclear export of Gag-RNA complexes. We envision that additional Gag proteins join the assembling multimeric complex in the cytoplasm to promote plasma membrane binding. Control of sequential dimerization, oligomerization, and multimerization must be finely tuned so that particles are assembled at the proper time and intracellular site. Thus, further efforts to determine the monomeric, dimeric, and oligomeric states of Gag proteins in the nucleus and cytoplasm and at the plasma membrane will be key steps in understanding the molecular basis of retrovirus assembly.
Acknowledgments
This research was supported by NIH grants R01CA76534 (L.J.P.) and T32CA60395 (T.L.L.).
We acknowledge the contributions of Alistair Barber of the Imaging Core and staff from the DNA Sequencing Core at the Penn State College of Medicine. We thank John Wills (Penn State College of Medicine), Volker Vogt (Cornell University), Chang-Deng Hu (Purdue University), Warner Greene (University of California at San Francisco), and Roger Tsien (University of California at San Diego) for generously providing reagents. We appreciate valuable insights contributed by Andrea Beyer, Nicole Gudleski, and Eileen Ryan.
Footnotes
Published ahead of print on 31 October 2007.
REFERENCES
- 1.Ako-Adjei, D., M. C. Johnson, and V. M. Vogt. 2005. The retroviral capsid domain dictates virion size, morphology, and coassembly of Gag into virus-like particles. J. Virol. 7913463-13472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrawiss, M., Y. Takeuchi, L. Hewlett, and M. Collins. 2003. Murine leukemia virus particle assembly quantitated by fluorescence microscopy: role of Gag-Gag interactions and membrane association. J. Virol. 7711651-11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett, R. P., T. D. Nelle, and J. W. Wills. 1993. Functional chimeras of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 676487-6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett, R. P., and J. W. Wills. 1999. Conditions for copackaging Rous sarcoma virus and murine leukemia virus Gag proteins during retroviral budding. J. Virol. 732045-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryant, M., and L. Ratner. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA 87523-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callahan, E. M., and J. W. Wills. 2000. Repositioning basic residues in the M domain of the Rous sarcoma virus Gag protein. J. Virol. 7411222-11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callahan, E. M., and J. W. Wills. 2003. Link between genome packaging and rate of budding for Rous sarcoma virus. J. Virol. 779388-9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell, S., and A. Rein. 1999. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 732270-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell, S., and V. M. Vogt. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 696487-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craven, R. C., and L. J. Parent. 1996. Dynamic interactions of the Gag polyprotein. Curr. Top. Microbiol. Immunol. 21465-94. [DOI] [PubMed] [Google Scholar]
- 11.Daelemans, D., S. V. Costes, E. H. Cho, R. A. Erwin-Cohen, S. Lockett, and G. N. Pavlakis. 2004. In vivo HIV-1 Rev multimerization in the nucleolus and cytoplasm identified by fluorescence resonance energy transfer. J. Biol. Chem. 27950167-50175. [DOI] [PubMed] [Google Scholar]
- 12.Datta, S. A., J. E. Curtis, W. Ratcliff, P. K. Clark, R. M. Crist, J. Lebowitz, S. Krueger, and A. Rein. 2007. Conformation of the HIV-1 Gag protein in solution. J. Mol. Biol. 365812-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derdowski, A., L. Ding, and P. Spearman. 2004. A novel fluorescence resonance energy transfer assay demonstrates that the human immunodeficiency virus type 1 Pr55Gag I domain mediates Gag-Gag interactions. J. Virol. 781230-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dupont, S., N. Sharova, C. DeHoratius, C. M. Virbasius, X. Zhu, A. G. Bukrinskaya, M. Stevenson, and M. R. Green. 1999. A novel nuclear export activity in HIV-1 matrix protein required for viral replication. Nature 402681-685. [DOI] [PubMed] [Google Scholar]
- 15.Engelsma, D., J. A. Rodriguez, A. Fish, G. Giaccone, and M. Fornerod. 2007. Homodimerization antagonizes nuclear export of survivin. Traffic 81495-1502. [DOI] [PubMed] [Google Scholar]
- 16.Feng, Y. X., T. Li, S. Campbell, and A. Rein. 2002. Reversible binding of recombinant human immunodeficiency virus type 1 Gag protein to nucleic acids in virus-like particle assembly in vitro. J. Virol. 7611757-11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gamble, T. R., S. Yoo, F. F. Vajdos, U. K. von Schwedler, D. K. Worthylake, H. Wang, J. P. McCutcheon, W. I. Sundquist, and C. P. Hill. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278849-853. [DOI] [PubMed] [Google Scholar]
- 18.Gottlinger, H. G. 2001. The HIV-1 assembly machine. AIDS 15(Suppl. 5)S13-S20. [DOI] [PubMed] [Google Scholar]
- 19.Himly, M., D. N. Foster, I. Bottoli, J. S. Iacovoni, and P. K. Vogt. 1998. The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 248295-304. [DOI] [PubMed] [Google Scholar]
- 20.Hu, C. D., Y. Chinenov, and T. K. Kerppola. 2002. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9789-798. [DOI] [PubMed] [Google Scholar]
- 21.Jares-Erijman, E. A., and T. M. Jovin. 2006. Imaging molecular interactions in living cells by FRET microscopy. Curr. Opin. Chem. Biol. 10409-416. [DOI] [PubMed] [Google Scholar]
- 22.Johnson, M. C., H. M. Scobie, Y. M. Ma, and V. M. Vogt. 2002. Nucleic acid-independent retrovirus assembly can be driven by dimerization. J. Virol. 7611177-11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karpova, T. S., C. T. Baumann, L. He, X. Wu, A. Grammer, P. Lipsky, G. L. Hager, and J. G. McNally. 2003. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. J. Microsc. 20956-70. [DOI] [PubMed] [Google Scholar]
- 24.Khokhlatchev, A. V., B. Canagarajah, J. Wilsbacher, M. Robinson, M. Atkinson, E. Goldsmith, and M. H. Cobb. 1998. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93605-615. [DOI] [PubMed] [Google Scholar]
- 25.Kjems, J., and P. Askjaer. 2000. Rev protein and its cellular partners. Adv. Pharmacol. 48251-298. [DOI] [PubMed] [Google Scholar]
- 26.Krishna, N. K., S. Campbell, V. M. Vogt, and J. W. Wills. 1998. Genetic determinants of Rous sarcoma virus particle size. J. Virol. 72564-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larson, D. R., Y. M. Ma, V. M. Vogt, and W. W. Webb. 2003. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J. Cell Biol. 1621233-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, X., B. Yuan, and S. P. Goff. 1997. Genetic analysis of interactions between Gag proteins of Rous sarcoma virus. J. Virol. 715624-5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lingappa, J. R., R. L. Hill, M. L. Wong, and R. S. Hegde. 1997. A multistep, ATP-dependent pathway for assembly of human immunodeficiency virus capsids in a cell-free system. J. Cell Biol. 136567-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malim, M. H., J. Hauber, S. Y. Le, J. V. Maizel, and B. R. Cullen. 1989. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature 338254-257. [DOI] [PubMed] [Google Scholar]
- 31.Morikawa, Y., T. Goto, and F. Momose. 2004. Human immunodeficiency virus type 1 Gag assembly through assembly intermediates. J. Biol. Chem. 27931964-31972. [DOI] [PubMed] [Google Scholar]
- 32.Muriaux, D., J. Mirro, D. Harvin, and A. Rein. 2001. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA 985246-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nandhagopal, N., A. A. Simpson, M. C. Johnson, A. B. Francisco, G. W. Schatz, M. G. Rossmann, and V. M. Vogt. 2004. Dimeric Rous sarcoma virus capsid protein structure relevant to immature Gag assembly. J. Mol. Biol. 335275-282. [DOI] [PubMed] [Google Scholar]
- 34.Ono, A., D. Demirov, and E. O. Freed. 2000. Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J. Virol. 745142-5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ono, A., J. M. Orenstein, and E. O. Freed. 2000. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J. Virol. 742855-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pante, N., and M. Kann. 2002. Nuclear pore complex is able to transport macromolecules with diameters of ∼39 nm. Mol. Biol. Cell 13425-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parent, L. J., R. P. Bennett, R. C. Craven, T. D. Nelle, N. K. Krishna, J. B. Bowzard, C. B. Wilson, B. A. Puffer, R. C. Montelaro, and J. W. Wills. 1995. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 695455-5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parent, L. J., T. M. Cairns, J. A. Albert, C. B. Wilson, J. W. Wills, and R. C. Craven. 2000. RNA dimerization defect in a Rous sarcoma virus matrix mutant. J. Virol. 74164-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parent, L. J., C. B. Wilson, M. D. Resh, and J. W. Wills. 1996. Evidence for a second function of the MA sequence in the Rous sarcoma virus Gag protein. J. Virol. 701016-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roldan, A., R. S. Russell, B. Marchand, M. Gotte, C. Liang, and M. A. Wainberg. 2004. In vitro identification and characterization of an early complex linking HIV-1 genomic RNA recognition and Pr55Gag multimerization. J. Biol. Chem. 27939886-39894. [DOI] [PubMed] [Google Scholar]
- 41.Sandefur, S., R. M. Smith, V. Varthakavi, and P. Spearman. 2000. Mapping and characterization of the N-terminal I domain of human immunodeficiency virus type 1 Pr55Gag. J. Virol. 747238-7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandefur, S., V. Varthakavi, and P. Spearman. 1998. The I domain is required for efficient plasma membrane binding of human immunodeficiency virus type 1 Pr55Gag. J. Virol. 722723-2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaufele, F., X. Carbonell, M. Guerbadot, S. Borngraeber, M. S. Chapman, A. A. Ma, J. N. Miner, and M. I. Diamond. 2005. The structural basis of androgen receptor activation: intramolecular and intermolecular amino-carboxy interactions. Proc. Natl. Acad. Sci. USA 1029802-9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheifele, L. Z., R. A. Garbitt, J. D. Rhoads, and L. J. Parent. 2002. Nuclear entry and CRM1-dependent nuclear export of the Rous sarcoma virus Gag polyprotein. Proc. Natl. Acad. Sci. USA 993944-3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheifele, L. Z., S. P. Kenney, T. M. Cairns, R. C. Craven, and L. J. Parent. 2007. Overlapping roles of the Rous sarcoma virus Gag p10 domain in nuclear export and virion core morphology. J. Virol. 8110718-10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheifele, L. Z., E. P. Ryan, and L. J. Parent. 2005. Detailed mapping of the nuclear export signal in the Rous sarcoma virus Gag protein. J. Virol. 798732-8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schliephake, A. W., and A. Rethwilm. 1994. Nuclear localization of foamy virus Gag precursor protein. J. Virol. 684946-4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sekar, R. B., and A. Periasamy. 2003. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 160629-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaner, N. C., R. E. Campbell, P. A. Steinbach, B. N. Giepmans, A. E. Palmer, and R. Y. Tsien. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 221567-1572. [DOI] [PubMed] [Google Scholar]
- 50.Sherer, N. M., M. J. Lehmann, L. F. Jimenez-Soto, A. Ingmundson, S. M. Horner, G. Cicchetti, P. G. Allen, M. Pypaert, J. M. Cunningham, and W. Mothes. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4785-801. [DOI] [PubMed] [Google Scholar]
- 51.Sherman, M. P., C. M. de Noronha, M. I. Heusch, S. Greene, and W. C. Greene. 2001. Nucleocytoplasmic shuttling by human immunodeficiency virus type 1 Vpr. J. Virol. 751522-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shkriabai, N., S. A. Datta, Z. Zhao, S. Hess, A. Rein, and M. Kvaratskhelia. 2006. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 454077-4083. [DOI] [PubMed] [Google Scholar]
- 53.Shyu, Y. J., H. Liu, X. Deng, and C. D. Hu. 2006. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. BioTechniques 4061-66. [DOI] [PubMed] [Google Scholar]
- 54.Siegel, R. M., F. K. Chan, D. A. Zacharias, R. Swofford, K. L. Holmes, R. Y. Tsien, and M. J. Lenardo. 2000. Measurement of molecular interactions in living cells by fluorescence resonance energy transfer between variants of the green fluorescent protein. Sci. STKE 2000pl1-pl6. [DOI] [PubMed] [Google Scholar]
- 55.Swanstrom, R., and J. Wills. 1997. Synthesis, assembly, and processing of retroviral proteins, p. 263-334. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- 56.Tritel, M., and M. D. Resh. 2000. Kinetic analysis of human immunodeficiency virus type 1 assembly reveals the presence of sequential intermediates. J. Virol. 745845-5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vogel, S. S., C. Thaler, and S. V. Koushik. 2006. Fanciful FRET. Sci. STKE 2006re2-re8. [DOI] [PubMed] [Google Scholar]
- 58.Vogt, V. M. 1997. Retroviral virions and genomes, p. 27-69. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- 59.Weldon, R. A., Jr., C. R. Erdie, M. G. Oliver, and J. W. Wills. 1990. Incorporation of chimeric Gag protein into retroviral particles. J. Virol. 644169-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weldon, R. A., Jr., and J. W. Wills. 1993. Characterization of a small (25-kilodalton) derivative of the Rous sarcoma virus Gag protein competent for particle release. J. Virol. 675550-5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wills, J. W., and R. C. Craven. 1991. Form, function, and use of retroviral Gag proteins. AIDS 5639-654. [DOI] [PubMed] [Google Scholar]
- 62.Wills, J. W., R. C. Craven, R. A. Weldon, Jr., T. D. Nelle, and C. R. Erdie. 1991. Suppression of retroviral MA deletions by the amino-terminal membrane-binding domain of p60src. J. Virol. 653804-3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilson, C. G., T. J. Magliery, and L. Regan. 2004. Detecting protein-protein interactions with GFP-fragment reassembly. Nat. Methods 1255-262. [DOI] [PubMed] [Google Scholar]
- 64.Xiang, Y., C. E. Cameron, J. W. Wills, and J. Leis. 1996. Fine mapping and characterization of the Rous sarcoma virus Pr76gag late assembly domain. J. Virol. 705695-5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang, F., L. G. Moss, and G. N. Phillips, Jr. 1996. The molecular structure of green fluorescent protein. Nat. Biotechnol. 141246-1251. [DOI] [PubMed] [Google Scholar]
- 66.Zhou, W., and M. D. Resh. 1996. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 708540-8548. [DOI] [PMC free article] [PubMed] [Google Scholar]