
FIG. 1.
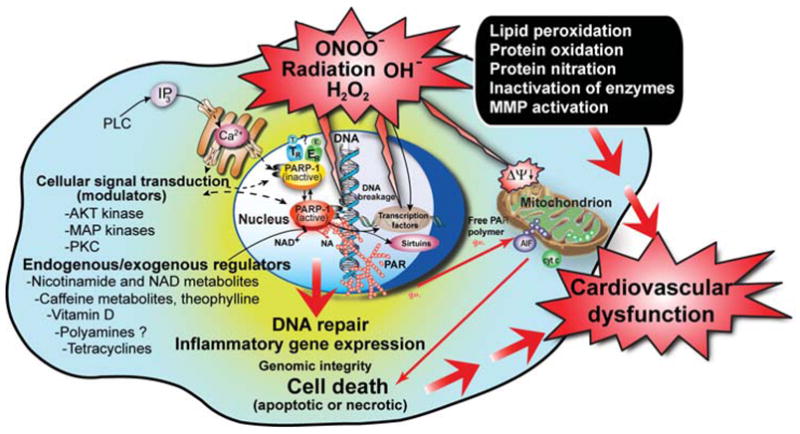
Triggers, mechanisms of PARP-mediated cell death, and exogenous/endogenous regulators/modulators of PARP activity. Reactive oxygen and nitrogen species (e.g., peroxynitrite)-dependent cytotoxicity in various cardiovascular diseases is mediated by a multitude of effects including lipid peroxidation, protein nitration and oxidation, DNA oxidative damage, activation of matrix metalloproteinases (MMPs), and inactivation of a series of enzymes. Mitochondrial enzymes are particularly vulnerable to attacks by peroxynitrite, leading to reduced ATP formation and induction of mitochondrial permeability transition by opening of the permeability transition pore, which dissipates the mitochondrial membrane potential (Δψm). These events lead to cessation of electron transport and ATP formation, mitochondrial swelling, and permeabilization of the outer mitochondrial membrane, allowing the efflux of several proapoptotic molecules, including cytochrome or C and apoptosis-inducing factor (AIF). In turn, cytochrome or C and AIF activate a series of downstream effectors, which eventually result in the fragmentation of nuclear DNA. In addition to its damaging effects on mitochondria, peroxynitrite inflicts more or less severe oxidative injury to DNA, resulting in DNA strand breakage, which in turn activates the nuclear enzyme poly(ADP-ribose) polymerase (PARP). Activated PARP consumes NAD to build up poly(ADP-ribose) polymers (PAR), which are themselves rapidly metabolized by the activity of poly(ADP-ribose) glycohydrolase (PARG). Some free PAR may exit the nucleus and travel to the mitochondria, where they amplify the mitochondrial efflux of AIF (nuclear to mitochondria crosstalk). Depending on the severity of the initial insult by peroxynitrite or other oxidants, the injured cell may either recover or die. In the latter case, the cell may be executed by apoptosis in the case of moderate PTP opening and PARP activation with preservation of cellular ATP, or by necrosis in case of widespread PTP opening and PARP overactivation, leading to massive NAD consumption and collapse of cellular ATP. Various endogenous factors can influence PARP activity either by inhibiting the binding of its substrate NAD+ to the active site of the enzyme or by forming a complex with PARP. An example for the latter may include estrogen (E) and thyroid hormones (T) and for the former nicotinamide (NA), NAD+ metabolites, caffeine metabolites, and vitamin D. PARP activity can also be modulated by various kinases by phosphorylation (e.g., MAP kinases and PKC), and PARP can modulate kinase (e.g., AKT) activity. Various exogenous factors such as caffeine and its endogenously formed metabolites, theophylline, and tetracycline antibiotics may also modulate PARP activity. Overall, PARP appears to be a subject of multiple lines of endogenous regulators, and it is conceivable that the processes regulated by PARP (e.g., DNA repair and cellular NAD homeostasis) are under a similarly dynamic control by a multitude of factors and influences.