

Abstract
“Self” melanocyte differentiation antigens are potential targets for specific melanoma immunotherapy. Vaccination against murine tyrosinase-related protein (TRP)-1/gp75 was shown recently to cause melanoma rejection, which was accompanied by autoimmune skin depigmentation (vitiligo). To further explore the linkage between immunotherapy and autoimmunity, we studied the response to vaccination with a related antigen, TRP-2. i.m. inoculation of plasmid DNA encoding murine trp-2 elicited antigen-specific CTLs that recognized the B16 mouse melanoma and protected the mice from challenge with tumor cells. Furthermore, mice bearing established s.c. B16 melanomas rejected the tumor upon vaccination with a recombinant vaccinia virus encoding trp-2. Depletion experiments showed that CD8+ lymphocytes and natural killer cells were crucial for the antitumor activity of the trp-2-encoding vaccines. Mice that rejected the tumor did not develop generalized vitiligo, indicating that protective immunity can be achieved in the absence of widespread autoimmune aggression.
Introduction
Tumor-specific human CD8+ and CD4+ lymphocytes respond to molecular complexes formed by MHC molecules and short peptides derived from the processing of tumor Ags3 expressed from either mutated or normal genes (1-3). Several melanoma Ags recognized by lymphocytes of melanoma patients in association with class I MHC molecules belong to the category of MDAs, which includes gp100/pmel-17, MART-1/Melan-A, tyrosinase, TRP-1/gp75, and TRP-2. Active immunization against these widely shared Ags represents an attractive and simple strategy for the therapy of cancer, and initial studies using mouse models have produced encouraging results. Thymic, central tolerance to MDAs is not complete, and peripheral tolerance can be broken by xenoimmunization or immunization with an altered source of the Ag. Administration of a rVV encoding human gp100/pmel-17 or with a plasmid encoding human TRP-1/gp75 elicited an immune response against the respective murine homologues (4, 5). Tolerance to murine TRP-1/gp75 was also broken by giving the murine Ag in an altered form; exposure to the protein expressed in insect cells or produced during in vivo infection with a rVV (6, 7) was able to trigger an immune response that fully recognized the naturally processed form of the native Ag and led to tumor eradication. The therapeutic effect required CD4+ lymphocytes, antibodies, NK1.1 cells, and the Fc receptor γ-chain, suggesting a scenario in which melanoma cells targeted by specific antibodies were eliminated by antibody-dependent cellular cytotoxicity-mediated lysis (6-9). Induction of an effective immune response against TRP-1/gp75 was associated with autoimmune manifestations consisting of diffuse depigmentation of skin patches (vitiligo) attributable to the destruction of normal melanocytes sharing the TRP-1/gp75 Ag. Additional studies are required to understand the association between vitiligo and melanoma therapy, because the mechanisms inducing vitiligo could be uncoupled from those causing tumor regression in one study but not in another (5, 7).
Although the immune response to TRP-1/gp75 is dominated by production of anti-TRP-1/gp75 antibodies, TRP-2 Ag is the main target of murine CTLs generated after immunization with irradiated melanoma cells (10); CTL lines raised from splenocytes by repeated in vitro stimulation with the “self,” immunodominant TRP-2 peptide showed therapeutic activity against established pulmonary metastases. On the other hand, active immunization with DNA encoding murine trp-2 administered using a gene gun in association with a plasmid encoding interleukin 12 was capable of eliciting CTLs recognizing B16 melanoma but induced only a weak protective response (11). The present study demonstrates that i.m. immunization with plasmid DNA encoding murine trp-2 conferred protection to challenge with B16 melanoma cells; moreover, administration of a rVV encoding murine trp-2 was therapeutic in tumor-bearing mice. In contrast to mice immunized with TRP-1/gp75, the main effector populations induced by DNA immunization with the trp-2 gene and involved in tumor protection were CD8+ lymphocytes and NK1.1+ cells. Furthermore, mice that rejected the tumor did not develop generalized vitiligo during the observation period but exhibited depigmentation and hair loss localized at the site of tumor inoculation. Active immunization with trp-2-encoding vectors may thus represent a promising immunotherapy strategy against melanoma.
Materials and Methods
Mice and Cell Lines
C57BL/6 × BALB/c mice, 5– 6 weeks of age, were purchased from Charles River (Calco, Como, Italy). Procedures involving animals and their care conformed with institutional guidelines that comply with national and international laws and policies [EEC Council Directive 86/609, OJ L 358, 1, Dec. 12, 1987; NIH Guide for the Care and Use of Laboratory Animals, NIH Publication 85-23, 1985; UKCCR Guidelines for Welfare of Animals in Experimental Neoplasia (12)]. Mice used for the in vivo tumor growth experiments were examined every other day and were euthanized when the tumor became ulcerated or reached a diameter >50 mm2.
Cell Lines
MBL-2 is a leukemia cell line (H-2b) derived from a Moloney murine leukemia virus-infected B6 mouse; C57BL/6 is a melanoma line (H-2b) spontaneously growing in C57BL/6 mice (kindly provided by Dr. I. J. Fidler, M. D. Anderson Cancer Center, Houston, TX). The cell lines were cultured in DMEM (Life Technologies, Inc., Gaithersburg, MD) supplemented with 2 mm l-glutamine, 10 mm HEPES, 20 μm 2-mercaptoethanol, 10 units of ampicillin/ml, and 5 or 10% heat-inactivated fetal bovine serum (Life Technologies).
Plasmid and Viruses
The cDNA coding for murine trp-2 was a kind gift of Dr. V. J. Hearing (Laboratory of Cell Biology, NIH, Bethesda, MD; Ref. 13). The trp-2 gene was cloned into the eukaryotic expression plasmid pcDNA3 (Invitrogen BV, Leek, the Netherlands), resulting in plasmid pcDNA3- trp-2, in which expression of the mouse trp-2 gene is driven by the cytomegalovirus promoter/enhancer. Endotoxin-free plasmids were purified by anion exchange chromatography (Qiagen GmbH, Hilden, Germany). rVV-trp-2 was generated by insertion of the trp-2 gene by homologous recombination as described previously by Moss and Earl (14) and was derived using plasmid pSC65, in which the completely synthetic early/late promoter pSE/L drives expression of the Ag and the early/late promoter p7.5E/L drives expression of the LacZ gene (14). Expression of TRP-2 protein by rVV-trp-2 and by pcDNA3-trp-2 was confirmed by immunostaining of infected/transfected cells using anti-TRP-2 serum (anti-PEP8 serum, kindly provided by Dr. V. J. Hearing).
DNA and Recombinant Virus Immunization Protocols
DNA immunization was performed according to commonly used protocols available at the “DNA vaccine web” site.4 Briefly, mice were anesthetized by ethyl ether inhalation and injected i.m. with 100 μl of 10 μm cardiotoxin (Latoxan, Rosans, France). Five to 9 days later, mice were injected i.m. with 100 μg of plasmid DNA in 100 μl of saline. rVV (5 × 106 PFU/mouse) was inoculated i.v. 5 days after tumor injection.
MLPC
Three weeks after plasmid DNA inoculation, spleens were removed, and 2.5 × 107 splenocytes were stimulated in vitro in a MLPC with 1 μm of a nonamer peptide corresponding to amino acids 180–188 of TRP-2 (SVYDFFVWL; Refs. 10 and 11); the peptide was synthesized and purified by Neosystem (Strasbourg, France). The cultures were set up in DMEM-10% fetal bovine serum, maintained for 5 days at 37°C under 5% CO2, and then tested in 51Cr and IFN-γ release assays. Cytotoxic activity of cultured splenocytes was tested in a short-term incubation 51Cr release assay by mixing 2 × 103 51Cr-labeled target cells with the effector cells at various E:T cell ratios in 96-well microplates; after 5 h of incubation at 37°C, supernatants were harvested, and radioactivity was counted in a microplate scintillation counter (Packard Instruments Co., Meriden, CT). For peptide pulsing, 106 51Cr-labeled cells were incubated for 30 min at 37°C with 1 μm peptide and then washed before use. The IFN-γ release assay was carried out by restimulating 105 splenocytes from MLPC for 24 h in triplicate wells with an equal number of target cells; the supernatant was then harvested and tested for released IFN-γ in a sandwich ELISA assay (Endogen, Boston, MA).
Tumor Protection and Therapy
Three weeks after DNA inoculation, mice were challenged s.c. with a lethal dose of B16 melanoma cells (i.e., 105 cells, 10-fold greater than the minimum tumorigenic dose), and then monitored for 120 days after tumor injection. Mice inoculated with mock plasmid and uninoculated mice were used as control groups. For tumor therapy experiments, 105 B16 cells were injected s.c. 5 days before rVV inoculation.
In Vivo Antibody-mediated Depletion
Mice were depleted of either CD4+, CD8+, or NK cells by four i.p. injections of 200 μg of GK1.5 (anti-CD4), 2.43 (anti-CD8), or PK136 (anti-NK1.1) monoclonal antibodies (mAb) prepared in 200 μl of endotoxin-free PBS (Sigma). Depleting mAb were given on days −2, 0, 4, and 8 with respect to the s.c. challenge with B16 melanoma cells. The mAbs were produced from hybridomas (obtained from American Type Culture Collection, Manassas, VA) grown in ascites and purified by ammonium sulfate precipitation, followed by protein-G Sepharose affinity chromatography (Pharmacia, Uppsala, Sweden). Depletion was monitored by cytofluorimetry of peripheral lymphocytes isolated from mouse blood and stained with FITC- or PE-conjugated anti-CD4, anti-CD8, or anti-NK1.1 (clones RM4-5, 53.6-7, and 2B4, respectively; all from PharMingen, San Diego, CA). Depletion was consistently >98%.
Statistical Analysis
The Wilcoxon-Mann-Whitney U test was used to examine the null hypothesis of rank identity between two sets of data. Kaplan-Maier plots and the Mantel-Haenszel test were used to compare survival of mice belonging to different treatment groups. All Ps presented are two sided.
Results
DNA Immunization Can Elicit an Immune Response against the Self Melanoma Ag TRP-2
To break tolerance against self MDAs, we constructed the eukaryotic expression vector pcDNA3-trp-2, coding for murine trp-2. After verifying the expression of TRP-2 from pcDNA3-trp-2 in transiently transfected cells by immunofluorescence with an antibody that recognized a COOH-terminal region of this protein (data not shown), we injected mice with the plasmid according the protocol reported in “Materials and Methods” and evaluated whether TRP-2 genetic immunization could elicit Ag-specific CTLs. Three weeks after DNA inoculation, splenocytes from injected mice were stimulated in a MLPC using the immunodominant, H-2Kb-restricted, TRP-2 peptide (TRP-2180–188). Five days later, cytotoxic activity against target cells was evaluated as shown in Fig. 1A. CTL activity against MBL-2 cells pulsed with TRP-2180–188 peptide was present only in mice injected with DNA expressing TRP-2. Peptide-specific CTLs were not elicited in control mice that received cardiotoxin alone or cardiotoxin followed by plasmid pcDNA3. Although CTLs specifically recognized the TRP-2 immunodominant peptide, they were not able to efficiently lyse B16 tumor cells.
Fig. 1.

Plasmid DNA encoding TRP-2 induces Ag-specific CTL. A, 3 weeks after i.m. inoculation of plasmid DNA, spleens were removed, and splenocytes were stimulated in vitro in an MLPC with 1 μm of TRP-2180–188 peptide. After 5 days of culture, cytotoxic activity of the splenocytes was tested in a 51Cr release assay carried out for 5 h against the following target cells: MBL-2, MBL-2 pulsed with TRP-2180–188 peptide, and B16. Each panel shows the CTL response generated in a single mouse after inoculation of either cardiotoxin alone or cardiotoxin followed by either the empty pcDNA3 vector or pcDNA3-trp-2. Results are representative of five experiments. The SD of the triplicate determinations for each effector/target cell ratio (E:T ratio) was <10%, and spontaneous release never exceeded 20%. B, splenocytes (105 cells) from MLPCs were restimulated for 24 h in triplicate wells with an equal amount of MBL-2 cells, MBL-2 cells pulsed for 1 h with 1 μm TRP-2180–188 peptide, or B16 melanoma cells; the supernatant was then harvested and tested for released IFN-γ in a sandwich ELISA assay. The SD of the triplicate determinations for each effector/stimulator combination was <10%, and IFN-γ measured in control wells containing either effectors or stimulators alone did not exceed the lowest amount of IFN-γ detectable in our assay (i.e., 547 pg/ml, determined using serial dilutions of IFN-γ). Two-tailed Ps calculated by the Mann-Whitney test are reported in the figure for those groups showing significant differences over their respective controls (filled symbols).
B16 melanoma cells express very low amounts of Kb molecules (<30% at low intensity in fluorescence-activated cell sorting analysis; data not shown). Such limited expression of Kb molecule can compromise the sensitivity of the 51Cr assay, which allows for a brief incubation of CTLs with their targets. One strategy to overcome this limitation is to expose the B16 cells to IFN-γ prior to the 51Cr assay (11); alternatively, the levels of IFN-γ released in the supernatant of CTL cultures stimulated for 24 h can be measured. Fig. 1B shows IFN-γ release assay of MLPC set up with splenocytes from mice immunized with pcDNA3-trp-2 or pcDNA3 and either B16 tumor cells or TRP-2180–188 peptide-pulsed MBL-2 cells as targets. Results showed that elevated amounts of IFN-γ were detected in the supernatants of lymphocytes cultured in the presence of either target, thus indicating that the DNA protocol is indeed capable of eliciting CTLs specifically recognizing the B16 melanoma.
DNA Immunization with pcDNA3-trp-2 Protects from Challenge with B16 Melanoma Cells
The release of IFN-γ by CTLs as an indicator of immune response was shown previously to correlate with the in vivo antitumor activity of the CTLs upon adoptive transfer (15). We thus asked whether the Ag-specific response demonstrated in mice immunized with pcDNA3-trp-2 could protect them from tumor challenge. Fig. 2 summarizes results of five experiments in which naïve mice or mice previously immunized with pcDNA3-trp-2 were injected with a lethal dose of B16 melanoma cells. Although control mice left untreated died within 3 weeks after challenge, almost complete protection against the lethal challenge was achieved in mice vaccinated with the plasmid DNA encoding trp-2. Data accumulated from the five experiments yielded an overall rate of tumor prevention of 86% (i.e., 36 of 42 mice were protected). Unexpectedly, untreated mice and mice injected with the empty pcDNA3 vector also showed a significant difference in survival, suggesting a marginal, Ag-independent effect of DNA immunization in prevention of tumor growth. This effect was related to the pcDNA3 vector, because it was not observed with a second construct derived from the plasmid VR1012 (Ref. 16; data not shown).
Fig. 2.
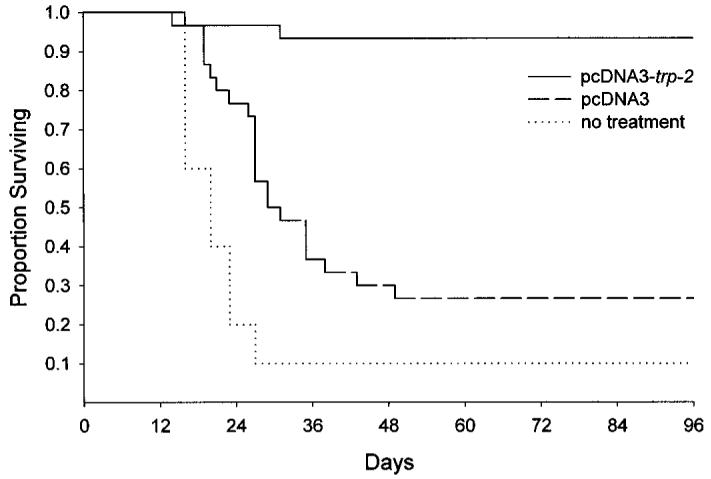
Immunization with a plasmid encoding TRP-2 prolongs survival of mice challenged with B16 melanoma cells. Three weeks after vaccination with pcDNA3-trp-2, mice were challenged s.c. in the right flank with 105 B16 melanoma cells. Untreated mice or mice immunized with the empty pcDNA3 vector were used as negative controls. Results represent the sums of two independently performed experiments with 5–10 mice in each treatment group. Immunization with pcDNA3-trp-2 resulted in a significant prolongation of survival (P < 0.0001). Inoculation of the empty pcDNA3 vector also caused a significant prolongation of survival (P = 0.006) as compared with untreated mice.
CD8+ Lymphocytes and NK Cells Are Involved in Tumor Eradication after DNA Immunization
To explore the role of T cells in the antitumor efficacy of the DNA vaccine, mice that had been immunized with pcDNA3-trp-2 received depleting doses of mAbs specific for either CD4+ lymphocytes, CD8+ lymphocytes, or NK cells. The inoculation schedule assured that the corresponding population was completely absent at the moment of tumor challenge and during the first 10 days of tumor growth, as assessed by cytofluorimetry (data not shown). As shown in Fig. 3A, DNA immunization increased the survival of both normal mice and mice depleted of CD4+ lymphocytes, indicating that CD4+ cells were not required to control B16 melanoma growth. In contrast, mice depleted of either CD8+ lymphocytes or NK cells were no longer protected and succumbed from tumor challenge. Thus, both CD8+ lymphocytes and NK cells acted in concert to eradicate melanoma cells in immune mice.
Fig. 3.
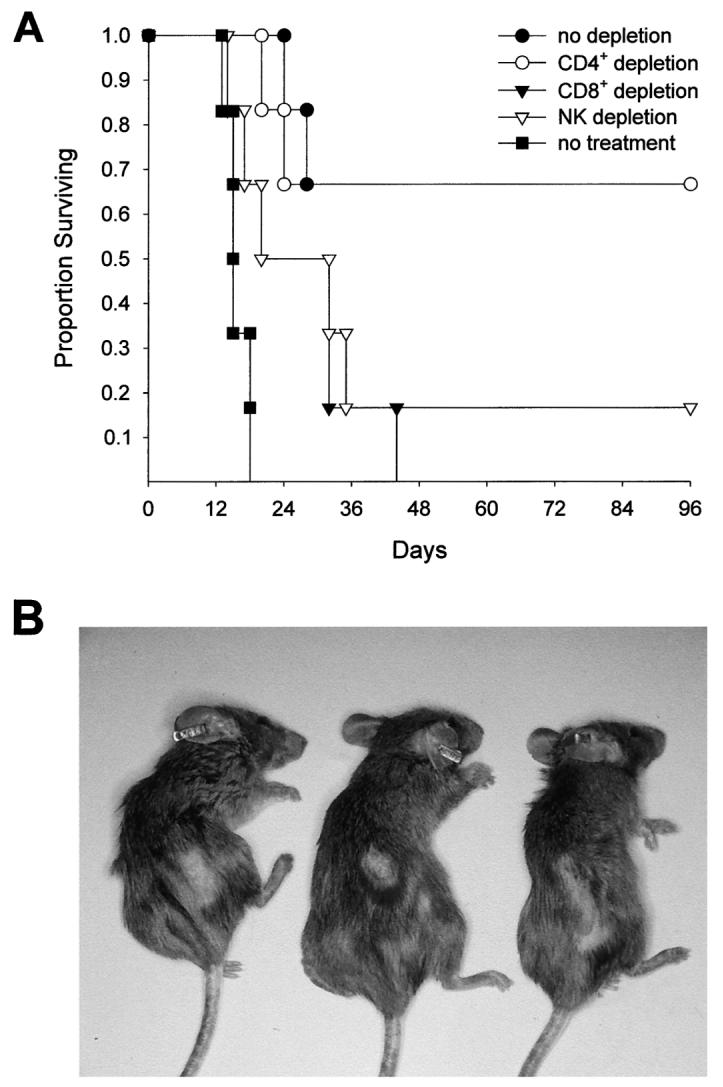
Protection against tumor challenge induced by DNA immunization depends on CD8+ lymphocytes and NK cells and is associated with local depigmentation and alopecia. A, to identify the effector population responsible for the antitumor effect, mice immunized with pcDNA3-trp-2 were depleted by i.p. injections of either anti-CD4, anti-CD8, or anti-NK1.1 mAbs as described in “Materials and Methods.” Mice not immunized with pcDNA3-trp-2 were used as controls (no treatment). Each treatment group contained six mice. The Mantel-Haenszel test gave P = 0.025 for the CD8+-depleted versus no depletion group and P = 0.09 for the NK-depleted versus no depletion group. Duplicate experiments confirmed these results. B, mice were routinely shaved on the right flank in preparation for the s.c. tumor challenge. Although the hair normally grew back in 3 weeks, some mice showed an area of persistent hair loss and depigmentation at the site of tumor injection. The photograph was taken 4 months after tumor challenge and shows mice (anesthetized for the photo) belonging to the “no depletion” and “CD4+ depletion” groups described in A.
Local Depigmentation and Alopecia but not Vitiligo Are Associated with Tumor Regression
On the basis of observations made after immunization with TRP-1/gp75 Ag (7, 8), the induction of a strong CTL and NK reactivity against TRP-2 was expected to give rise to melanocyte destruction and manifestations of vitiligo. However, mice that had rejected the tumor after challenge did not develop vitiligo during the follow-up period of >1 year, even when given two consecutive i.m. injections of pcDNA3-trp-2 (data not shown). Localized hair loss and depigmentation in the area surrounding the site of tumor inoculation was evident in some mice, including those that had been depleted of CD4+ lymphocytes by antibody treatment (Fig. 3B). Haircoat loss was clearly evident 3 weeks after tumor challenge when the hair grew back in the shaved left flank, with the exception of the tumor injection site. The haircoat alterations were observed in ∼40% (15 of 36) of all of the mice that survived the challenge, and they remained stable for the observation period of 1 year.
Immunization with rVV Encoding trp-2 Can Cure Established Tumors
The high rate of protection against B16 melanoma conferred by a DNA vaccine coding for TRP-2 Ag prompted us to investigate its therapeutic ability against established tumors. However, application of the same DNA immunization protocol used in the protection experiment did not result in tumor regression (data not shown). We speculated that the time required for optimal CTL generation after DNA immunization (2 weeks, not shown) was too long to allow a successful treatment of a swiftly growing tumor such as B16 melanoma, which is able to kill the host in 3–4 weeks. Therefore, we turned to an rVV vector as a means of expressing the TRP-2 Ag. In the hope that it might reduce the lag phase between vaccination and CTL production, mice were injected with a lethal dose of B16 tumor cells and 5 days later received 5 × 106 PFU of either mock rVV or rVV-trp-2 (Fig. 4). As expected, negative control mice that were untreated or injected with the mock rVV died within 3–4 weeks after tumor challenge. In contrast, 50% of the mice that were treated with rVV-trp-2 remained tumor free 3 months after inoculation with B16 cells. These mice did not show any signs of vitiligo and did not exhibit the changes in pigmentation and haircoat noted in pcDNA3-trp-2-vaccinated mice. The reasons for these differences are not clear. We hypothesize that NK cell activation induced by DNA immunization might help in causing more extensive tissue destruction. Moreover, immunization after tumor implantation might affect a population of CTL preprimed by the encounter with the tumor cells that is already highly specific for TRP-2. Immunization before tumor challenge might expand largely cross-reactive CTLs responsible for an higher degree of cross-killing and tissue devastation.
Fig. 4.
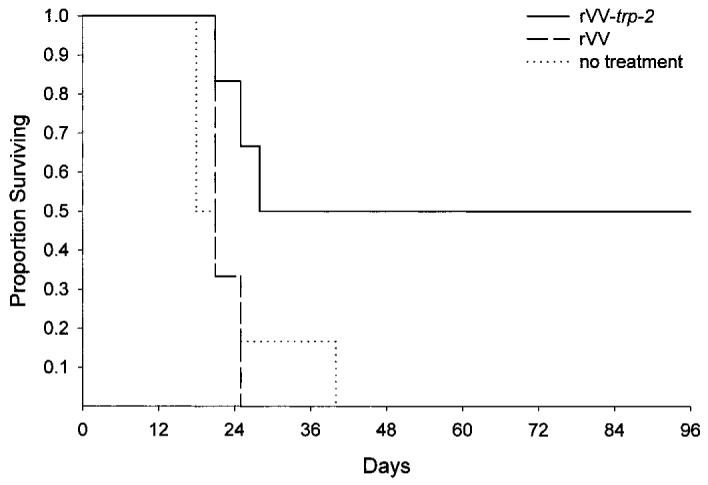
rVV encoding TRP-2 has a therapeutic effect on tumor-bearing mice. Mice were injected s.c. with 105 B16 melanoma cells. Five days later, they received a single i.v. inoculation of 5 × 106 PFU of an rVV encoding TRP-2 (rVV-trp-2). Untreated mice and mice injected with the same amount of mock virus (rVV) were used as negative controls. Results represent the sums of two independently performed experiments with five to six mice in each treatment group. The statistical analysis was performed according to the Mantel-Haenszel test (P = 0.04, no treatment versus rVV-trp-2; P = 0.001, rVV versus rVV-trp-2).
Discussion
Immune recognition of melanoma cells by T and B lymphocytes has been studied extensively, and several melanoma Ags have been defined (1-3). These Ags belong to three main categories: (a) mutated or aberrantly expressed Ags (CDK4, β-catenin, and Casp8); (b) cancer/testis-specific Ags (MAGE, BAGE, GAGE, PRAME, and NYESO-1); and (c) MDAs, which include tyrosinase, Melan-A/MART-1, gp100, TRP-1/gp75 and TRP-2. This last group comprises the most prevalent Ags recognized by T lymphocytes on human melanomas. The development of an immune response to MDAs is quite surprising, because it occurs despite the mechanisms of central and peripheral tolerance to self Ags. Indeed, the immune response to these MDAs is presumably limited by the existence of tolerance mechanisms that spare only T lymphocytes with low affinity T-cell receptors (17). Low-affinity lymphocytes can nonetheless be used therapeutically, and among the various MDAs, TRP-1/gp75 and gp100 were clearly shown to be tumor-regression Ags in mice and humans (4, 7, 18). The present study broadens the number of potential targets of specific immunotherapy by showing that TRP-2 represents a melanoma rejection Ag in mice.
The choice of the vaccine is as important as the choice of the tumor Ag, because the formulation and the route of delivery may profoundly influence the immune responses. We sought to understand whether the therapeutic responses induced by rVV or by naked DNA were qualitatively different. DNA vaccines are thought to be less efficient than recombinant viruses in the therapy of established tumors expressing a model tumor Ag (19, 20). However, we urge caution in interpreting the finding of the reduced efficacy of DNA vaccination in a therapeutic setting (as described in the present report). Clearly, the decreased efficacy after vaccination with DNA could be entirely attributable to the long time required to elicit TRP-2-specific CTLs.
The association between autoimmune disorders, i.e., vitiligo and tumor regression described in mice immunized with TRP-1/gp75 Ag and in melanoma patients undergoing therapy with high doses of interleukin 2, has led to the suggestion that a deliberate induction of autoimmunity against tissue Ags may be an acceptable side effect of tumor therapy (21), especially in the case of tumors arising from nonessential tissues. Our study indicates that diffuse vitiligo is not invariably associated with tumor eradication caused by immunization with MDAs and instead appears to depend on the specific Ag used.
Some mice immunized with TRP-2 showed localized hair loss and depigmentation, likely sequelae to the inflammatory response and residual fibrosis accompanying the immune destruction of tumor cells and adjacent tissues. The observation that the presence of TRP-2-specific CTLs did not lead to generalized destruction of normal melanocytes supports the hypothesis that the pathogenesis of human vitiligo in melanoma patients is related more to antibodies recognizing melanoma proteins such as tyrosinase or TRP-1/gp75 rather than CTL activity (8, 22). Cutaneous lesions similar to those described in the present study were described recently in mice treated with a protocol consisting of immunization with a granulocyte/macrophage-colony stimulating factor-expressing melanoma vaccine, followed by inoculation with an mAb blocking the activity of the CTL-associated antigen 4; the combined treatment caused melanoma rejection which, analogous to our findings, was dependent on CD8+ and NK1.1+ cells but independent of CD4+ T cells (23). About half of the mice surviving tumor challenge after combination treatment developed depigmentation, starting at the site of vaccination or challenge and spreading to distant sites; the initial lesions were similar to those observed after immunization with TRP-2-encoding plasmids. Because we did not observe vitiligo progression, it is conceivable that the CTL-associated antigen 4 blockade used in the previous study enhanced T-cell activation and triggered a massive proliferation of autoreactive T cells, the numbers of which are normally contained by the mechanisms of peripheral tolerance. Thus, although unleashing the mechanisms controlling the magnitude of the immune response may be relevant for enhancement of therapeutic efficacy, it must be weighed against the cost of an autoimmunity sequela.
We used CB6 F1 mice in the present experiments for several reasons: (a) C57BL/6 mice lack a portion of the genome that contains the α chain of the I-E molecule, a condition that related to the human beings would translate in the loss of the DR locus and might affect the immune response to melanoma Ags; and (b) F1 hybrids are heterozygous at the H-2 locus and thus closer to patients who display an extreme polymorphism in HLA loci. Experimentally, F1 mice showed anti-TRP-2 immune responses stronger than that observed in C57BL/6 mice after DNA immunization. The autoimmune and anti-tumor responses are currently being compared in C57BL/6 and CB6F1 mice. Preliminary results indicate that differences in T-lymphocyte repertoire directed against the Kb-TRP-2 peptide complex might explain the different magnitudes of the immune responses in the two strains.
In contrast to the highly efficient protection afforded by immunization with artificial model tumor-associated antigens such as β-galactosidase (19, 20), protection against tumor challenge was not observed in the totality of the mice immunized with the trp-2-expressing plasmid. These differences almost certainly reflect the stronger immunogenicity of the model tumor Ags as compared with self Ags. Similar findings were made in our previous studies with the murine Ag P1A, which is naturally expressed in P815 mastocytoma cells (24); although vaccination of mice with a plasmid expressing P1A considerably increased their survival rate after challenge with P815 cells, it did not confer complete protection. Several strategies to increase the efficacy of DNA immunization are currently being evaluated. Although repeatedly boosting the immune response is the simplest approach, it cannot be based on repeated i.m. inoculation of DNA in cardiotoxin-pretreated muscles. In fact, we did not observe any increase in the percentage of mice rejecting a B16 challenge after two DNA inoculations instead of a single immunization (data not shown). A promising approach is the use of a DNA vaccine for priming and the modified vaccinia virus Ankara for boosting, a protocol that has been used successfully to increase the CTL response against malaria Ags (25). Alternatively, combining different weak, self Ags in the same immunization protocol might increase the overall immune response against the tumor. This hypothesis is supported by preliminary results of a study, indicating that a mixture of two plasmids encoding gp100 and trp-2 increases the rate of protection in C57BL/6 mice from challenge with B16 melanoma cells.5
Acknowledgments
We thank Susanna Mandruzzato for helpful discussions and for critical reading of the manuscript, Pierantonio Gallo for assistance with graphics, Donna D'Agostino for editing the manuscript, Vito Barbieri for technical assistance in mouse studies, and Anna Cabrelle for assistance with cytofluorimetry.
Footnotes
Supported in part by the Italian Association for Cancer Research and by the Istituto Superiore SanitàItaly-USA cooperative program for the therapy of cancer (Grant 981/A.14). E. A. is supported by a fellowship from The Italian Foundation for Cancer Research.
The abbreviations used are: Ag, antigen; TRP, tyrosinase-related protein; MDA, melanocyte lineage differentiation antigen; rVV, recombinant vaccina virus; NK, natural killer; mAb, monoclonal antibody; MLPC, mixed leukocyte peptide culture; PFU, plaque-forming unit(s).
Internet address: http://www.genweb.com/Dnavax/dnavax.html.
A. Singhal, G. Thai, N. Thull, N. Eslashi, M. Matar, V. Bronte, and F. Pericle, manuscript in preparation.
References
- 1.Henderson RA, Finn OJ. Human tumor antigens are ready to fly. Adv. Immunol. 1996;62:217–256. doi: 10.1016/s0065-2776(08)60431-9. [DOI] [PubMed] [Google Scholar]
- 2.Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr. Opin. Immunol. 1997;9:684–693. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10:281–287. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- 4.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber LW, Bowne WB, Wolchok JD, Srinivasan R, Qin J, Moroi Y, Clynes R, Song P, Lewis JJ, Houghton AN. Tumor immunity and autoimmunity induced by immunization with homologous DNA. J. Clin. Investig. 1998;102:1258–1264. doi: 10.1172/JCI4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naftzger C, Takechi Y, Kohda H, Hara I, Vijayasaradhi S, Houghton AN. Immune response to a differentiation antigen induced by altered antigen: a study of tumor rejection and autoimmunity. Proc. Natl. Acad. Sci. USA. 1996;93:14809–14814. doi: 10.1073/pnas.93.25.14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Overwijk WW, Lee DS, Surman DR, Irvine KR, Touloukian CE, Chan CC, Carroll MW, Moss B, Rosenberg SA, Restifo NP. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc. Natl. Acad. Sci. USA. 1999;96:2982–2987. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hara I, Takechi Y, Houghton AN. Implicating a role for immune recognition of self in tumor rejection: passive immunization against the brown locus protein. J. Exp. Med. 1995;182:1609–1614. doi: 10.1084/jem.182.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clynes R, Takechi Y, Moroi Y, Houghton A, Ravetch JV. Fc receptors are required in passive and active immunity to melanoma. Proc. Natl. Acad. Sci. USA. 1998;95:652–656. doi: 10.1073/pnas.95.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bloom MB, Perry-Lalley D, Robbins PF, Li Y, el-Gamil M, Rosenberg SA, Yang JC. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J. Exp. Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuting T, Gambotto A, DeLeo A, Lotze MT, Robbins PD, Storkus WJ. Induction of tumor antigen-specific immunity using plasmid DNA immunization in mice. Cancer Gene Ther. 1999;6:73–80. doi: 10.1038/sj.cgt.7700020. [DOI] [PubMed] [Google Scholar]
- 12.Workman P, Balmain A, Hickman JA, McNally NJ, Rohas AM, Mitchison NA, Pierrepoint CG, Raymond R, Rowlatt C, Stephens TC. UKCCCR guidelines for the welfare of animals in experimental neoplasia. Lab. Anim. 1988;22:195–201. doi: 10.1258/002367788780746467. [DOI] [PubMed] [Google Scholar]
- 13.Winder A, Kobayashi T, Tsukamoto K, Urabe K, Aroca P, Kameyama K, Hearing VJ. The tyrosinase gene family–interactions of melanogenic proteins to regulate melanogenesis. Cell. Mol. Biol. Res. 1994;40:613–626. [PubMed] [Google Scholar]
- 14.Moss B, Earl PL. Expression of proteins in mammalian cells using vaccinia viral vector. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. Vol. 2. John Wiley and Sons, Inc.; New York: 1998. pp. 16.15.1–16.19.11. [Google Scholar]
- 15.Barth RJ, Jr., Mule JJ, Spiess PJ, Rosenberg SA. Interferon γ and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp. Med. 1991;173:647–658. doi: 10.1084/jem.173.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartikka J, Sawdey M, Cornefert-Jensen F, Margalith M, Barnhart K, Nolasco M, Vahlsing HL, Meek J, Marquet M, Hobart P, Norman J, Manthorpe M. An improved plasmid DNA expression vector for direct injection into skeletal muscle. Hum. Gene Ther. 1996;7:1205–1217. doi: 10.1089/hum.1996.7.10-1205. [DOI] [PubMed] [Google Scholar]
- 17.Ashton-Rickardt PG, Bandeira A, Delaney JR, Van Kaer L, Pircher HP, Zinkernagel RM, Tonegawa S. Evidence for a differential avidity model of T cell selection in the thymus. Cell. 1994;76:651–663. doi: 10.1016/0092-8674(94)90505-3. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, Wunderlich JR, Parkhurst MR, Kawakami Y, Seipp CA, Einhorn JH, White DE. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irvine KR, Rao JB, Rosenberg SA, Restifo NP. Cytokine enhancement of DNA immunization leads to effective treatment of established pulmonary metastases. J. Immunol. 1996;156:238–245. [PMC free article] [PubMed] [Google Scholar]
- 20.Bronte V, Tsung K, Rao JB, Chen PW, Wang M, Rosenberg SA, Restifo NP. IL-2 enhances the function of recombinant poxvirus-based vaccines in the treatment of established pulmonary metastases. J. Immunol. 1995;154:5282–5292. [PMC free article] [PubMed] [Google Scholar]
- 21.Pardoll DM. Inducing autoimmune disease to treat cancer. Proc. Natl. Acad. Sci. USA. 1999;96:5340–5342. doi: 10.1073/pnas.96.10.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merimsky O, Baharav E, Shoenfeld Y, Chaitchik S, Tsigelman R, Cohen-Aloro D, Fishman P. Anti-tyrosinase antibodies in malignant melanoma. Cancer Immunol. Immunother. 1996;42:297–302. doi: 10.1007/s002620050286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosato A, Zambon A, Milan G, Ciminale V, D'Agostino DM, Macino B, Zanovello P, Collavo D. CTL response and protection against P815 tumor challenge in mice immunized with DNA expressing the tumor-specific antigen P815A. Hum. Gene Ther. 1997;8:1451–1458. doi: 10.1089/hum.1997.8.12-1451. [DOI] [PubMed] [Google Scholar]
- 25.Schneider J, Gilbert SC, Blanchard TJ, Hanke T, Robson KJ, Hannan CM, Becker M, Sinden R, Smith GL, Hill AV. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat. Med. 1998;4:397–402. doi: 10.1038/nm0498-397. [DOI] [PubMed] [Google Scholar]