

Abstract
Antitumor T cells often recognize targets that are nonmutated “self” tissue differentiation Ags, but the relative impact of Ag expression by normal and transformed tissue for a human self/tumor Ag has not been studied. To examine the influence of self-tolerance mechanisms on the function of self/tumor-specific T cell responses in humans, we sought to identify an Ag that was expressed, processed, and presented in an MHC-restricted fashion by tumor cells, but for which there was the human equivalent of a “knockout.” In this study, we report the first immunological characterization of a melanoma/melanocyte differentiation Ag, called OA1, which meets these criteria. This Ag, an X chromosome-encoded melanoma/melanocyte differentiation Ag, was completely deleted in a male patient. Using a newly identified HLA-A*2402-restricted epitope (LYSACFWWL) to study T cell tolerance, we found that OA1-specific T cell reactivity was more than five SD higher in the knockout patient that in normal controls. These data provide compelling evidence for T cell tolerance to OA1 in humans. Most surprisingly, we found elevated levels of OA1-specific T cells in patients with metastatic malignant melanoma, indicating that the tumor-bearing state partially reversed tolerance observed in normal (non-“knockout”) individuals. Taken together, these findings indicated that tolerance can exist for self/tumor Ags in humans, and that this tolerance could be partially abrogated by the growth of the tumor, increasing the reactivity of tumor Ag-specific T cells. Thus, the tumor-bearing state reverses, in part, the tolerance of T cells that results from the normal expression of tissue differentiation Ags.
Tumor-specific T cells frequently recognize nonmutated “self” Ags that are expressed in both normal tissues and in growing tumor deposits (1, 2). These Ags may be useful as vaccine targets, and T cells targeted against them can cause tumor regression upon adoptive transfer (2, 3). However, T cells recognizing self/tumor Ags are potentially susceptible to the mechanisms of self-tolerance that can alter, suppress, or abrogate their functions. At the same time, interactions of these T cells with tumor cells expressing cognate Ags may also influence their activities. Many workers have asserted that tumor cells tolerize host T cells (4-7), although this reigning paradigm has recently been called into question (8).
Animal models form much of the foundation of our understanding of host-tumor immune interactions, but it is difficult to use models to gauge the relative impact of Ag expression by normal and transformed tissues on T cell function in humans. For example, it may be difficult to accurately model many aspects of the recognition of tumor/host interactions, including key aspects regarding Ag expression and affinity and precursor T cell frequencies. It may be difficult to even approximate in mice the impact of the chronic tumor-bearing state on host immunity, which in humans can last for years. However, the relative impact on T cell functionality of expression by normal and tumor cells in humans remains unelucidated.
To examine the influence of self-tolerance mechanisms on the function of self/tumor-specific T cell responses in humans, we needed to identify an Ag that was expressed, processed, and presented in an MHC-restricted fashion by tumor cells, but for which there was the human equivalent of a “knockout.” Ideally, such an Ag-loss variant would be a total loss of gene expression, such as a full-length deletion of the gene that would result in the complete absence of the targeted protein throughout development. Most known melanocyte differentiation Ag-loss variants are point mutations in the active sites of enzymes. One of the most common forms of albinism is characterized by point mutations of tyrosinase, a rate-limiting enzyme in the biosynthesis of the pigment melanin (9). Furthermore, for autosomal genes to be immunologically relevant, both alleles of a codominantly expressed gene must be deleted, a condition that is prohibitively rare. In males, a self-Ag encoded on the X chromosome would circumvent this problem, but no such X-linked melanoma/melanocyte differentiation Ag (MDA)2 had been described as a vaccine candidate.
In this study, we report the first immunological characterization of a melanoma/melanocyte differentiation Ag that meets these criteria. This Ag, called OA1, is the ocular albinism type I gene product and has previously been reported to be an integral endolysosomal membrane protein that may function as a G protein-coupled receptor (10, 11). We identified a patient with a human Ag-loss variant that completely lacked the expression of the OA1 target Ag. We then identified a target epitope restricted by one of the patient's HLA class I molecules (A*2402-restricted) and found it to be a shared self/tumor Ag, processed and presented by tumor cells derived from several other HLA-A*2402+ patients with malignant melanoma.
To experimentally address the complex question of the function and survival of autoreactive T cells in the tumor-bearing host, we first studied the impact of Ag expression by normal (nontransformed) tissue on the function of T cells specific for self/tumor Ags. This study represents the first published immunological analysis of a human self/tumor Ag knockout. To study how chronic Ag expression by transformed (tumor) tissue affected OA1-specific T cells, we compared T cell responses from normal individuals and melanoma patients. We found that while expression of the OA1 tissue differentiation Ag by normal melanocytes can partially tolerize specific T cells, the tumor can reverse, in part, these tolerizing influences.
Materials and Methods
HLA typing of PBMC and tumor lines
HLA serotypes and DNA genotypes of fresh human PBMC and tumor cell lines were determined by the National Institutes of Health HLA Laboratory (Bethesda, MD). Nine normal volunteers, five melanoma patients, and the OA1 patient were all found to be positive for HLA-A*2402. OA1 patients were anonymously contacted and invited to donate their PBMC after informed consent (National Institutes of Health protocol: 98C128). PBMC from melanoma patients were acquired before the administration of any form of immunotherapy. The HLA-DRB1* genotypes of tumor lines used in the following experiments included 526 Mel (0401, 1401); 624 Mel (0401, 0701); 697 EBV-B (0401, 1501); 888 Mel (1502, 1601); 1088 Mel and EBV-B (0301, 0401). The HLA-A* genotypes of tumor lines used included 397 Mel (01, 25); 501 Mel (0201, 2402); 624 Mel (0201, 03); 526 Mel (0201, 03), 1088 Mel (01, 0201), 888 Mel and EBV-B (01, 2402). All T cells and tumor lines were maintained in culture media as previously described (12).
Cloning of full-length OA1 gene
The full-length OA1 gene was amplified from cDNA generated from neonatal melanocyte line 9F1966 (Clonetics, Walkersville, MD) by PCR using primers containing a forward-EcoRI restriction site (position 1; GCGG AATTCCACCATGGCCTCCCCGCGCCTAGG), and a reverse-NotI restriction site (position 1409; AAGGCGGCCGCGCTGGTGATGAGAG CAAGGT). Amplification of the 1409-bp product (1212 bp of which is coding) was performed using 5% DMSO, 25 μg of BSA, and Platinum Pfx DNA Polymerase (Life Technologies, Rockville, MD) under the following reaction conditions: one cycle at 94°C for 2 min; 35 cycles at 94°C for 15 s, 55°C for 30 s, 68°C for 2 min using conditions designed to abrogate inhibition of PCR by melanin (13). The purified product was subcloned into a ZeroBlunt vector (Invitrogen, Carlsbad, CA), and then transferred into a high-expression plasmid (VR1012) with a CMV-based promoter (Vical, San Diego, CA). The product was then sequenced with a ABI Prism 310 Genetic Analyzer (PerkinElmer, Foster City, CA) and found to be identical with the nucleotide sequence accessioned on GenBank (no. Z48804). Functionality of the OA1 plasmid was then confirmed by transient transfection into human fibroblast line 888F followed by RT-PCR using OA1-specific primers (not shown).
Northern and Southern blotting
For Northern blot, 10 μg of total RNA from 15 normal tissues–brain, heart, kidney, liver, lung, mammary gland, pancreas, placenta, prostate, skeletal muscle, spleen, stomach, testis, thymus, and uterus (Clontech, Palo Alto, CA)–and from neonatal melanocyte line 9F1966 (prepared as above) were separated by gel electrophoresis and transferred to a positively charged nylon membrane using standard techniques. For Southern blot, genomic DNA from OA1 patients OAP-24, OAP-46, and a control male were prepared from autologous EBV-B cells with a Genomic DNA Purification Kit (Promega, Madison, WI) according to manufacturer's instructions. Fifteen micrograms of DNA were separated by gel electrophoresis following an overnight digest with 150 U of PstI and transferred to a positively charged nylon membrane using standard techniques. Blots were incubated with salmon-sperm DNA and 32P-labeled full-length OA1 or β-actin probes.
Peptides and binding affinity assay
Peptides were synthesized using a solid phase method based on fluorenylmethoxycarbonyl (F-moc) chemistry on a multiple peptide synthesizer (Model AMS 422; Gilson, Worthington, OH). The molecular masses of peptides were verified by laser desorption mass spectrometry (Biosynthesis, Lewisville, TX). To measure the binding affinity of HLA-A*2402 peptides, purified human A*2402 molecules (5–15 nM) were incubated with 1–10 nM 125I-radiolabeled probe peptide, iodinated by the Chloramine T method, for 48 h at room temperature in the presence of 1 μM human β2-microglobulin (Scripps Laboratories, San Diego, CA), and a mixture of protease inhibitors (14). Class I peptide complexes were separated from free peptide by gel filtration on TSK2000 columns (Tosohaus, Montgomeryville, AL), and the fraction of bound peptide was calculated as previously described (14). In preliminary experiments, the HLA class I preparation was titered in the presence of fixed amounts of radiolabeled peptides to determine the concentration of class I molecules necessary to bind 10–20% of the total radioactivity. Using an inhibition assay to measure binding affinity, candidate peptides were tested at concentrations ranging from 120 μg/ml to 1.2 ng/ml. The data were plotted and the dose yielding IC50 was measured. Peptides were tested in three completely independent experiments because under these conditions (label) < (MHC) and (IC50) ≥ (MHC), the measured IC50s are reasonable approximations of the true KD values. A consensus peptide (sequence AYIDNYNKF) was used as the standard control and radiolabeled probe for the A*2402 binding assay. The average IC50 level of the control peptide was 12 nM.
Generation of human T cell lines
All studies were done in accordance with Institutional Review Board-approved protocols. To generate OA1-specific T cells, a list of the 10 most theoretically avid peptide binders for HLA-A*2402 from the OA1 protein was generated using an established computer-assisted, allele-specific epitope binding forecast algorithm (http://bimas.dcrt.nih.gov/molbio/hla_bind/). Following an RMA-S-A*2402 stabilization assay, five peptides (originally ranked 1, 2, 3, 5, 6) were selected for further analysis by in vitro sensitization (not shown). Fresh PBMC from melanoma patient SE were cultured as above except that peptide was added at 10 μM. Again, 13 days after the first restimulation, all 48 wells for each peptide from the original 96-well plates were tested for specific peptide reactivity using 888 EBV-B cells (A*2402+) pulsed with either a control peptide from β-catenin or the specific peptide in question. Only one peptide (ranked number 3; LYSACFWWL; positions 126–134) was found to induce peptide-specific T cells, and the most reactive wells were selected and expanded with autologous, irradiated, pulsed (as above) PBMC in 24-well flat-bottom plates at a 10:1 ratio (5 × 106 APCs per well). Following the fourth stimulation, one line, SE OA1-3 2/6, was selected as the most optimal and tested against multiple targets. To evaluate the T cell responsiveness, 1 × 105 T cells were cocultured in duplicate with 105 APC per well (peptide pulsed 888 EBV-B's or tumor cells) in U-bottom 96-well plates for 24 h. Culture supernatants were assayed for IFN-γ using commercially available ELISA kits (Endogen, Woburn, MA). All experiments were performed between two and four times with similar results, using in some cases different, but comparable, targets.
Assessment of human T cell responses
To evaluate human T cell lines or clones, EBV-B cells or activated B cells were incubated for 5–18 h with peptide, protein, or with freeze-thaw ly-sates of tumor cells (105 cell equivalents per well). Whole tumor cells or pulsed APC (105 cells per well) were cocultured with 105 T cells per well in U-bottom 96-well plates for 24 h. In both cases, culture supernatants were assayed for either IFN-γ or GM-CSF using commercially available ELISA kits (Endogen, Woburn, MA).
Comparative in vitro sensitization assay
For analysis of OA1 reactivity, PBMC from nine normal volunteers, three melanoma patients, and one OA1 patient, OAP-46, were plated at 6 × 106 per well in four wells in 24-well plates with either OA1126–134, tyrosinase 205–213, or mutated β-catenin 29–37 at 10 μM (15, 16). Seven days later, individual wells from each peptide for each sample were pooled, counted, and incubated with targets. To evaluate T cell responsiveness, 2 × 105 T cells were cocultured in duplicate with 105 APC per well (888 EBV-Bs pulsed with peptide or with OKT3, 1 μg/ml) in U-bottom 96-well plates for 24 h. Culture supernatants were assayed for IFN-γ using commercially available ELISA kits (Endogen). The IFN-γ stimulation index (IFN-γ-SI) was generated by dividing the IFN-γ secretion produced to experimental antigen against secretion produced against the control Ag. Assays were performed two to three times with similar results, using different, but comparable targets.
Cytotoxic T cell assay
51Cr-release assays were performed as previously described (12). Briefly, target cells pulsed with 200 μCi 51Cr for 60 min were distributed onto U-bottom 96-well plates at 104 cells per well with effector T cells added at varying E:T ratios and incubated for 4 h. Percent lysis was calculated using the standard formula: ((experimental51Cr release − spontaneous release)/(total release − spontaneous release)) × 100. The experiment was performed three times with similar results.
Results
Studying T cell tolerance to the OA1 self/tumor Ag-loss variant
To study tissue expression of OA1, we performed Northern blot analysis on mRNA obtained from 16 normal tissues, verifying that the probe hybridized at 1.6 kb, the predicted size of the OA1 message (Fig. 1A). Specific hybridization was only observed for mRNA derived from melanocytes (lane 16). OA1 expression was analyzed in tumor cells using RT-PCR. OA1 expression was found in six of six melanomas tested, although one of these was relatively weak. No OA1 expression was found in other tested tumor histologies (Fig. 1B). Thus, OA1 appears to be specifically expressed only in melanocytes and in melanoma cells, which is consistent with a report demonstrating high OA1-RNA levels only in pigmented cells, specifically retinal pigment and melanoma (17).
FIGURE 1.
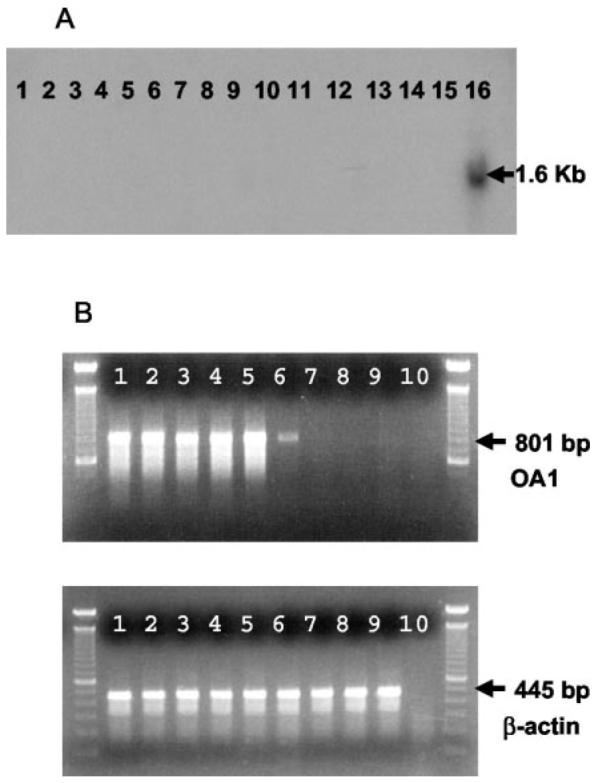
Analysis of OA1 in normal tissues and in tumor. A, Northern blot demonstrates specific expression of OA1 in melanocyte mRNA. Total RNA from 16 normal tissues (brain, heart, kidney, liver, lung, mam-mary gland, pancreas, placenta, prostate, skeletal muscle, spleen, stomach, testis, thymus, uterus, and melanocyte; lanes 1–16) was assayed with a full-length OA1 32P-labeled probe. Specific hybridization at 1.6 kb was observed for melanocyte mRNA only (lane 16). B, OA1 gene expression in tumor cells. RT-PCR was performed with OA1- and β-actin-specific primers on six different melanoma lines (397, 501, 526, 624, 888, 1102; lanes 1–6), one breast carcinoma line (MDA-231; lane 7), one colon carcinoma line (SW-480; lane 8), and one EBV-B line (1359 EBV-B; lane 9). Specific amplification of OA1 was observed (801-bp product) for all melanoma lines, but not for nonmelanoma lines and control water sample (lane 10). Control β-actin expression (445-bp product) was observed for all samples.
To study immune tolerance in humans, we sought to identify individuals in which OA1 expression was completely absent. The Nettleship Falls ocular albinism, also known as ocular albinism type I, is associated with the functional absence of functional OA1 and is characterized by reduced visual acuity, foveal hypoplasia, retinal hypopigmentation, congenital nystagmus, and photophobia (17). Nettleship Falls ocular albinism can be caused by a variety of mutations within the X chromosome-encoded OA1 gene, including frameshifts, missense, and nonsense mutations, frequent large intragenic deletions and less frequent whole gene deletions (18, 19). Several males from a large cohort of OA1 gene-loss patients were selected for analysis because they were known or suspected to have large intragenic OA1 deletions. Three of these patients were found to express prevalent HLA-A* alleles after HLA genotyping: patient OAP-23, A*03,25; patient OAP-24, A*01,03; and patient OAP-46, A*2402,68. To evaluate the extent of the OA1 deletion, genomic DNA from patient OAP-46 and OAP-24 and a control volunteer were digested with PstI, separated electrophoretically, and then hybridized with a 32P-labeled full-length OA1 probe. Both of these patients had large genetic deletions of OA1: patient OAP-24 was shown to have only a deletion of exon 2–8 which was consistent with a previously published report (17), while patient OAP-46 was found to have a complete OA1 gene deletion with no hybridization detected to any of the nine exons when compared with a control male sample (Fig. 2A). Based on these and the previously reported results, patient OAP-46 was chosen for additional studies and consented to undergo leukapheresis at the National Institutes of Health. A fundoscopic photograph from OA1 patient OAP-46 compared with a normal volunteer revealed phenotypically characteristic changes including retinal hypopigmentation and foveal hypoplasia (Fig. 2B).
FIGURE 2.
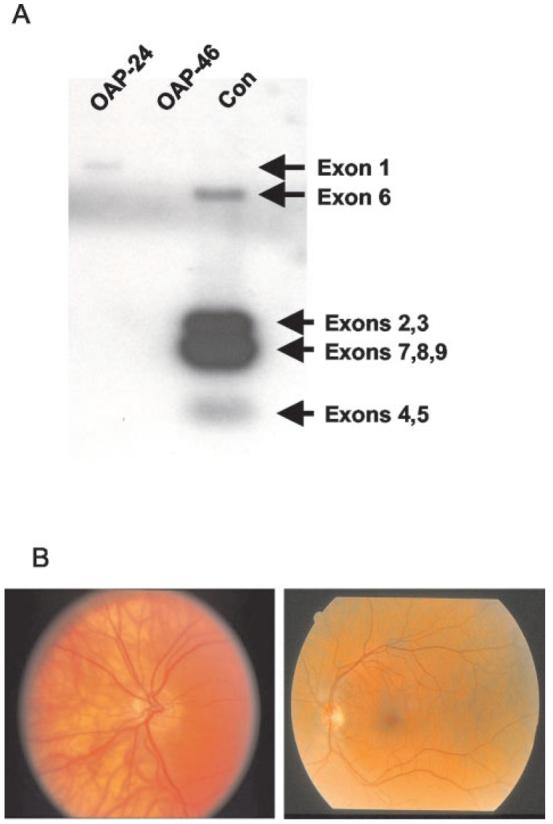
Analysis of genotype and phenotype of an Ag-loss variant patient. A, Volunteer OAP-46 has a full-length OA1 gene deletion. Southern blot of PstI-digested DNA from OA1 patients and a control male, separated electrophoretically, then hybridized with a full-length 32P-labeled OA1 cDNA probe. Patient OAP-46 had a full-length deletion spanning exons 1–9 confirming previous findings (17). All samples were positive for the β-actin probe (not shown). Note that hybridization with Exon 1 is only visible in the sample from patient OAP-24 (who was excluded from further immunological analysis), but not the sample from the control volunteer, presumably because the exons that are present on the blot compete for the labeled probe. The fragment from Exon 9 in the unused subject OAP-24, although present in this patient (as reported elsewhere, Ref.17), is not visible in the blot shown here. B, Fundoscopic photograph of an OA1-Ag-loss variant volunteer OAP-46 (left panel) showing characteristic hypopigmentation on retinal exam. That of a normal volunteer is shown on the right panel for comparison.
The study of immune responses to OA1 first required the identification of a T cell epitope. Approximately 20% of all Caucasians express the HLA-A24 allele—most (97%) of those seen at the National Cancer Institute with melanoma express the A*2402 sub-type (20). The top nine HLA-A*2402 binding peptides of OA1, as predicted by allele-specific epitope forecasting (http://www.bimas.dcrt.nih.gov/molbio/hla_bind/index.html) were synthesized and an HLA-A*2402-transfected RMA-S stabilization assay (21) was used to identify the five best actual binding peptides (not shown). One of these (LYSACFWWL; ranked number 3, positions 126–134) could be used to elicit HLA-A*2402-restricted T cells from a melanoma patient (SE) that were capable of specific peptide recognition at subnanomolar concentrations (Fig. 3A). In a competition assay, the ability of OA1126–134 to form stable complexes with HLA-A*2402 was similar to other known epitopes derived from tumor and viruses (Fig. 3B). The most reactive T cell microcultures stimulated with OA1126–134 were restimulated with peptide-pulsed autologous human PBMC. Following the fourth stimulation, line SE OA1-3 2/6 was found to recognize A*2402+/OA1+ melanomas (501 and 888 Mel), but not A*2402−/OA1+ melanoma lines (397 and 624 Mel), nor did it recognize the control peptide (mutated β-catenin 29–37) pulsed onto 888 EBV-B (Fig. 4A). This and all other OA1-specific T cell lines were >90% CD8+ as determined by flow cytometry (not shown). Although growth of these OA-specific T cells required at least four ex vivo stimulations with Ag, the function of OA1-specific T cells shows for the first time that an OA1 peptide epitope is processed and presented on the surface of melanoma cells for recognition by Ag-specific T cells. OA1 is thus a potential target for melanoma vaccines. Recognition of the Ag is not only associated with cytokine production by the OA1-specific T cell. In a standard 51Cr-release assay, a cell line derived by repeated stimulation of PBMC from the OA1-negative patient OAP-46 was able to specifically lyse A*2402+/OA1+ melanoma 888, but not A*2402−/OA1+ melanoma 624 mel (Fig. 4B).
FIGURE 3.
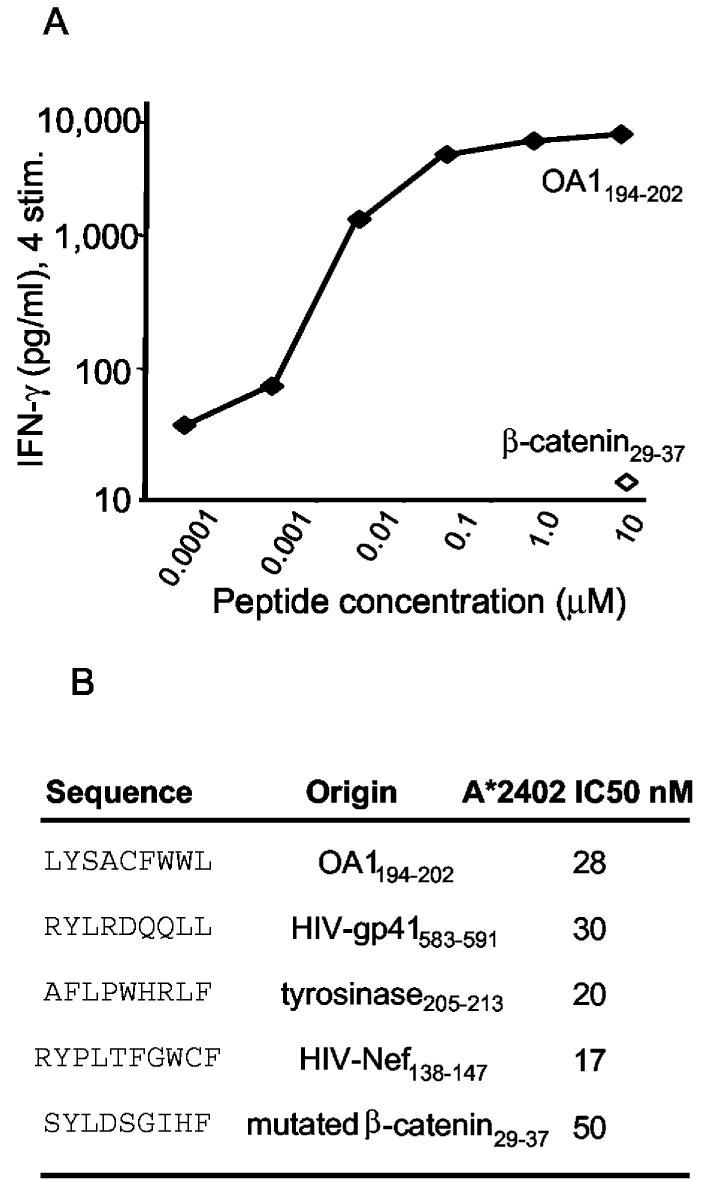
A, Recognition of OA1126–134 peptide-pulsed A*2402+ EBV-B cells (888 EBV-B) by CD8+ T cell line SE OA1-3 2/6, following the fourth stimulation. B, The binding affinity of the OA1 epitope to HLAA*2402 was equivalent to both HIV-derived peptides and both melanoma-derived peptides (mutated β-catenin and tyrosinase).
FIGURE 4.
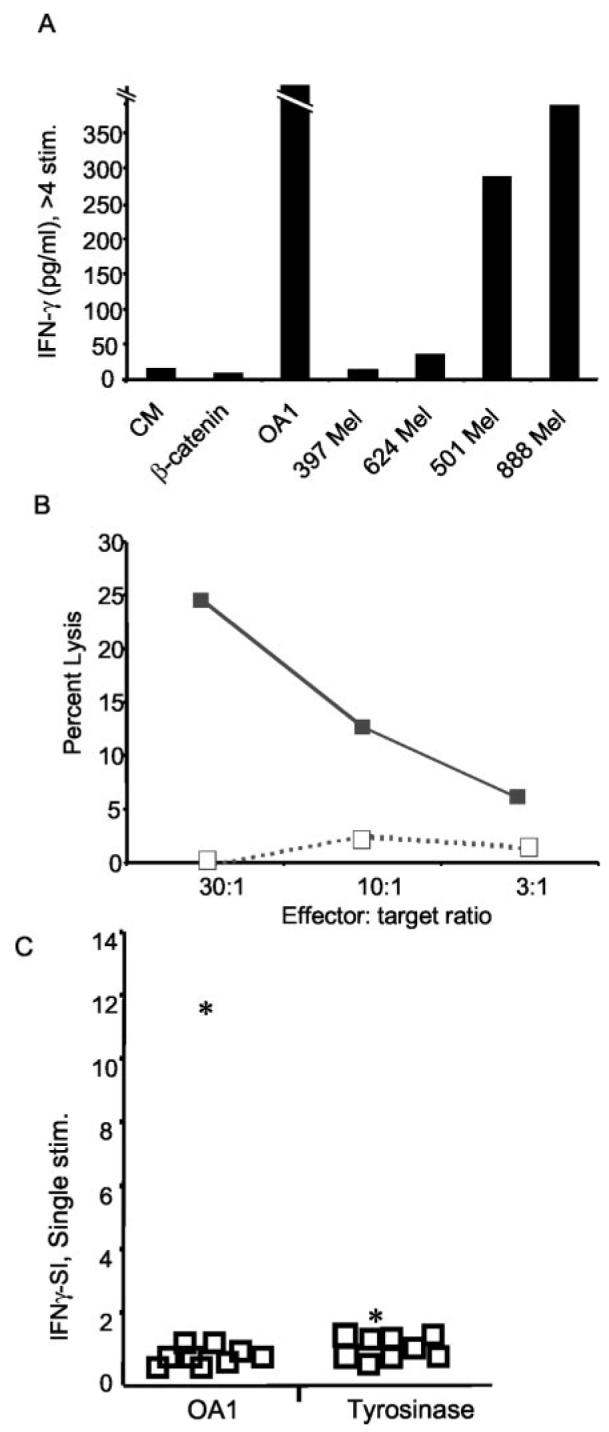
T cells from the OA1 Ag-loss variant patient avidly respond to OA1 and recognize melanoma. A, Long-term CD8+ T cells from line SE OA1-3 2/6, generated by four or more in vitro stimulations with OA1126–134, recognize OA1 peptide and A*2402+/OA1+ melanoma lines (501 and 888 Mel), but not A*2402−/OA1+ melanoma lines (397 and 624 Mel), nor the control peptide (mutated β-catenin29–37) pulsed onto 888 EBV-B. B, T cell line obtained by repeated stimulation of PBMC from patient OAP-46 with peptide OA1126–134 lyse A*2402+/OA1+ melanoma 888 (■), but not A*2402−/OA1+ melanoma 624 mel (□) in a 4-hour 51Cr-release assay. C, T cells from patient OAP-46 have superior reactivity to OA1. T cells from patient OAP-46 react with OA1126–134, but not tyrosinase 205–213, while T cells from normal volunteers and melanoma patients lack reactivity to both Ags. PBMC from nine normal volunteers (□) and OA1 patient OAP-46 (*) were stimulated for 7 days in vitro with either OA1126–134 or tyrosinase 205–213. To evaluate responses from all samples to both Ags, T cells were stimulated with OKT3 (anti-CD3), control peptide (mutated β-catenin 29–37 pulsed at 10 μM), or the specific peptide in question (either OA1126–134 or tyrosinase 205–213 also pulsed at 10 μM) both pulsed onto 888 EBV-B. All lines secreted IFN-γ in response to OKT3 (not shown). The IFN-γ-SI was generated by dividing the IFN-γ secretion produced to OA1126–134 or tyrosinase 205–213 against the secretion produced against mutated β-catenin 29–37. Experiments in all panels were performed three times with similar results, using different but comparable targets to demonstrate the generalizability of the findings. CM, culture media.
To determine whether the OA1 Ag induces tolerance in normal individuals, we elicited epitope-specific T cells from nine normal HLA-A*2402+ volunteers and compared them to the OA1-Ag-loss variant patient, OAP-46, using the newly identified epitope as an experimental self- and nonself-Ag. An HLA-A*2402-restricted epitope from tyrosinase, corresponding to aa 205–213, served as a control self/tumor Ag for all subjects. All cell preparations were found to be strongly reactive to anti-CD3 Ab (not shown).
The greatest difference in OA1126–134-specific T cell reactivity between the OA1-Ag-loss variant patient and the normal volunteers was observed after a single in vitro stimulation (Fig. 4C). This difference was more than five SD, generating a confidence interval of 99.9% in three of three independently performed experiments.
Significant differences could also be observed after the second round of stimulation, where OA1126–134-specific reactivity could be elicited in cells derived from three of nine normal volunteers with IFN-γ-SIs that ranged from 3–10 whereas in the same assay, the IFN-γ-SI in the OA1-Ag-loss variant patient, OAP-46, was 183 (as discussed below). Note that after four or more in vitro stimulations, OA1126–134-specific T cells could be generated out of normal volunteers (Fig. 5B). Taken together, these data provide compelling evidence for T cell tolerance to OA1 in humans.
FIGURE 5.

Ag expressed by tumor primes low-avidity T cells. A, T cell cultures generated after a single in vitro stimulation are peptide reactive: T cells from patient MG react with mutated β-catenin 29–37 and cells from patient OAP-46 react with OA1126–134. PBMC from melanoma patient MG (▲), OA1 KO patient OAP-46 (*), melanoma patient SE (●), and two normal volunteers (□) were stimulated for 7 days in vitro with mutated β-catenin 29–37. To evaluate responses from all samples, T cells were stimulated with either OKT3 1 μg/ml, control peptide (tyrosinase 205–213) or peptide. The IFN-γ-SI was generated by dividing the IFN-γ secretion produced to mutated β-catenin29–37against the secretion produced against the control peptide (tyrosinase 205–213). B, OA1-specific T cells generated by two in vitro OA1 peptide stimulations from melanoma patients can react to OA1 peptide. A comparative in vitro sensitization assay was performed with PBMC from four DR4+ normal volunteers (□), five DR4+ melanoma patients (●), and the OA1 KO patient (*) as a positive control. IFN-γ release was then measured in a 24-h coculture with EBV-B cells pulsed with β-catenin or OA1-peptide. The IFN-γ-SI was calculated based on the background reactivity to β-catenin at 10 μg/ml. C, The same OA1-specific T cells from melanoma patients, stimulated two times in vitro, do not recognize melanoma. T cells from the same individuals as above were stimulated in vitro with OA1126–134 as described in B and their reactivity to 888 mel was measured in a 24-h coculture assay followed by IFN-γ ELISA. The IFN-γ-SI was calculated based on the background-reactivity to the A*2402− line 624 mel. Experiments in all panels were performed between two and four times with similar results, using different but comparable targets to demonstrate the generalizability of the findings.
Impact of the tumor-bearing state on T cells with specificity for a “self” tumor Ag or “foreign” tumor neoantigen
Tumors have been reported to depress T cell responses locally and systemically, through a diversity of mechanisms (4-7, 22, 23). One patient (MG) enabled us to study immune responses to a mutated Ag expressed by her tumor cells. MG's tumor expressed a single point mutation in β-catenin, which resulted in an amino acid change in an MHC anchor position yielding a heteroclitic peptide that was recognized by autologous T cells in an HLA-A*2402-restricted fashion at concentrations that were 1 million-fold lower than the nonmutated peptide (16). We then compared the levels of Ag-specific T cell reactivities found in MG to those found in non-tumor-bearing controls using an in vitro sensitization assay in which all patients were verified to be HLA-A*2402-positive.
We found that after a single in vitro stimulation, only melanoma patient MG had significant reactivity to the β-catenin epitope (Fig. 5A). By contrast, β-catenin-specific T cell reactivity was not significantly higher than background in two normal volunteers and the OA1-Ag-loss variant patient, OAP-46. Nor were increased levels of β-catenin-specific T cells found in a patient with metastatic melanoma whose tumor did not express the mutated β-catenin epitope.
Results from patient MG indicated that tumor expression of a neoantigen is capable of priming a T cell response, but many of the tumor-associated Ags identified thus far are nonmutated self-Ags (1, 2). This prompted us to compare T cell responses to OA1 in normal and melanoma patients, where the additional comparison to an individual with no expression of the Ag in normal tissue was possible.
OA1126–134-specific T cell reactivity was poor in normal volunteers after a single in vitro stimulation (Fig. 5A). Reactivity to OA1126–134 remained poor in normal volunteers after an additional in vitro stimulation, that is after a total of two in vitro stimulations, while cells from the patient with OA1-deficiency yielded an SI of 183 (Fig. 5B). Patients with melanoma, whose tumors were confirmed to express OA1, were found to have variable responses to OA1 peptide, with three of five patients showing strong reactivity to OA1 after two in vitro stimulations (Fig. 5B). Note that one patient had reactivity at levels comparable to those seen in the Ag-loss variant patient. Parallel to the IFN-γ-release assay, ELISPOT analysis was performed to determine the number of cytokine-producing cells. The numbers obtained correlated with cytokine levels detected by ELISA, thus confirming the presence of OA1-reactive T cells not only in an Ag-knockout situation, but also in normal melanoma patients (not shown). However, when we assessed the abilities of OA1126–134-specific T cells to recognize tumor cells in the same assay (Fig. 5C), we found that only the OA1-deficient patient had T cells that were capable of specifically recognizing melanoma. Neither normal volunteer nor tumor-bearing patients had T cells that recognized Ag expressed by tumor. This indicated that although tolerance to self/tumor Ags could be abrogated by the growth of the tumor, increasing the reactivity of tumor-specific T cells, any reversal of tolerance might be only partial.
Discussion
Murine models are often used to address tolerance to surrogate tumor Ags, but the important question of tolerance to self/tumor Ags in humans has not previously been addressed. The use of Ag-loss variants conclusively distinguishes immunological tolerance from other mechanisms that may account for the poor immunogenicity of any given antigenic epitope including poor expression of the target protein, inefficient Ag processing (24, 25), poor MHC binding of an epitope (26), or insufficiencies in the T cell repertoire not deriving from central or peripheral tolerance mechanisms. Using a novel human OA1 Ag-loss variant, we demonstrate in this study for the first time that immune tolerance indeed plays a major role in poor reactivity to this particular self/tumor Ag. Thus, poor apparent immunogenicity of the target epitope is derived not from the intrinsic qualities of the target peptide epitope, but instead is a consequence of a decrement in T cell functionality that is attributable to Ag expression in normal tissues.
It has been hypothesized that tolerance mechanisms could result in the deletion of the T cell repertoire specific for the highest binding peptides, thus resulting in a “reversal” of the immunodominance hierarchy (27) and deletion of T cells specific for the highest MHC-binding epitopes. Our results indicate that deletion is not the primary mechanism of tolerance induction for the OA1 epitope, although we cannot exclude deletion of some OA1-specific T cells. However, our data clearly show that OA1-specific T cells are not completely deleted in OA1+ individuals, but rather are expanded in melanoma patients. The presence of OA1-specific T cell reactivity in the OA1 knockout patient serves in large part as a positive control for the analysis because it indicates that OA1-specific T cell reactivity is greater in the absence of the self-Ag. Taken together, our data is most consistent with the notion that OA1-specific T cells are partially inactivated in normal (OA1+) individuals (i.e., T cell reactivity to a high binding self/tumor epitope was preserved and not deleted).
Short-term cultured T cells from the Ag-loss variant patient were far more reactive than those of normal controls (Fig. 5B). The reasons for the lower functionality of T cells in Ag bearing vs Ag-negative individuals could be due either to anergy of T cells, which may be reversed ex vivo with time by culture of the cells, or to the systematic elimination of T cells bearing high affinity receptors. These mechanisms are not mutually exclusive and both may contribute to the observations made in this report. Both mechanisms are in evidence in published experimental models: high-avidity T cells directed against tyrosinase could be isolated from mice despite the presence of the Ag in vivo (28), suggesting a nondeletional mechanism for T cell tolerance, such as anergy, that was the result from signal 1 in the absence of signal 2 or anergy that is enforced by regulatory CD4+ T cells (29, 30).
The ability to ultimately obtain high-avidity cells after multiple in vitro stimulations does not conclusively indicate a mechanism involving anergy, but may instead reflect a highly significant yet incomplete deletion of precursor T cells. Others have highlighted a deletional mechanism, which results in the elimination of high-avidity T cells and skewing of the T cell repertoire in wild-type vs transgenic or Ag-loss variant mice (31, 32). It is possible that, in addition to simple differences in interpretation of the data, the different experimental systems are responsible for apparent discrepancies in the findings.
Our findings conclusively establish that even the highly tissue-restricted expression of “peripheral” Ags can have a profound impact on the T cell repertoire. It was initially difficult to reconcile this conclusion with the observation that T cells specific for self-tissue differentiation Ags are frequently isolated from tumors. Therefore, we sought to systematically examine the impact of tumor on the state of immunological tolerance that is established by the expression of Ag by normal tissue. Many workers have asserted that the tumor-bearing state only exacerbates immunological unresponsiveness seen as a result of expression in normal tissues (4-6, 32), while others claim that tumors release soluble immunosuppressive cytokines such IL-10 and TGF-β (7, 33) or express death receptor ligands such as TRAIL (34) or FasL, although aspects of these reports have been disputed (35). In contrast to these reports, we detected increased T cell reactivity to mutated β-catenin but only in a patient whose tumor expressed that particular mutation (Fig. 5A) (16). We also found increased T cell reactivity to the self-Ags tyrosine-related protein-1 (36) and OA1 (Fig. 5B) in patients with metastatic melanoma, which constitutively express these Ags. This is consistent with previously reported elevations in T cells specific for melanoma Ag recognized by T cells-1, another nonmutated MDA (37). We do not yet understand the mechanisms underlying activation of the immune system by tumors when the response had been suppressed by normal tissue expressing the same Ag, although it has recently been suggested in a mouse model that the mechanism involves increased cross-presentation (8).
If tumor cells lead to expanded T cell precursors that are more rather than less functionally reactive, why does T cell stimulation by the tumor not result in the rejection of melanoma in patients? To address this paradoxical issue, we tested the reactivity of OA1-peptide stimulated T cells to melanoma. In contrast to the peptide-pulsed target, T cells from melanoma patients in short term cultures did not recognize intact tumor cells, which were well recognized and lysed by T cells from the OA1 Ag-negative individual. The findings presented in this study help to explain the apparent paradox of tumor priming of T cells in the face of continued tumor growth in melanoma patients: expression of a given Ag in normal tissue suppressed T cell responses while expression of the same target Ags by tumor resulted in the expansion of T cell precursor populations. Thus, a key challenge in immunotherapeutic strategies that target self/tumor Ags is how to fully activate T cells whose encounter with Ag expressed by tumor has already resulted in a partial reversal of self tolerance.
Acknowledgments
We thank P. Lehmann, V. J. Hearing, S.J. Orlow, R.W. Nowakowski, and A.V. Levin for helpful advice and their assistance in obtaining critical reagents and patients and F. M. Marincola for HLA genotyping of patients.
Footnotes
Abbreviations used in this paper: MDA, melanoma/melanocyte differentiation Ag; SI, stimulation index.
References
- 1.Overwijk WW, Lee DS, Surman DR, Irvine KR, Touloukian CE, Chan CC, Carroll MW, Moss B, Rosenberg SA, Restifo NP. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA. 1999;96:2982. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA. Progress in human tumour immunology and immuno-therapy. Nature. 2001;411:380. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 3.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998;188:277. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staveley-O'Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, Pardoll D, Levitsky H. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA. 1998;95:1178. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizoguchi H, O'Shea JJ, Longo DL, Loeffler CM, McVicar DW, Ochoa AC. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science. 1992;258:1795. doi: 10.1126/science.1465616. [DOI] [PubMed] [Google Scholar]
- 6.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 1999;5:677. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 7.Seo N, Hayakawa S, Takigawa M, Tokura Y. Interleukin-10 expressed at early tumour sites induces subsequent generation of CD4+ T-regulatory cells and systemic collapse of antitumour immunity. Immunology. 2001;103:449. doi: 10.1046/j.1365-2567.2001.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen LT, Elford AR, Murakami K, Garza KM, Schoenberger SP, Odermatt B, Speiser DE, Ohashi PS. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J. Exp. Med. 2002;195:423. doi: 10.1084/jem.20010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toyofuku K, Wada I, Spritz RA, Hearing VJ. The molecular basis of oculocutaneous albinism type 1 (OCA1): sorting failure and degradation of mutant tyrosinases results in a lack of pigmentation. Biochem. J. 2001;355:259. doi: 10.1042/0264-6021:3550259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samaraweera P, Shen B, Newton JM, Barsh GS, Orlow SJ. The mouse ocular albinism 1 gene product is an endolysosomal protein. Exp. Eye Res. 2001;72:319. doi: 10.1006/exer.2000.0962. [DOI] [PubMed] [Google Scholar]
- 11.Schiaffino MV, d'Addio M, Alloni A, Baschirotto C, Valetti C, Cortese K, Puri C, Bassi MT, Colla C, De Luca M, et al. Ocular albinism: evidence for a defect in an intracellular signal transduction system. Nat. Genet. 1999;23:108. doi: 10.1038/12715. [DOI] [PubMed] [Google Scholar]
- 12.Touloukian CE, Leitner WW, Topalian SL, Li YF, Robbins PF, Rosenberg SA, Restifo NP. Identification of a MHC class II-restricted human gp100 epitope using DR4-IE transgenic mice. J. Immunol. 2000;164:3535. doi: 10.4049/jimmunol.164.7.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giambernardi TA, Rodeck U, Klebe RJ. Bovine serum albumin reverses inhibition of RT-PCR by melanin. BioTechniques. 1998;25:564. doi: 10.2144/98254bm03. [DOI] [PubMed] [Google Scholar]
- 14.Sette A, Buus S, Colon S, Miles C, Grey HM. Structural analysis of peptides capable of binding to more than one Ia antigen. J. Immunol. 1989;142:35. [PubMed] [Google Scholar]
- 15.Kang X, Kawakami Y, El-Gamil M, Wang R, Sakaguchi K, Yannelli JR, Appella E, Rosenberg SA, Robbins PF. Identification of a tyrosinase epitope recognized by HLA-A24-restricted, tumor-infiltrating lymphocytes. J. Immunol. 1995;155:1343. [PubMed] [Google Scholar]
- 16.Robbins PF, El-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, Rosenberg SA. A mutated β-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 1996;183:1185. doi: 10.1084/jem.183.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bassi MT, Schiaffino MV, Renieri A, De Nigris F, Galli L, Bruttini M, Gebbia M, Bergen AA, Lewis RA, Ballabio A. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat. Genet. 1995;10:13. doi: 10.1038/ng0595-13. [DOI] [PubMed] [Google Scholar]
- 18.Bassi MT, Bergen AA, Bitoun P, Charles SJ, Clementi M, Gosselin R, Hurst J, Lewis RA, Lorenz B, Meitinger T, et al. Diverse prevalence of large deletions within the OA1 gene in ocular albinism type 1 patients from Europe and North America. Hum. Genet. 2001;108:51. doi: 10.1007/s004390000440. [DOI] [PubMed] [Google Scholar]
- 19.Schnur RE, Gao M, Wick PA, Keller M, Benke PJ, Edwards MJ, Grix AW, Hockey A, Jung JH, Kidd KK, et al. OA1 mutations and deletions in X-linked ocular albinism. Am. J. Hum. Genet. 1998;62:800. doi: 10.1086/301776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bettinotti MP, Norris RD, Hackett JA, Thompson CO, Simonis TB, Stroncek D, Marincola FM. Frequency of human leukocyte antigen-A 24 alleles in patients with melanoma determined by human leukocyte antigen-A sequence-based typing. J. Immunother. 2000;23:282. doi: 10.1097/00002371-200003000-00013. [DOI] [PubMed] [Google Scholar]
- 21.Oiso M, Eura M, Katsura F, Takiguchi M, Sobao Y, Masuyama K, Nakashima M, Itoh K, Ishikawa T. A newly identified MAGE-3-derived epitope recognized by HLA-A24-restricted cytotoxic T lymphocytes. Int. J. Cancer. 1999;81:387. doi: 10.1002/(sici)1097-0215(19990505)81:3<387::aid-ijc12>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 22.Shevach EM, McHugh RS, Thornton AM, Piccirillo C, Natarajan K, Margulies DH. Control of autoimmunity by regulatory T cells. Adv. Exp. Med. Biol. 2001;490:21. doi: 10.1007/978-1-4615-1243-1_3. [DOI] [PubMed] [Google Scholar]
- 23.Finke J, Ferrone S, Frey A, Mufson A, Ochoa A. Where have all the T cells gone: mechanisms of immune evasion by tumors. Immunol. Today. 1999;20:158. doi: 10.1016/s0167-5699(98)01435-2. [DOI] [PubMed] [Google Scholar]
- 24.Restifo NP, Esquivel F, Kawakami Y, Yewdell JW, Mule JJ, Rosenberg SA, Bennink JR. Identification of human cancers deficient in antigen processing. J. Exp. Med. 1993;177:265. doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Restifo NP, Bacik I, Irvine KR, Yewdell JW, McCabe BJ, Anderson RW, Eisenlohr LC, Rosenberg SA, Bennink JR. Antigen processing in vivo and the elicitation of primary CTL responses. J. Immunol. 1995;154:4414. [PMC free article] [PubMed] [Google Scholar]
- 26.Van den Eynde BJ, Morel S. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr. Opin. Immunol. 2001;13:147. doi: 10.1016/s0952-7915(00)00197-7. [DOI] [PubMed] [Google Scholar]
- 27.Sercarz EE, Lehmann PV, Ametani A, Benichou G, Miller A, Moudgil K. Dominance and crypticity of T cell antigenic determinants. Annu. Rev. Immunol. 1993;11:729. doi: 10.1146/annurev.iy.11.040193.003501. [DOI] [PubMed] [Google Scholar]
- 28.Colella TA, Bullock TN, Russell LB, Mullins DW, Overwijk WW, Luckey CJ, Pierce RA, Restifo NP, Engelhard VH. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J. Exp. Med. 2000;191:1221. doi: 10.1084/jem.191.7.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 30.Shevach EM. Regulatory T cells in autoimmunity. Annu. Rev. Immunol. 2000;18:423. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 31.Theobald M, Biggs J, Hernandez J, Lustgarten J, Labadie C, Sherman LA. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J. Exp. Med. 1997;185:833. doi: 10.1084/jem.185.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan DJ, Kreuwel HT, Fleck S, Levitsky HI, Pardoll DM, Sherman LA. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J. Immunol. 1998;160:643. [PubMed] [Google Scholar]
- 33.Nemunaitis J, Fong T, Shabe P, Martineau D, Ando D. Comparison of serum interleukin-10 (IL-10) levels between normal volunteers and patients with advanced melanoma. Cancer Invest. 2001;19:239. doi: 10.1081/cnv-100102550. [DOI] [PubMed] [Google Scholar]
- 34.Cappello P, Novelli F, Forni G, Giovarelli M. Death receptor ligands in tumors. J. Immunother. 2002;25:1. doi: 10.1097/00002371-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Restifo NP. Not so Fas: re-evaluating the mechanisms of immune privilege and tumor escape. Nat. Med. 2000;6:493. doi: 10.1038/74955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Touloukian CE, Leitner WW, Robbins PF, Li YF, Kang X, Lapointe R, Hwu P, Rosenberg SA, Restifo NP. Expression of a “self-” antigen by human tumor cells enhances tumor antigen-specific CD4+ T-cell function. Cancer Res. 2002;62:5144. [PMC free article] [PubMed] [Google Scholar]
- 37.Marincola FM, Rivoltini L, Salgaller ML, Player M, Rosenberg SA. Differential anti-MART-1/MelanA CTL activity in peripheral blood of HLA-A2 melanoma patients in comparison to healthy donors: evidence of in vivo priming by tumor cells. J. Immunother. Emphasis Tumor Immunol. 1996;19:266. doi: 10.1097/00002371-199607000-00003. [DOI] [PubMed] [Google Scholar]