

Abstract
Cholecystokinin (CCK) is the most abundant neuropeptide in the central nervous system. In the hippocampal CA1 region, CCK is co-localized with GABA in a subset of interneurons that synapse on pyramidal cell somata and apical dendrites. CCK-containing interneurons also uniquely express a high level of the cannabinoid receptor, CB1, and mediate the retrograde signaling process called DSI. Reported effects of CCK on inhibitory post-synaptic potentials (IPSPs) in hippocampus are inconsistent, and include both increases and decreases in activity. Hippocampal interneurons are very heterogeneous, and these results could be reconciled if CCK affected different interneurons in different ways. To test this prediction, we used sharp microelectrode recordings from pyramidal cells with ionotropic glutamate receptors blocked, and investigated the effects of CCK on pharmacologically distinct groups of IPSPs during long-term recordings. We find that CCK, acting via the CCK2 receptor, increases some IPSPs and decreases others, and most significantly, that the affected IPSPs can be classified into two groups by their pharmacological properties. IPSPs that are increased by carbachol (CCh-sIPSPs), are depressed by CCK, ω-conotoxin GVIA, and endocannabinoids. IPSPs that are enhanced by CCK (CCK-sIPSPs) are blocked by ω-agatoxin IVA, and are unaffected by carbachol or endocannabinoids. Interestingly, a CCK2 antagonist enhances CCh-sIPSPs, suggesting normally they may be partially suppressed by endogenous CCK. In summary, our data are compatible with the hypothesis that CCK has opposite actions on sIPSPs that originate from functionally distinct interneurons.
Keywords: CCK2, GABAB, neuronal rhythms, endocannabinoid, muscarinic, LY225910
Introduction
Cholecystokinin (CCK) is the most abundant neuropeptide in the central nervous system (CNS) (Beinfeld et al., 1981), and is highly expressed in a subset of GABAergic interneurons of the hippocampus (Dockray, 1976; Innis et al., 1979). CCK is released mainly as CCK8-S, and also, at low concentrations, as CCK4 or CCK8-U (Rehfeld, 1985). CCK-releasing interneurons in the hippocampus all contain GABA (Somogyi et al., 1984) and most express cannabinoid receptors (Katona et al., 1999; Freund, 2003). The axons of many CCK-positive neurons terminate on hippocampal pyramidal cell somata in stratum (s.) pyramidale and their proximal dendrites of s. radiatum (Freund & Buzsaki, 1996). Central (CCK2) receptors are widely distributed throughout the CNS (Zarbin et al., 1983) and modulate stress, anxiety and exploratory behaviors (Singh et al., 1991; Matto et al., 1997).
Physiological actions of CCK in the hippocampus have been attributed to CCK2 (Bohme et al., 1988; Carlberg et al., 1992); however CCK2 has not yet been localized to specific neuronal sub-types in hippocampus or elsewhere (cf. Mercer et al., 2000). Reports of CCK’s physiological actions are inconsistent, with both excitation (Dodd & Kelly, 1979; Boden & Hill, 1988; Bohme et al., 1988; Shinohara & Kawasaki, 1997), and inhibition (MacVicar et al., 1987; Perez de la Mora et al., 1993) of pyramidal cells having been demonstrated. CCK may inhibit pyramidal cells indirectly (Perez de la Mora et al., 1993) by increasing GABA release from interneurons (Miller & Lupica, 1994; Miller et al., 1997; Ferraro et al., 1999; Deng & Lei, 2006). Some discrepancies have been ascribed to dosage and application method, or to different effects of CCK on interneurons and pyramidal cells (Miller et al., 1997). The close association of CCK only with certain interneurons suggests that some of the reported discrepancies in CCK effects might reflect its actions on distinct classes of interneurons (Freund & Buzsaki, 1996).
The primary aim of the present study was to test the hypothesis that CCK affects different interneurons in different ways, by using pharmacological tools to identify classes of interneuron outputs. Focusing on the rat hippocampal CA1 region, we show that CCK2 activation mediates the effects of CCK agonists, and directly stimulates persistent spontaneous (sIPSP) activity in control conditions. However, other sIPSPs are initiated in the presence of carbachol (CCh) and activation inhibits the CCh-sIPSPs. This does not represent opposing effects of CCK and CCK2 CCh on the same interneurons however, because the IPSPs in these two different conditions are sharply distinguished by their sensitivity to endocannabinoids, calcium channel antagonists, muscarinic agonists, and GABAB agonists. We also report the first evidence that endogenously released CCK suppresses CCh-sIPSPs. Our data are consistent with the hypothesis that the disparate actions of CCK on inhibition reflect opposite effects on distinct interneuron classes. Indeed, the pharmacological profiles of these two classes of sIPSPs correspond well with the properties of PV- and CCK-expressing interneurons as described in the literature (see Table 1). We suggest that CCK could thereby link the actions of different interneurons, a hypothesis that may have implications for understanding some of the oscillatory electrical activity in hippocampus (Buzsaki, 2002; Baraban & Tallent, 2004; Freund, 2003; Whittington & Traub, 2003).
Table 1.
Comparison of hippocampal CCK and PV basket interneurons
| Properties | PV | CCK | References |
|---|---|---|---|
| Spike Timing | Fast-Spiking | Regular Spiking | Reviewed in Freund, 2003; Hefft & Jonas, 2005; Glickfeld & Scanziani, 2006 |
| GABA release | Quantal Release Synchronous | Quantal Release Asynchronous | Hefft & Jonas, 2005 |
| Ca Channels mediating GABA release | P/Q-Type | N –Type | Reviewed in Freund, 2003 |
| CB1R | Absent | Present | Tsou et al., 1998; Marsicano & Lutz, 1999; Freund, 2003 |
| mAChR | M2 on terminals (no M1 or M3) | M1 and M3 on soma, (no M2) | Reviewed in Freund 2003; Fukudome et al, 2004 |
| Pre-synaptic GABABRs | Low concentration | High | Reviewed in Freund, 2003 |
Materials and Methods
Male Sprague-Dawley Rats, 5–7 wks old (Charles River Laboratories) were deeply anaesthetized with halothane and decapitated in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee of the University of Maryland, School of Medicine. The brain was rapidly removed from the skull and both hippocampi dissected free. Transverse hippocampal sections (400 μm thick) were cut on a Vibratome (Series 1000, Technical Products International). Slices were kept in a holding chamber at room temperature at the interface of artificial cerebro-spinal fluid (ACSF) and a humidified gas mixture of 95%O2 and 5% CO2 for ≥ 1 hr and then transferred to a submersion chamber (Nicoll & Alger, 1981) that was continuously perfused with ACSF at 29–31°C, and positioned under a dissecting microscope. The submerged chamber permits rapid, thorough access of bath-applied drugs to the entire slice, and cells remain in healthy condition (as judged by active and passive cell properties) for 6–8 hours. ACSF contained (in mM): 120 NaCl, 3 KCl, 2 MgSO4, 1 NaH2PO4, 25 NaHCO3, 10 glucose, and 2.5 CaCl2, and was continuously bubbled with 95%O2 and 5% CO2 (pH 7.4). The ionotropic glutamate antagonists D-AP5 (20μM) and NBQX (10μM, both from Tocris) were present in all experiments to block EPSPs. CCK8-S, LY225910, YM022 CGP55845 and WIN55212-2 were obtained from Tocris. Carbachol (CCh), CCK4, ω-agatoxin GVIA (agatoxin), ω-conotoxin GVIA (conotoxin) and all other chemicals were obtained from Sigma. All drugs were bath applied. To avoid desensitization induced by repeated applications of CCK, each slice was limited to a single application of CCK.
Electrophysiology
Conventional high resistance intracellular (“sharp”) electrode recordings were carried out in CA1. The CA1 stratum pyramidale was visualized under a dissecting microscope at 4X (the objective does not touch the solution, and the tip of the electrode can readily be positioned directly over the layer), the pyramidal cells were then impaled by lowering the electrode “blindly” into the layer. Microelectrodes (50 –150 MΩ) were filled with a 3M KCl solution to facilitate the observance of GABAA-mediated sIPSPs. Acceptable cells had resting potentials of > −60mV. When necessary, a modest holding current (< −0.5 nA) was used to maintain a slightly hyperpolarized membrane potential at −70 mV, to suppress action potential firing, and enhance sIPSP size. The negative holding potential also prevented CCh from depolarizing the cell because many of the currents affected by CCh are activated only at more depolarized levels. The holding current used was constant during the experiment. In some cases, as noted, we stimulated cells with current injections through the microelectrode in a ‘theta burst’ pattern, where one theta burst equaled 5 depolarizing pulses, 10 msec each in duration, given at 100 Hz; bursts separated by 200 msec. Signals were digitized at 5 kHz (Digidata 1200A, Axon Inst., Foster City, CA), filtered at 2 kHz, and analyzed with pClamp 8.0 or 9.0 software (Axon Inst.). For miniature IPSC (mIPSC) experiments, whole-cell patch clamp recordings were performed. Pyramidal cells were held under whole-cell voltage clamp at −70mV and cells with low, stable holding current (<300pA) were used. Whole cell intracellular solution contained in mM: 90 CsCH3SO3, 1 MgCl2, 1 CsCl, 2 MgATP, 0.2 Cs4-BAPTA, 10 HEPES, 0.3 tris-GTP, and 5 QX-314 (Lidocaine-N-ethyl Bromide). The electrode access resistance measured in a cell was ≤ 30 MΩ and did not change by more than 15% in acceptable experiments.
For analysis of theta rhythms, the data were filtered at 200 Hz with a low-pass, eight pole Bessel filter (Frequency Devices, Haverhill, MA). Power spectrum analysis and autocorrelations were done in Clampfit 9.0. We calculated a value of “relative theta power” for each cell by summing the spectral power between 4–14 Hz, and dividing this by the total spectral power between 1–50 Hz during 10 seconds of sIPSP activity (Reich et al., 2005). We also measured peak theta power as the largest peak spectral power within the theta range of frequencies. Both measures led to the same conclusions, therefore we generally refer to peak power in the text.
MiniAnalysis software (Synaptosoft Inc, Decatur, GA) was used to perform sIPSP amplitude and kinetics analyses of sIPSPs and mIPSCs. The MiniAnalysis program is not a simple ‘window discriminator’ that detects events solely by their amplitudes. Rather it employs a sophisticated fitting routine that takes into account amplitude, rise and decay times and area under curve, in effect creating a template of the target event, in its detection routine. A large sample (n = 10036 events from 25 cells) of CCK- and CCh-sIPSPs had amplitudes of 4.17 ± 0.13 mV (mean ± s.e.m.), rise times of 4.6 ± 0.02 ms, decay times 23.8 ± 0.16 ms, and areas (ms*mV) of 60.9 ± 3.31. Visual checking confirmed the absence of aberrant events in the records. Hence, detectability of even small synaptic potentials is much more accurate with MiniAnalysis than if only amplitude measurements are made.
Data are presented as mean ± s.e.m. Unless noted, paired-sample t-tests were used to determine statistical significance, although for display purposes, group data are shown in bar graphs. Komolgorov-Smirnov tests of sIPSP amplitudes in three cells (total of 1490 sIPSPs) showed distributions did not differ significantly from normal distributions (p > 0.1), further supporting use of parametric tests. Statistical analysis was performed using Sigmastat (Jandel Scientific) at the significance level of p < 0.05, except for K-S tests, for which the significance level was p < 0.005.
Results
CCK analogs initiate sIPSP activity in control conditions by activating CCK2
We used high resistance microelectrodes for pyramidal cell recording under current clamp conditions because the experiments, involving multiple pharmacological tests on single cells, demanded long-term (>2 hour), stable recording conditions. The advantages of these electrodes – less disruption of cell internal environment and long lasting maintenance of normal cell properties – outweighed the drawback of somewhat noisier recordings. With D-AP5 and NBQX present, the average pyramidal cell resting membrane potential was −70.4 ± 1.90 mV (n=33). Bath application of 1 μM CCK8-S for 15 min did not alter the membrane potential (−71.6 ± 0.99 mV, n=33, p>0.05) or input resistance (82.4 ± 2.7 MΩ in CCK, and 82.2 ± 2.3 MΩ, pre-CCK, n=33, p>0.05). CCK8-S reduced the amplitude of the slow AHP elicited by “theta-burst” pattern of dc stimulation given through the recording electrode (see Methods). The AHP was reversibly reduced by ~ 30% by CCK8-S (from 7.3 ± 1.4 mV in control to 4.9 ± 1.1 mV in CCK8-S, p<0.01, n=16), although its duration was not significantly affected (6.3 ±1.4 s and 7.0 ± 2.1 s in CCK, n.s., n=16). Hence, although the agonist does reduce the AHP, the pyramidal cell passive properties are insensitive to CCK8-S. This finding is similar to observations by Miller et al. (1997), who observed no effect of CCK on passive membrane properties of pyramidal cells. However, Miller et al., (1997) did not observe an effect of CCK on the AHP, while others (e.g. Dodd and Kelly, 1979; Shinohara and Kawasaki, 1997) reported the AHP reduction that we see. There is no obvious explanation for this discrepancy, and the issue deserves further study. In any event, our focus in this investigation was on synaptic inhibition, and the CCK effects on the slow AHP that occurred only after strong pyramidal cell activation do not interact with the effects on sIPSPs.
We observed that, in control solution, CCK8-S induced a steady barrage of sIPSPs that were significantly larger and more frequent than sIPSPs prior to CCK8-S application (1.7± 0.29 mV at 2.2 ± 0.25 Hz in control conditions, 4.0 ± 0.96 mV at 4.2 ± 0.47 Hz in CCK8-S, p<0.0001, n=8, for both comparisons, see Fig. 1A). For the sake of clarity, sIPSPs that were increased by any CCK analog will be referred to as “CCK-sIPSPs”. The CCK-sIPSPs often had abrupt onset and occurred rhythmically. A power spectral analysis of 10-sec stretches of sIPSPs in 8 cells revealed a small peak power in the theta frequency range, ~4–14 Hz (power in this range was 0.4 ± 0.02 mV2/Hz in control, and 0.5 ± 0.02 mV2/Hz in CCK8-S, p<0.006, n=8, Fig. 1B). The CCK8-S-enhanced sIPSP activity in our young adult rats diminished only slightly over tens of minutes: the initial CCK-sIPSPs were 4.1 ± 0.31 mV in amplitude and occurred at 4.0 ± 0.29 Hz, and 25 min later were 4.1 ± 0.62 mV at 3.6 ± 0.31 Hz (n = 5 cells, n.s.),. This is unlike the transient effect that CCK has in cells from younger animals (Miller et al., 1997; Deng & Lei, 2006), suggesting developmental changes may affect CCK responses.
Figure 1. CCK receptor agonist enhances sIPSP activity (CCK-sIPSPs) in CA1 pyramidal cells.

Recordings are shown for the same cells before and during the experimental treatment, unless otherwise stated. A1), Upper - representative trace of sIPSPs before (Con) and during wash-in of 1 μM CCK8-S. Lower - representative trace of CCK-sIPSPs in same cell. Calib. =10 mV, 2 sec. A2) Group data (n = 8 cells, 10-sec traces from each cell) showing that CCK8-S increased sIPSP amplitudes and frequencies (paired-t tests). B) sIPSP spectral power in the ‘theta’ frequency range (4–14 Hz) is enhanced by CCK8-S (control spectrum in inset). Graphs represent data from one cell, and are representative of spectra from all 16 cells. C1) Traces (same cell throughout) showing sIPSPs in control solution (upper trace), 15 min after addition of the CCK2-selective antagonist, LY225910 (2 μM), to the solution (middle trace), and 20 min after adding CCK8-S to the solution (lower trace). Calib. = 5mV, 1 sec. Note LY225910 does not affect control sIPSPs, but prevents increase in sIPSPs by CCK8-S. C2) Group data (n = 9, same cells in all conditions).
The first question we addressed was which CCK receptor mediates the effects of CCK8-S. We found that the effects of CCK8-S were replicated by the highly specific CCK2 receptor agonist, CCK4 (1 μM), (1.3 ± 0.1 mV at 2.5 ± 0.05 Hz in control, 2.0 ± 0.05 mV at 4.2 ± 0.29 Hz in CCK4, p < 0.05, n=5 for both comparisons). Relative theta power was significantly increased by CCK4 (from 0.5 ± 0.07 to 0.6 ± 0.03 mV2/Hz, p<0.005, n = 5, data not shown). CCK4 also reduced the pyramidal cell AHP from 7.4 ± 1.1 mV to 5.5 ± 0.9 mV (n=5, p < 0.05). Selective CCK2 antagonists LY225910 (2 μM) or YM022 (1 μM), were bath-applied for > 1 hr before the subsequent addition of CCK agonists. The antagonists did not affect the pyramidal cell membrane potential (n.s.) or the baseline sIPSPs, but fully prevented CCK8-S from increasing sIPSP amplitude or frequency, example shown in Fig. 1C (LY225910, n=9; YM022, n=6; p>0.05, each comparison). The selective CCK1 antagonist, devazepide, had no effect on CCK-sIPSPs (n = 4, data not shown) further supporting the conclusion that the observed effects are mediated via the CCK2 receptor.
These experiments revealed that activation of CCK2 mediates the effects of CCK8-S, and triggers a persistent barrage of sIPSPs in control conditions.
CCK analogs suppress sIPSPs activated by mAChR activation
It is well established that agonists of mAChR, including CCh, also induce the occurrence of persistent, rhythmic sIPSPs (Pitler & Alger, 1992a; Martin & Alger, 1999; Fischer et al., 2002; Gillies et al., 2002; Reich et al., 2005), probably via the M1 and M3 receptor subtypes (Martin & Alger, 1999; Fukudome et al., 2004). These sIPSPs will be designated “CCh-sIPSPs.” To determine if CCK-sIPSPs and CCh-sIPSPs originate from the same class of interneurons, we compared a number of their key features.
We confirmed that 5 μM CCh induced persistent sIPSP activity in the 22 cells tested (e.g. Fig 2A). Once CCh-sIPSPs were initiated, we added 1 μM CCK8-S to the bath solution and observed its effects on them. The mean membrane potential of CCh-treated CA1 pyramidal cells was maintained at −69.8 ± 1.11 mV (n=18). If both agonists affect the same cells, CCh, having already strongly initiated the sIPSP activity, should reduce or occlude the relative ability of CCK8-S to increase the activity. Contrary to this prediction, however, we found that CCK2 agonists strongly suppressed the CCh-sIPSPs (Fig 2A). CCh-sIPSP amplitudes were reduced from 8.2 ± 0.60 mV, to 5.6 ± 0.46 mV by CCK8-S (p<0.001, n=18 cells) and their frequency from 9.4 ± 0.36 Hz to 5.6 ± 0.48 Hz in CCK8-S (p<0.0001, n=18 cells) (Figs. 2A and 2B). Reich et al. (2005) showed that CCh application induces sIPSPs that occur at theta-rhythm frequency in CA1 pyramidal cells, independent of activation of glutamate receptors or of inputs from other hippocampal regions. CCK8-S significantly reduced, but did not abolish, the increase in peak spectral power of CCh-sIPSPs across the theta range (from 7.9 ± 2.15 mV2/Hz to 3.3 ± 1.76 mV2/Hz, n=22, Fig. 2B). CCK4 also reduced the CCh-sIPSP amplitudes and frequency (CCh 5.6 ± 0.41 mV, reduced to 4.5 ± 0.27 mV in CCh plus CCK4; sIPSP frequency in CCh was 7.3 ± 0.60 Hz, reduced to 2.7 ± 0.34 Hz in CCh plus CCK4, n = 4, p<0.003 for both comparisons, data not shown).. The CCK2 antagonists LY225910 and YM022 prevented the suppressive effects of CCK8-S on CCh induced activity. The sIPSPs were 10.0 ± 1.31 mV in CCh plus LY225910, and 9.8 ± 1.23 mV in CCh plus LY225910 and CCK8-S, n=5 cellsp>0.05. CCh-sIPSP frequency was not affected by LY225910 or subsequent addition of CCK8-S (8.2 ±1.25 Hz in CCh, 8.7 ± 0.58 Hz in CCh plus LY225910, 8.3 ±1.41 Hz in CCh plus LY225910 plus CCK 8-S, n=5 cells, p>0.05). In CCh plus YM022 the sIPSPs were 5.3 ± 0.62 mV, 5.4 ± 0.49 mV and in CCh plus YM022 plus CCK8-S they were 5.3 ± 0.56 mV, n=7, p>0.05. The mean sIPSP frequency in CCh was 7.1 ± 0.98 Hz, 7.0 ± 1.1 Hz in CCh plus YM022, and 7.2 ± 0.99 in CCh plus YM022 plus CCK8-S, n=7, (p>0.05 for all comparisons). Hence, CCK8-S suppressed CCh-sIPSPs by activating CCK2. The significance of the larger sIPSPs in LY225910 will be discussed in a later section.
Figure 2. sIPSP activity induced by carbachol (CCh-sIPSP) is reduced by CCK8-S.
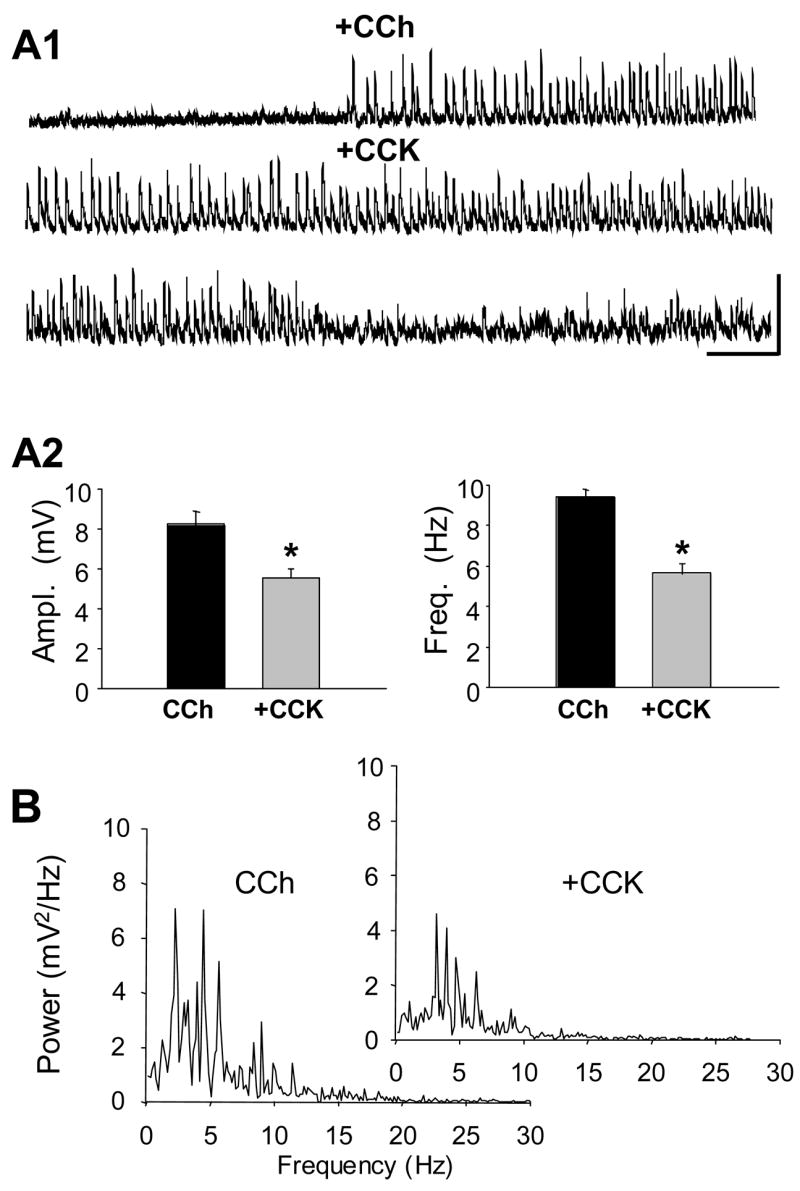
A1) All traces from the same cell. Upper trace - wash-in of 5 μM CCh and onset of robust CCh-sIPSPs. Middle trace - CCh sIPSPs in same cell after 5 min and during start of wash-in of CCK8-S. Lower trace - subsequent cessation of CCh-sIPSPs caused by CCK8-S. Calib. = 10mV, 2 sec. A2) Group data (n = 18) showing significant reduction in CCh-sIPSP amplitude and frequency caused by CCK8-S; paired t-tests. B) Power spectra for a cell first recorded in CCh (left), and then in CCh plus CCK8-S (inset). Note the peak spectral power occurs between 3–10 Hz in both cases; results typical of 18 cells.
When CCh was added after CCK8-S, there were small increases in sIPSP amplitude (from 3.5 ± 0.16 mV in CCK8-S alone to 4.2 ± 0.32 mV when CCh was added, n = 5, p<0.005), frequency (from 3.9 ± 0.27 Hz in CCK alone to 4.4 ± 0.1 when CCh added, n=5, p<0.05) and theta power (from 0.6 ± 0.03 mV2/Hz in CCK alone to 4.0 ± 1.32 mV2/Hz when CCh was added, n=5, p<0.001). However, the sIPSPs were smaller, less frequent, and had less theta power in these cells than in those treated with CCh alone (n=5, p<0.01 for each comparisons), illustrating that, in this case also, CCK opposed the effects of CCh.
Thus, contrary to our initial hypothesis that CCK and CCh would mutually occlude each other because of similar actions on the same interneurons, they had opposite effects on sIPSPs. Moreover, CCK8-S suppressed CCh-induced sIPSPs by activating CCK2 so different CCK receptor subtypes could not explain the results. We considered two other explanations: CCK2 and M1/M3 mAChR agonists could produce opposing effects on the same interneurons, or they could each primarily affect a different type of interneuron. Predictions of these hypotheses are tested below.
Different interneurons originate CCK-sensitive and CCh-sensitive sIPSPs
Previous work has shown that the large sIPSPs in the hippocampal CA1 cells arise from perisomatic synapses, almost certainly from basket cell interneurons (Banks et al., 1998; Martin & Alger, 1999). There are two major classes of GABA-ergic basket interneurons in hippocampus. Both predominantly innervate the perisomatic region of pyramidal cells in CA1 and CA3 (Pawelzik et al., 2002; Hefft & Jonas, 2005) which enables them to control pyramidal cell firing patterns (Cobb et al., 1995; Freund, 2003, for review). Nevertheless, the two classes of basket cells are not identical. For example, they express different complements of receptors and channels, and have distinctive physiological properties. Hence, the IPSPs they produce can be distinguished by means of pharmacological tools and physiological characteristics (see Table 1). We made use of several of these distinguishing characteristics to compare the CCh- and CCK-sIPSPs, and determine if they arise from the same or different cell types.
Cannabinoid Sensitivity
CB1 receptors in the hippocampal CA1 region exist in very high concentrations on the terminals of CCK-expressing interneurons, and are either absent or in very low concentration elsewhere (Katona et al., 1999). DSI dramatically reduces CCh-sIPSP/Cs (Pitler & Alger, 1992b; Alger et al., 1996; Martin & Alger, 1999; Wilson & Nicoll, 2001; Fortin et al., 2004; Reich et al., 2005) an effect we confirmed in all 18 cells that were tested (e.g., Fig. 3A1). We initiated DSI using a single, 1-second, depolarizing voltage-step from the resting potential to 0 mV, or by 3 brief ‘theta-burst’ trains of intracellular current injection (see Methods). Thus our data agree with previous reports that CCh-sIPSPs originate from CB1-expressing interneurons.
Figure 3. Short-term endocannabinoid effect (DSI) reduces CCh-sIPSPs, but not CCK- sIPSPs.
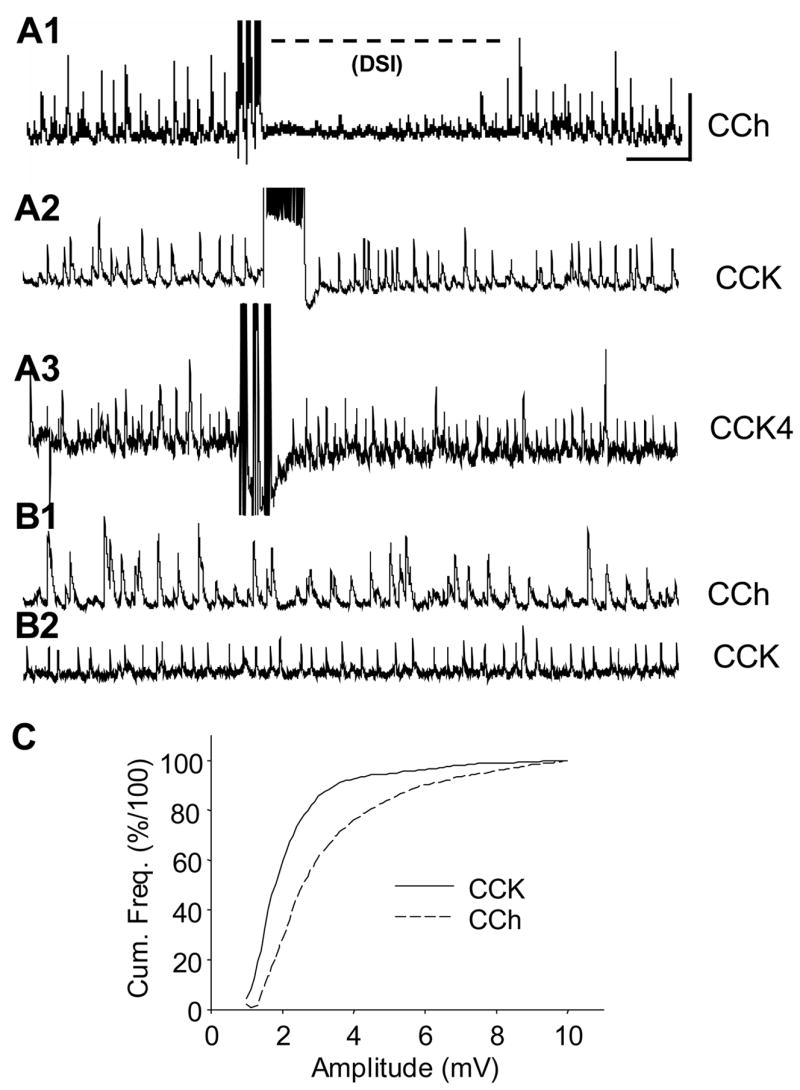
A1) Typical trace from a cell treated with 5 μM CCh for 20 min. Following three ‘theta burst’ trains of action potentials (see Methods), the sIPSPs are reversibly abolished. Calib. = 5 mV, 1 sec, applies to all traces in this figure. A2) and A3) illustrate typical results from two cells to which either CCK8-S or CCK4 are applied. Neither a 1-sec long depolarizing current pulse that triggered a train of action potentials (A2), nor a theta-burst protocol (A3) suppresses the CCK-sIPSPs. Results are typical of 9 cells. B1) and B2) compare sIPSPs from two cells treated either with CCh, or CCK8-S. Note that relative uniformity in amplitude of the CCK-sIPSPs compared with the CCh-sIPSPs. C) Cumulative frequency plots of data from 10 cells (10 sec of data per cell). A K-S two-sample test shows that the difference in the two curves is highly significant (p<0.001, D1293,2463=99.17). Note that ~70% of the sIPSPs induced by CCK2 agonists had amplitudes between 1.0 – 2.35 mV, whereas the range for 70% of the CCh-induced sIPSPs was 1.6 – 4.20 mV (i.e., twice as broad).
In contrast, CCK-sIPSPs are resistant to suppression by DSI (e.g. Figs. 3A2 and 3A3). There was a hint of DSI of CCK-sIPSPs in only 1 of 9 cells, and furthermore, bath application of the CB1 agonist, WIN55212-2 (5 μM) had no effect on CCK-sIPSP amplitude or frequency (2.4 ± 0.04 mV at 4.6 ± 0.72 Hz, compared to 2.5 ± 0.06 mV at 4.4 ± 0.52 Hz in WIN 55212-2, n=3 cells). In contrast, CCh-sIPSPs were significantly reduced by WIN 55212-2 (from 7.21 ± 0.92 mV and 8.43 ± 1.72 Hz in control to 4.33 ± 0.86 mV and 3.41 ± 1.65 Hz, in WIN 55212-2, n=3, p<0.05). DSI of CCh-sIPSPs was similarly abolished in WIN-treated cells (data not shown). This confirms previous findings that CCh-sIPSPs are predominantly generated by interneurons with the CB1 receptor (Wilson & Nicoll, 2001; Varma et al., 2001; Fortin et al., 2004), and represents the first evidence that CCK-sIPSPs are generated by interneurons lacking the CB1 receptor.
Variability
We observed that the amplitudes of the CCK-sIPSPs are often much more uniform than are those of CCh-sIPSPs (compare e.g., 3B1 and 3B2). The mean cumulative frequency plots of sIPSPs (10 CCK8-S treated and 10 CCh-treated) were significantly different (p< 0.001 by K-S test, Fig. 3C). Differences in firing patterns of groups of hippocampal interneurons have often been reported (Freund & Buzsaki, 1996; Pawelzik et al., 2002; Hefft & Jonas, 2005; Glickfeld & Scanziani, 2006). In particular, IPSPs from CB1-expressing interneurons are much more variable in amplitude and temporal dispersion than IPSPs from CB1-lacking neurons (Glickfeld & Scanziani, 2006). Hence amplitude variability measurements also support the interpretation that different classes of interneurons mediate the different responses resulting from CCK2 activation.
Ca2+ channel antagonists
GABA release from different classes of interneurons is mediated by either P/Q, or N-type, Ca2+ channels (Poncer et al., 1997). IPSPs that are inhibited by CB1 agonists are entirely dependent on N-type Ca2+ channel influx (Lenz et al., 1998; Hoffman & Lupica, 2000; Wilson et al., 2001); whereas IPSPs that are insensitive to CB1 agonists are dependent on the P/Q-dependent, or mixed (N and P/Q) Ca2+ channels (Lenz et al., 1998; Wilson et al., 2001). Thus, sensitivity of sIPSPs to agatoxin (selective P/Q-type channel blocker) or conotoxin (selective N-type channel blocker) can help ascertain the class of the interneuron releasing GABA. In slices pre-treated for ≥ 1 hr with the P/Q-type Ca2+ channel blocker ω-agatoxin IVA (250 nM), CCK8-S did not induce any spontaneous activity (e.g. Fig. 4A). Agatoxin did not directly interfere with CCK2 activation, however, as CCK8-S continued to reduce the pyramidal cell AHP in agatoxin (data not shown). Thus, CCK-sIPSPs depend on activation of P/Q-type Ca2+ channels for their occurrence.
Figure 4. CCK-induced sIPSPs are generated by interneurons that depend on P/Q- and not N-type Ca2+ channels for GABA release.

Agatoxin (AgTx) prevents occurrence of CCK-sIPSPs, but not occurrence of CCh-sIPSPs. A) Pre-treatment of a slice for 60 min with 1 μM ω-agatoxin prevents CCK8-S from inducing sIPSPs. B1) Agatoxin does not prevent induction of sIPSPs by CCh. Subsequent CCK8-S application does not reduce the CCh-sIPSPs in agatoxin. Calib. = 5 mV, 1 sec in A and B. B2) Group data (n=7) showing that agatoxin blocks the CCK reduction of CCh-sIPSPs. C1) Slices were pretreated for 60 min with 250 nM conotoxin, which was also present in the bath solution. Conotoxin does not prevent CCK8-S from inducing sIPSPs, but prevents large CCh-sIPSPs. Calib. 5 mV, 2 sec. C2) Group data (n=4) showing effects of conotoxin.
On the other hand, CCh-sIPSP activity is robust in agatoxin (Fig. 4B), and statistically indistinguishable from the CCh-sIPSP activity in control solution (6.9 ± 0.5 mV in agatoxin versus 8.2 ± 0.60 mV in control; n=7, p>0.05) consistent with the evidence that the DSI-sensitive, CB1-expressing interneurons release GABA via the activation of N-type, but not P/Q-type Ca2+ channels (Lenz et al., 1998; Hoffman & Lupica, 2000; Wilson et al., 2001). Importantly, in the presence of agatoxin, CCK8-S did not reduce CCh-sIPSPs (Fig. 4B2). Note also that agatoxin generally abolished the sIPSPs that occurred in the absence of CCh, suggesting that interneurons releasing GABA via P/Q channels are predominantly active in control conditions. This would agree with previous reports that these baseline sIPSPs are generally insensitive to DSI (e.g., Martin & Alger, 1999; Martin et al., 2001).
The N-type Ca2+ channel blocker, ω-conotoxin GVIA (250 nM; pre-treatment to the slices and present in the bath solution) did not prevent induction of CCK-sIPSPs (Fig. 4C1). In conotoxin, CCK8-S increased both sIPSP amplitudes and frequency (n=4, p<0.01 for each comparison). Conotoxin also prevented the CCh-induced increase in sIPSP amplitude, although not the increase in frequency of small sIPSPs (to 5.7 ± 1.1 Hz from 2.2 ± 0.3 Hz in conotoxin alone, n=3, p<0.05, Fig. 4C2), suggesting that CCh can also induce the occurrence of small sIPSPs from conotoxin-insensitive interneurons.
The results indicate that CCK-sIPSPs are generated by interneurons that depend on P/Q- but not N-type Ca2+ channels for GABA release. Large, DSI-sensitive sIPSPs induced by CCh are released from N-channel dependent interneurons, although a population of smaller CCh-sIPSP originates from cells that are partly or wholly dependent on P/Q channel activation.
TTX-resistant mIPSCs
CCK could affect sIPSPs solely by altering the action potential firing of interneurons, or it could affect action potential-independent quantal release of GABA, or both. To determine where CCK acts, we recorded miniature IPSCs (mIPSCs) under whole-cell voltage clamp in the presence of 1 μM tetrodotoxin to block action potential-dependent responses (Edwards et al., 1990). We measured mIPSCs during 4-minute intervals before, during and after bath-application of CCK8-S (n = 100 mIPSCs per condition, total of 300 mIPSCs per cell for 5 cells) and found that CCK8-S slightly and transiently increased their amplitude and frequency (mean amplitude in TTX: 14.5 ± 0.43 pA, mean amplitude in TTX plus CCK8-S; 17.6 ± 0.68 pA, p < 0.005; mean frequency in TTX: 0.6 ± 0.15 Hz; mean frequency in TTX plus CCK8-S 1.0 ± 0.15 Hz, p<0.05; n = 5 cells, Fig 5A, 5B1 and 5B2). In four additional cells, we bath-applied the selective CCK2 antagonist, LY225910, and found that CCK8-S had no effect in its presence (p>0.05), confirming that the enhancement of mIPSCs was mediated by CCK2 (Fig 5B1 and 5B2).
Figure 5. CCK8-S increases mIPSCs in TTX.

CCK application increases miniature IPSCs (mIPSCs) recorded under whole-cell voltage clamp in the presence of 1 μM tetrodotoxin. A) Representative 30-sec traces of mIPSCs before (upper trace) and in the same cell in CCK8-S (lower trace). Calib. = 20 pA, 1 sec. Group data (n=5 cells) show that mIPSC frequency (B1) and amplitude (B2) are transiently increased during application of CCK8-S, and return to baseline levels after wash-out of the peptide (black circles). The increase in mIPSCs is blocked by the CCK2 receptor antagonist LY225910 (2μM; grey circles, n=4 cells).
Increase in mIPSCs could contribute to the initial onset of CCK-sIPSPs, although the magnitude and transient nature of the increases demonstrate that they cannot account for the entire effect of CCK8-s on sIPSPs in the absence of TTX. Most importantly, the results imply that that the suppression of CCh-sIPSPs by CCK8-S does not reflect a post-synaptic effect on GABAA receptors, since mIPSC amplitudes do not decrease. We consider the mechanism of the decrease in CCh-sIPSPs in more detail below.
Relationship between CCK-activated and CCK-inhibited interneurons
The results strongly argue that CCK- and CCh-sIPSPs originate from different interneurons, but they do not address the issue of how CCK affects the two responses. CCK could directly excite some interneurons and inhibit others, if CCK2 were present on all interneurons, but coupled to different effectors on the different cells. Although possible, this seems unlikely. An alternative hypothesis would be that CCK affects some cells directly, and others only indirectly. Heterosynaptic effects of CCK have been reported in the n. accumbens (Kombian et al., 2005). There, CCK initiates the release of GABA from interneurons, which activates GABAB receptors on glutamate or GABA terminals, and thereby inhibits transmitter release indirectly.
Extrapolation of this concept to our system would mean that the CCK might directly excite certain interneurons in control conditions, and via GABABR activation, indirectly inhibit those excited by CCh. To test this mechanism, we observed the effects of CCK application in the presence of the GABAB antagonist, CGP55845 (1 μM).
In control conditions, bath-application of CGP55485 slightly increased the baseline sIPSPs (1.5 ± 0.13 mV at 0.6 ± 0.1Hz in control, and 1.9 ± 0.09 mV at 2.2 ± 0.5 Hz in CGP55485, n = 3, p<0.05), but did not alter the ability of CCK8-S to increase sIPSP activity (3.8 ± 0.9 mV at 4.2 ± 0.3 Hz in CGP55485 plus CCK8-S, n=3, p<0.05 each comparison, Fig. 6A). Similarly, CCK- sIPSPs were unaffected by subsequent addition of CGP55485 to the bath (4.0 ± 0.41 mV at 4.2 ± 0.09 Hz in CCK8-S, compared with 4.1 ± 0.46 mV at 4.3 ± 0.16 Hz, in CCK plus CGP55485, n.s., n=3).
Figure 6. CCK8-S cannot reduce CCh- sIPSPs if GABAB receptors are blocked.
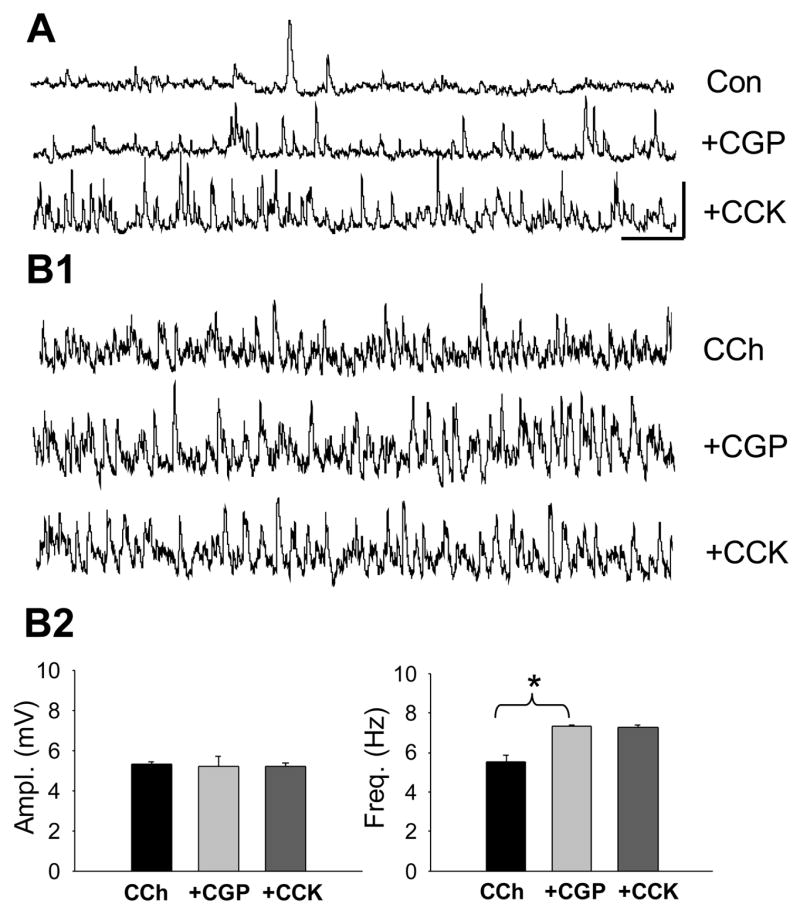
A) The GABAB antagonist, CGP55845 slightly increases the baseline sIPSP activity, suggesting a resting inhibitory GABAB –mediated inhibitory ‘tone’. CCK8-S does not affect the GABAB increased sIPSPs. Calib. = 5 mV, 1 sec, applies to all traces in this figure. B1) Sample CCh-sIPSPs treated with CGP55845. There is a slight increase in CCh-sIPSP frequency, and subsequent addition of CCK8-S has no effect. B2) Group data (n = 3) showing effects of CGP55485 and CCK8-S on CCh-sIPSPs.
In contrast, CGP55845 markedly enhanced the large CCh-sIPSP frequency, but not amplitudes (from 5.3 ± 0.14 mV at 5.5 ± 0.34 Hz in CCh, to 5.21 ± 0.51 mV at 7.3 ± 0.06 Hz in CCh plus CGP55845, n=4, p<0.05 for frequency comparison). Most importantly, in the presence of CGP55845, CCK8-S had no effect on the CCh-sIPSPs, (p>0.05, n=3, e.g., Figs. 6B1 and 6B2). Therefore the ability of CCK8-S to suppress CCh-sIPSPs is not a direct effect, but is dependent on GABAB receptor activation, in full agreement with the proposal of CCK-mediated heterosynaptic inhibition (Kombian et al. 2005).
Activity dependent release of endogenous CCK
The results described in the previous section imply that exogenously applied CCK can release GABA and cause the subsequent suppression of CCh-sIPSPs by activation of GABAB receptors. There is essentially no information on the effects of endogenously released CCK, however. Therefore we considered whether a CCK2 antagonist would affect sIPSPs at a time when CCK release was expected, i.e., while interneurons are being strongly activated by CCh. This was suggested during the identification of the CCK2 receptor, when, in the presence of LY225910, we noted that the CCh-sIPSPs were very large. Indeed, we observed that bath application of the CCK2 antagonist LY225910 markedly increased the amplitudes of CCh-induced sIPSPs, (from 4.5 ± 1.1 mV in CCh, to 10.0 ± 1.3 mV in CCh plus LY225910, p<0.001, n=5 cells), without affecting sIPSP frequency (p>0.05, Figs. 7A and 7B. As noted earlier, the CCK2 antagonists applied alone in control conditions (total n = 15 cells) have no effects on baseline sIPSPs, and therefore the increase in CCh-sIPSPs by LY225910, demonstrates a significant link between the increase in sIPSP activity by CCh and the endogenous action of CCK itself. The spontaneous activity was completely abolished by bicuculline (10 μM, Fig. 7A), confirming that it represents GABAA sIPSP firing. Importantly, the large LY225910-enhanced sIPSPs were highly susceptible to DSI (Fig. 7C, typical of 4 cells tested) arguing that the sIPSPs originate from DSI-sensitive, i.e., CB1- expressing interneurons. Enhancement of CCh-sIPSPs by a CCK2 antagonist suggests that endogenously released CCK partially counteracts the interneurons’ ability to release transmitter, and that CCK participates in a complex feedback mechanism among interneurons.
Figure 7. Endogenous CCK release caused by bath-applied CCh.

A) Representative traces of CCh-sIPSPs (top) and their increase by LY225910, 2 μM (middle). Subsequent addition of bicuculline methochloride (bottom) abolishes all activity, confirming the large events were CCh-sIPSPs. Calib. = 10 mV, 1 sec. B) Group data (n=5) showing that LY225910 significantly (p<0.001) increases the mean CCh-sIPSP amplitudes without changing sIPSP frequency. C) The CCh-sIPSPs that are enhanced by LY225910 are susceptible to DSI. Top trace is in CCh, middle and bottom traces are in CCh plus LY225920 (30-sec traces). Three theta-burst trains of action potentials were given at the point shown by the upward arrow. Marked suppression of sIPSPs reflects DSI. Calib. = 10 mV, 3 sec.
Summary and hypothetical model
While the data may appear complex, they can be succinctly summarized by the schematic model shown in Fig. 8. The model is consistent with a great deal of prior work by other investigators, although definitive testing must await recordings from interneuron-pyramidal cell pairs under our experimental conditions, followed by immunocytochemical identification of the interneurons. For ease of comparison with prior work, we excerpted some key features of the literature on interneurons, as presented in the review by Freund (2003) and other references, and illustrated them in Table 1. Note that the characteristics of the CCK-expressing basket cells predict that they would be the source of IPSPs that are sensitive to CCh, conotoxin, endocannabinoids and GABAB activation; i.e., ideal candidates for the origin of CCh-sIPSPs. In contrast, PV-expressing basket cells could be the source of IPSPs that are sensitive to agatoxin, but insensitive to direct stimulation by CCh, or inhibition by endocannabinoids. Our data suggest that CCK may directly excite PV interneurons (probably via the inhibition of an resting K+ conductance as suggested by Cox et al., 1995 and Miller et al, 1997), while indirectly inhibiting the CCK-interneurons via GABA (since the inhibition was blocked by a GABAB antagonist). An unresolved but important issue is exactly where the CCK2 receptors are located. We (unpub. obs.) have been unsuccessful in unambiguously localizing the CCK2 receptors to single cells with commercially (CalBiochem) or provided (Mercer et al., 2000) CCK2 antibodies. In view of the general paucity of published work on this topic, it is likely that unknown difficulties prevent the ready staining of CCK2 in CNS.
Figure 8. Summary and schematic model.

The data have shown that there are two distinct groups of interneuronal responses that we propose originate from functionally distinct interneuron classes, types 1 and 2. Type 1 interneurons are stimulated by CCh, and IPSPs originating from these neurons are inhibited by endocannabinoids, conotoxin and activation of GABAB receptors. Type 2 interneurons are activated by CCK, and IPSPs originating from these neurons are inhibited by agatoxin, but are insensitive to endocannabinoids or conotoxin. Our data also suggest that CCh causes the liberation of CCK, perhaps from type 1 cells. Activation of type 2 cells by CCK feeds back GABA, which by activation of GABAB receptors, partially suppresses the type 1 interneurons. Because their pharmacological profiles are consistent with the identification of type 1 as CCK-expressing interneurons and type 2 with PV interneurons, we tentatively propose this conclusion. Predictions of the model remain to be tested.
Discussion
Despite the close association of CCK with GABAergic interneurons, prior reports of its actions on synaptic inhibition have not yielded a coherent picture. Our findings can reconcile some of the previous contradictory results, and in addition, suggest that CCK may mediate interactions between different classes of interneurons. The main observation was that CCK has opposite effects on two kinds of pyramidal cell sIPSPs, suppressing the endocannabinoid-sensitive, CCh-sIPSPs, but inducing the occurrence of CCK-sIPSPs. These sIPSPs arise from interneurons with distinct functional properties. The data show that CCK triggers GABA release from interneurons that are not excited by CCh, or inhibited by endocannabinoids, but that express P/Q- type Ca2+ channels. Other interneurons that are excited by CCh, but inhibited by CCK, release GABA via N-type Ca2+ channels and produce IPSPs that are suppressed by endocannabinoids. The inhibition of the CCh-sIPSPs by CCK appears to be an indirect effect, because it is blocked by a GABAB receptor antagonist. A simple interpretation is that, when CCK stimulates the first group of interneurons, the released GABA then heterosynaptically activates GABAB receptors on the second group of interneurons and inhibits their release. The main features of our work are parsimoniously represented by the model in Fig. 8. Comparison of our results with the published properties of CCK-expressing and PV-expressing interneurons (see Table 1) supports our tentative identification of the interneurons. Testing the predications of this model must await paired, interneuron-pyramidal cell recordings followed by immunocytochemical staining for the interneuronal markers.
It might appear that CCK could reduce CCh-sIPSPs by directly decreasing the ability of CCh to excite the interneurons. However, both agatoxin and CGP55845 prevented the reduction of CCh-sIPSPs by CCK, and there is no reason to think that these agents would prevent an interaction between CCK and mAChRs. A more likely interpretation is that CCK-sIPSPs arise from CB1-lacking interneurons, from which GABA release is P/Q channel dependent. Agatoxin blocks release from these interneurons, thereby preventing CCK from stimulating GABA release from them. CGP55845, on the other hand, by blocking activation of GABAB receptors on the CCK interneurons (where they exist in higher numbers than on PV interneurons, see Freund, 2003), prevents nearby release of GABA from inhibiting the CCh-sIPSPs. These two non-obvious results support the conclusion that the CCK-mediated inhibition of CCh-sIPSPs is an indirect effect. Our results with Ca2+ channel blockers and interneuron output inhibited by cannabinoids are exactly in line with previous reports (Hoffman & Lupica, 2000; Wilson et al., 2001).
Our observations also provide the first evidence that endogenously released CCK can affect synaptic inhibition. We observed that the CCK2 antagonist, LY225910, increased CCh-sIPSP amplitudes. A likely explanation is that activation by CCh caused the interneurons to release CCK as well as GABA. Like exogenous CCK, endogenous CCK evidently exerted a suppressive influence on the CCh-sIPSPs, which was relieved when the CCK2 antagonist was applied by itself in the presence of CCh (i.e., an agonist of CCK2 was not also applied). As noted, the CCK2 antagonist did not affect baseline sIPSPs in the absence of CCh or CCK, so its ability to enhance CCh-sIPSPs was linked to the heightened activity induced by CCh. Endogenous CCK may have caused reduced release from the interneuron terminals, rather than interneuron action potential firing, because only the amplitude and not the frequency of the CCh-sIPSPs was altered by the antagonist.
We also noted that CCK-sIPSPs were less variable than CCh-sIPSPs, which appears to reflect a difference in their release properties. Hefft and Jonas (2005) have recently found that in the dentate gyrus CCK- and PV- cells can be distinguished by the degree of synchrony with which they release GABA quanta. Recently, Glickfeld and Scanziani (2006) found that CB1-expressing and CB1--lacking basket cells in hippocampus differ in their synaptic inputs and spike timing properties. Specifically, these authors showed that the CB1-negative interneurons responded quickly and reliably to synaptic inputs, while the CB1-positive cells required higher levels of stimulation and did not reliably follow high (20 Hz) frequency stimulation. These properties enabled the interneurons to serve different functions in the hippocampal circuit. By inference from the previous work of others, the results of Glickfeld and Scanziani (2006) can be attributed to PV- and CCK-expressing interneurons, although was not shown directly. Thus, our observation that CCK-sIPSPs were less variable than CCh-sIPSPs is in line with the hypothesis that they are derived CB1-negative and CB1-positive respectively, although it is emphasized that the work of Hefft and Jonas was done in the dentate gyrus.
We have focused on the large, CCh-sIPSPs that are produced by perisomatic synapses and originate from CB1-expressing cells (Martin & Alger, 1999; Martin et al., 2001; Wilson et al., 2001). Nevertheless, mAChR agonists can activate a number of hippocampal interneuron subtypes (McQuiston & Madison, 1999; Chapman & Lacaille, 1999; McMahon et al., 1998; Cobb & Davies, 2005), in addition to the perisomatic targeting ones. It is, therefore, no surprise that in conotoxin, when the large CCh-sIPSPs are blocked, CCh-sIPSPs with different properties are revealed (they are small, insensitive to DSI, and their frequency but not amplitude was reduced by CCK8-S). Identifying the cells from which these small CCh-sIPSPs originate must await further work.
Networks of interneurons can generate rhythmic oscillations (e.g. gamma and theta), thereby inducing IPSPs, which tightly control the timing of pyramidal cell action potentials (Cobb et al., 1995; Fisahn et al., 1998; Gillies et al., 2002; Freund, 2003). Our results are compatible with these interpretations, but may in addition, point to a novel mechanism for potential functional linkage between the interneuron classes. The release of CCK could trigger GABA release from some interneurons, which in turn would transiently inhibit others. Strong persistent activation of the former interneurons (e.g., by mAChR activation) would continuously regenerate the cycle. GABAB-mediated heterosynaptic inhibition of glutamate release occurs in the hippocampus (Isaacson et al., 1993) when vigorous stimulation generates enough GABA spillover to reach the GABAB receptors on the glutamate terminals. In our case, this would imply that the postulated interactions between the different interneurons would not be instantaneous, and the kinetic details would influence the properties of the rhythms involved.
Despite the rhythmic behaviors of the interneuronal output induced by CCh and CCK, clearly they would not solely be responsible for hippocampal oscillations. Other factors, including interneuronal rhythms initiated by mGluRs (Gillies et al., 2002; Palhalmi et al., 2004), excitatory inputs (Buzsaki, 2002), autapses (Cobb et al., 1997), and gap junctions (Blatow et al., 2003; Whittington & Traub, 2003) play critical roles. Nevertheless, the importance of inhibition in modulation of rhythms (Fisahn et al., 1998; Cobb & Davies, 2005), together with the endocannabinoid- and CCh-sensitivity of the IPSPs, suggests that the regulation of inhibitory synaptic rhythms by CCK will be important for behaviorally relevant oscillations. By suppressing the CCh-sIPSPs, CCK will modulate the functional roles of endocannabinoids in such network activity.
Acknowledgments
We thank Scott Thompson, Celine Dinocourt, David Edwards, and Carlos Lafourcade for their comments on a draft of this manuscript. This work was supported by NIH grants R01 DA140625, R01 MH077277, R01 NS30219 and T32 NS007375 to B.E.A. M.A.K. was supported in part by the Cellular and Integrative Neurosciences Training Grant (NS07275) to the University of Maryland.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alger BE, Pitler TA, Wagner JJ, Martin LA, Morishita W, Kirov SA, Lenz RA. Retrograde signaling in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. Journal of Physiology (London) 1996;496:197–209. doi: 10.1113/jphysiol.1996.sp021677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks MI, Li TB, Pearce RA. The synaptic basis of GABAA(slow) Journal of Neuroscience. 1998;18:1305–1317. doi: 10.1523/JNEUROSCI.18-04-01305.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Tallent MK. Interneuronal neuropeptides -- endogenous regulators of neuronal excitability. Trends in Neurosciences. 2004;27:135–142. doi: 10.1016/j.tins.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Beinfeld MC, Meyer DK, Eskay RL, Jensen RT, Brownstein MJ. The distribution of cholecystokinin immunoreactivity in the central nervous system of the rat as determined by radioimmunoassay. Brain Research. 1981;212:51–57. doi: 10.1016/0006-8993(81)90031-7. [DOI] [PubMed] [Google Scholar]
- Blatow M, Rozov A, Katona I, Hormuzdi SG, Meyer AH, Whittington MA, Caputi A, Monyer H. A novel network of multipolar bursting interneurons generates theta frequency oscillations in neocortex. Neuron. 2003;38:805–817. doi: 10.1016/s0896-6273(03)00300-3. [DOI] [PubMed] [Google Scholar]
- Boden P, Hill RG. Effects of cholecystokinin and pentagastrin on rat hippocampal neurons maintained in vitro. Neuropeptides. 1988;12:95–103. doi: 10.1016/0143-4179(88)90037-6. [DOI] [PubMed] [Google Scholar]
- Bohme GA, Stutzmann JM, Blanchard JC. Excitatory effects of cholecystokinin in rat hippocampal slices inhibits potassium-evoked cholecystokinin release, a possible mechanism contributing to the spatial memory defects produced by cannabinoids. Brain Research. 1988;451:309–318. doi: 10.1016/0006-8993(88)90776-7. [DOI] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Carlberg M, Gundlach AL, Mercer LD, Beart PM. Autoradiographic Localization of Cholecystokinin A and B Receptors in Rat Brain Using [125I]d-Tyr25 (Nle28,31)-CCK 25 - 33S. European Journal of Neuroscience. 1992;4:563–573. doi: 10.1111/j.1460-9568.1992.tb00906.x. [DOI] [PubMed] [Google Scholar]
- Chapman CA, Lacaille JC. Cholinergic induction of theta-frequency oscillations in hippocampal inhibitory interneurons and pacing of pyramidal cell firing. The Journal of Neuroscience. 1999;19:8637–8645. doi: 10.1523/JNEUROSCI.19-19-08637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Davies CH. Cholinergic modulation of hippocampal cells and circuits. Journal of Physiology (London) 2005;562:81–88. doi: 10.1113/jphysiol.2004.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Halasy K, Vida I, Nyiri G, Tamas G, Buhl EH, Somogyi P. Synaptic effects of identified interneurons innervating both interneurons and pyramidal cells in the rat hippocampus. Neuroscience. 1997;79:629–648. doi: 10.1016/s0306-4522(97)00055-9. [DOI] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Cholecystokinin depolarizes rat thalamic reticular neurons by suppressing a K+ conductance. J Neurophysiology. 1995;74:990–1000. doi: 10.1152/jn.1995.74.3.990. [DOI] [PubMed] [Google Scholar]
- Deng PY, Lei S. Bi-directional modulation of GABAergic transmission by cholecystokinin in hippocampal dentate gyrus granule cells of juvenile rats. Journal of Physiology. 2006;572:425–442. doi: 10.1113/jphysiol.2005.104463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockray GJ. Immunochemical evidence of cholecystokinin-like peptides in the brain. Nature. 1976;274:711–713. doi: 10.1038/264568a0. [DOI] [PubMed] [Google Scholar]
- Dodd J, Kelly JS. Excitation of CA1 pyramidal neurones of the hippocampus by the tetra- and octapeptide C-terminal fragments of cholecystokinin. Journal of Physiology. 1979;295:61P–62P. [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B. Quantal analysis of inhibitory synaptic transmission in the dentate gyrus of rat hippocampal slices: a patch-clamp study. Journal of Physiology. 1990;430:213–249. doi: 10.1113/jphysiol.1990.sp018289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro L, Beani L, Trist D, Reggiani A, Bianchi C. Effects of cholecystokinin peptides and GV150013, a selective cholecystokinin B receptor antagonist, on electrically evoked endogenous GABA release from rat cortical slices. Journal of Neurochemistry. 1999;73:1973–1981. [PubMed] [Google Scholar]
- Fisahn A, Pike FG, Buhl EH, Paulsen O. Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nature. 1998;394:186–189. doi: 10.1038/28179. [DOI] [PubMed] [Google Scholar]
- Fischer Y, Wittner L, Freund TF, Gahwiler BH. Simultaneous activation of gamma and theta network oscillations in rat hippocampal slice cultures. Journal of Physiology (London) 2002;539:857–868. doi: 10.1113/jphysiol.2001.013050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DA, Trettel J, Levine ES. Brief trains of action potentials enhance pyramidal neuron excitability via endocannabinoid-mediated suppression of inhibition. Journal of Neurophysiology. 2004;92:2105–2112. doi: 10.1152/jn.00351.2004. [DOI] [PubMed] [Google Scholar]
- Freund TF. Interneuron Diversity series: Rhythm and mood in perisomatic inhibition. Trends in Neurosciences. 2003;26:489–495. doi: 10.1016/S0166-2236(03)00227-3. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M, Manabe T, Kano M. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signaling. European Journal of Neuroscience. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Gillies MJ, Traub RD, LeBeau FEN, Davies CH, Gloveli T, Buhl EH, Whittington MA. A model of atropine-resistant theta oscillations in rat hippocampal area CA1. Journal of Physiology. 2002;543:779–793. doi: 10.1113/jphysiol.2002.024588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Scanziani M. Distinct timing in the activity of cannabinoid-sensitive and cannabinoid- insensitive basket cells. Nature Neuroscience. 2006;9:807–815. doi: 10.1038/nn1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nature Neuroscience. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. The Journal of Neuroscience. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innis RB, Correa FM, Uhl GR, Schneider B, Snyder SH. Cholecystokinin octopeptide- like immunoreactivity: histochemical localization in the rat brain. Proceedings of the National Academy of Sciences of the USA. 1979;77:6917–6921. doi: 10.1073/pnas.76.1.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. The Journal of Neuroscience. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kombian SB, Ananthalakshmi KVV, Parvathy SS, Matowe WC. Cholecystokinin inhibits evoked inhibitory postsynaptic currents in the rat nucleus accumbens indirectly through γ-aminobutyric acid and γ-aminobutyric acid type B receptors. Journal of Neuroscience Research. 2005;79:412–420. doi: 10.1002/jnr.20349. [DOI] [PubMed] [Google Scholar]
- Lenz RA, Wagner JJ, Alger BE. N- and L-type calcium channel involvement in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. Journal of Physiology (London) 1998;512:61–73. doi: 10.1111/j.1469-7793.1998.061bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA, Kerrin J, Davidson J. Inhibition of synaptic transmission in the hippocampus by cholecystokinin (CCK) and its antagonism by a CCK analog (CCK-27-33) Brain Research. 1987;406:130–135. doi: 10.1016/0006-8993(87)90777-3. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. European Journal of Neuroscience. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Martin LA, Alger BE. Muscarinic facilitation of the occurrence of depolarization-induced suppression of inhibition in rat hippocampus. Neuroscience. 1999;92:61–71. doi: 10.1016/s0306-4522(98)00745-3. [DOI] [PubMed] [Google Scholar]
- Martin LA, Wei DS, Alger BE. Heterogeneous susceptibility of GABAA receptor-mediated IPSCs to depolarization-induced suppression of inhibition in rat hippocampus. Journal of Physiology (London) 2001;532:685–700. doi: 10.1111/j.1469-7793.2001.0685e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matto V, Harro J, Allikmets L. The effects of cholecystokinin A and B receptor antagonists on exploratory behaviour in the elevated zero-maze in rat. Neuropharmacology. 1997;36:389–396. doi: 10.1016/s0028-3908(97)00011-7. [DOI] [PubMed] [Google Scholar]
- McMahon LL, Williams JH, Kauer JA. Functionally distinct groups of interneurons identified during rhythmic carbachol oscillations in hippocampus in vitro. The Journal of Neuroscience. 1998;18:5640–5651. doi: 10.1523/JNEUROSCI.18-15-05640.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Muscarinic receptor activity has multiple effects on the resting membrane potentials of CA1 hippocampal interneurons. The Journal of Neuroscience. 1999;19:5693–5702. doi: 10.1523/JNEUROSCI.19-14-05693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer LD, Le VQ, Nunan J, Jones NM, Beart PM. Direct visualization of cholecystokinin subtype2 receptors in rat central nervous system using anti-peptide antibodies. Neuroscience Letters. 2000;293:167–170. doi: 10.1016/s0304-3940(00)01504-4. [DOI] [PubMed] [Google Scholar]
- Miller KK, Hoffer A, Svoboda KR, Lupica CR. Cholecystokinin increases GABA release by inhibiting a resting K+ conductance in hippocampal interneurons. The Journal of Neuroscience. 1997;17:4994–5003. doi: 10.1523/JNEUROSCI.17-13-04994.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KK, Lupica CR. Morphine-induced excitation of pyramidal neurons is inhibited by cholecystokinin in the CA1 region of the rat hippocampal slice. The Journal of Pharmacology and Experimental Therapeutics. 1994;268:753–761. [PubMed] [Google Scholar]
- Nicoll RA, Alger BE. A simple chamber for recording from submerged brain slices. Journal of Neuroscience Methods. 1981;4:153–156. doi: 10.1016/0165-0270(81)90049-2. [DOI] [PubMed] [Google Scholar]
- Palhalmi J, Paulsen O, Freund TF, Hajos N. Distinct properties of carbachol- and DHPG- induced network oscillations in hippocampal slices. Neuropharmacology. 2004;47:381–389. doi: 10.1016/j.neuropharm.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Pawelzik H, Hughes DI, Thomson AM. Physiological and morphological diversity of immunocytochemically defined parvalbumin- and cholecystokinin-positive interneurones in CA1 of the adult rat hippocampus. Journal of Comparative Neurology. 2002;443:346–367. doi: 10.1002/cne.10118. [DOI] [PubMed] [Google Scholar]
- Perez de la Mora M, Hernandez-Gomez AM, Mendez-Franco J, Fuxe K. Cholecystokinin-8 increases K+-evoked [3H]y-aminobutyric acid release in slices from various brain areas. European Journal of Pharmacology. 1993;250:423–430. doi: 10.1016/0014-2999(93)90029-h. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Cholinergic excitation of GABAergic interneurons in the rat hippocampal slice. Journal of Physiology (London) 1992a;450:127–142. doi: 10.1113/jphysiol.1992.sp019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. The Journal of Neuroscience. 1992b;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncer JC, McKinney RA, Gahwiler BH, Thompson SM. Either N- or P-type calcium channels mediate GABA release at distinct hippocampal inhibitory synapses. Neuron. 1997;18:463–472. doi: 10.1016/s0896-6273(00)81246-5. [DOI] [PubMed] [Google Scholar]
- Rehfeld JF. Neuronal cholecystokinin: one or multiple transmitters? Journal of Neurochemistry. 1985;44:1–10. doi: 10.1111/j.1471-4159.1985.tb07105.x. [DOI] [PubMed] [Google Scholar]
- Reich CG, Karson MA, Karnup SV, Jones L, Alger BE. Regulation of IPSP theta rhythms by muscarinic receptors and endocannabinoids in hippocampus. Journal of Neurophysiology. 2005;94:4290–4299. doi: 10.1152/jn.00480.2005. [DOI] [PubMed] [Google Scholar]
- Shinohara S, Kawasaki K. Electrophysiological changes in rat hippocampal pyramidal neurons produced by cholecystokinin octapeptide. Neuroscience. 1997;78:1005–1016. doi: 10.1016/s0306-4522(96)00653-7. [DOI] [PubMed] [Google Scholar]
- Singh L, Lewis AS, Field MJ, Hughes J, Woodruff GN. Evidence for an involvement of the brain cholecystokinin B receptor in anxiety. Proceedings of the National Academy of Sciences of the USA. 1991;88:1130–1133. doi: 10.1073/pnas.88.4.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Hodgson AJ, Smith AD, Nunzi MG, Gorio A, Wu JY. Different populations of GABAergic neurons in the visual cortex and hippocampus of cat contain somatostatin- or cholecystokinin-immunoreactive material. Journal of Neuroscience. 1984;4:2590–2603. doi: 10.1523/JNEUROSCI.04-10-02590.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. The Journal of Neuroscience. 2001;21(1–5):RC188. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA, Traub RD. Interneuron Diversity series: Inhibitory interneurons and network oscillations in vitro. Trends in Neurosciences. 2003;26:676–682. doi: 10.1016/j.tins.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Zarbin MA, Innis RB, Wamsley JK, Snyder SH, Kuhar MJ. Autoradiographic localization of cholecystokinin receptors in the rodent brain. The Journal of Neuroscience. 1983;3:877–906. doi: 10.1523/JNEUROSCI.03-04-00877.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]