

Abstract
In the brain, the extensively studied FAD-dependent enzyme D-amino acid oxidase (DAO) degrades the gliotransmitter D-serine, a potent activator of N-methyl-D-aspartate type glutamate receptors, and evidence suggests that DAO, together with its activator G72 protein, may play a key role in the pathophysiology of schizophrenia. Indeed, its potential clinical importance highlights the need for structural and functional analyses of human DAO. We recently succeeded in purifying human DAO, and found that it weakly binds FAD and shows a significant slower rate of flavin reduction compared with porcine DAO. However, the molecular basis for the different kinetic features remains unclear because the active site of human DAO was considered to be virtually identical to that of porcine DAO, as would be expected from the 85% sequence identity. To address this issue, we determined the crystal structure of human DAO in complex with a competitive inhibitor benzoate, at a resolution of 2.5 Å. The overall dimeric structure of human DAO is similar to porcine DAO, and the catalytic residues are fully conserved at the re-face of the flavin ring. However, at the si-face of the flavin ring, despite the strict sequence identity, a hydrophobic stretch (residues 47–51, VAAGL) exists in a significantly different conformation compared with both of the independently determined porcine DAO–benzoate structures. This suggests that a context-dependent conformational variability of the hydrophobic stretch accounts for the low affinity for FAD as well as the slower rate of flavin reduction, thus highlighting the unique features of the human enzyme.
Keywords: D-amino acid oxidase, Homo sapiens, X-ray crystallography, structurally ambivalent peptides, conformational variability
D-amino acid oxidase (DAO) (EC 1.4.3.3) was first identified by Hans Krebs in 1935 and was later recognized to be the first enzyme known to use FAD as a cofactor (Krebs 1935). DAO noncovalently binds FAD as a prosthetic group and catalyzes the oxidative deamination of D-amino acids to their corresponding imino acids with concomitant reduction of FAD. The reduced flavin is subsequently reoxidized by molecular oxygen generating H2O2, and the imino acid is released into the solvent where it nonenzymatically hydrolyzes, yielding the corresponding α-keto acid and ammonia. DAO exhibits optimal activity toward neutral amino acids and marginal activity toward basic ones; acidic D-amino acids are oxidized by another flavoprotein, D-aspartate oxidase. DAO has been the subject of numerous studies over the past 70 yr, becoming a model for the class of flavin-dependent oxidases (for review, see Pilone 2000).
We previously determined the primary structures of DAO mRNAs isolated from pig kidney (Fukui et al. 1987) and human kidney (Momoi et al. 1988) and also detected a single mRNA species in the brain (Fukui et al. 1988). In addition, we isolated genomic clones of the entire gene from human placental genomic libraries and localized the human gene to chromosome 12 (Fukui and Miyake 1992). Two groups have independently reported the same crystal structure for pig kidney DAO in complex with a competitive inhibitor benzoate at resolutions of 2.6 Å (PDB code 1KIF; Mattevi et al. 1996) and 2.5 Å (PDB code 1VE9; Mizutani et al. 1996). The crystal structure of yeast DAO from Rhodotorula gracilis was determined at a higher resolution of 1.2 Å (Umhau et al. 2000), followed by the structure in complex with o-aminobenzoate (Pollegioni et al. 2002). These three-dimensional structures reported between 1996 and 2002 (six from porcine DAO, four from yeast DAO) have provided us with the molecular basis for our understanding of the mechanism via which this FAD-dependent enzyme acts (Mattevi et al. 1996; Mizutani et al. 1996, 2000; Miura et al. 1997; Todone et al. 1997; Pollegioni et al. 2002; for review, see Pilone 2000). Biochemical characterization of human DAO was not achieved until recently, mainly because of the difficulty of expressing it in a heterologous system such as Escherichia coli (Raibekas et al. 2000). However, we recently succeeded in purifying human DAO and investigating its main functional properties. We found that, in contrast to other known DAO enzymes, human DAO binds FAD only weakly and exists as a stable homodimer, even in the apoprotein form (Molla et al. 2006). The molecular basis for the difference between human DAO and other forms remains unclear because the three-dimensional structure of human DAO was considered to be virtually identical to that of the porcine enzyme, as would be expected from their 85% sequence identity.
From a clinical point of view, new data on the three-dimensional structure of human DAO are highly important because activation of human DAO by G72, which leads to enhanced degradation of the gliotransmitter D-serine, a potent activator of N-methyl-D-aspartate (NMDA)-type glutamate receptors, has been implicated in the pathophysiology of schizophrenia (Chumakov et al. 2002). Indeed, inhibitors acting selectively on human DAO are being intensively sought for clinical purposes (Brandish et al. 2006). Historically, a major advance in the treatment of schizophrenia was achieved in the early 1950s with the introduction of chlorpromazine, a dopamine D2 receptor antagonist (for review, see Sawa and Snyder 2002). It is noteworthy, however, that chlorpromazine also inhibits DAO activity by competing with FAD (Yagi et al. 1956). Because gene variants that result in expression of mutant DAO or G72 proteins have not been identified, it is conceivable that pathogenic mutations at the susceptibility loci or in regulatory regions (e.g., modified expression of DAO or G72) affect degradation of D-serine by DAO, leading to NMDA dysfunction. Our approach to modifying NMDA neurotransmission is to alter the availability of synaptic D-serine by modulating intracellular DAO activity. We previously confirmed that extracellular D-serine is metabolized by DAO expressed in astrocytes and that such activity can be inhibited by application of chlorpromazine (Park et al. 2006). In the present study, we determined the crystal structure of recombinant human DAO in complex with benzoate at a resolution of 2.5 Å. Comparison with the known structure of porcine DAO revealed a remarkable difference in the conformation of the VAAGL hydrophobic stretch, which is located at the si-face of the flavin ring.
Results and Discussion
Purification and crystallization of recombinant human DAO
With an estimated yield of as little as 1 mg of enzyme per liter of culture, human DAO has proven to be a difficult protein to express in recombinant form, making isolation of the pure enzyme a very difficult task (Raibekas et al. 2000). In contrast, 16 mg of recombinant porcine DAO can be obtained per liter of IPTG-induced culture (Setoyama et al. 1996). Nevertheless, we recently succeeded in purifying recombinant human DAO to 85% purity (estimated by SDS-PAGE) in amounts of ∼4.2 mg of enzyme per liter of culture, with an overall purification yield of 60% (Molla et al. 2006). Although this work enabled us to functionally characterize the human enzyme, we were unable to produce crystals of human DAO (T. Kawazoe, H. Tsuge, and K. Fukui, unpubl.). In order to prepare enough enzyme for crystallization, we further explored a wide range of conditions including the IPTG concentration (Fig. 1A) and the purification steps (data not shown). As a result of this effort, we were ultimately able to purify 5.0 mg of enzyme per liter of culture to 95% purity (estimated by SDS-PAGE; Fig. 1B) with an overall purification yield of 55% (Table 1A).
Figure 1.

Purification and crystallization of recombinant human DAO. (A) Western blot of recombinant human DAO produced in E. coli. (B) SDS-PAGE of purified DAO isolated from E. coli. The gel (10%) was stained with Coomassie blue. (Lane 1) Whole cell (containing 20 μg of protein), (lane 2) after heat treatment (59°C, 3 min) and 70% ammonium sulfate fractionation (20 μg of protein), (lane 3) DEAE Sepharose CL-6B column eluate (5 μg of protein), (lane 4) hydroxylapatite column eluate (2 μg of protein). Values indicate the molecular weight of the marker proteins: phosphorylase b (97 kDa), BSA (66 kDa), aldolase (42 kDa), carbonic anhydrase (30 kDa), and soybean trypsin inhibitor (20 kDa). (C) Crystal of human DAO.
Table 1.
Purification and apparent kinetic parameters

To assess the functional characteristics of the purified enzyme, we used an oxygen electrode to detect the consumption of oxygen during catalysis (Table 1B). The gliotransmitter D-Ser, a potent physiological substrate of human DAO, was oxidized by the purified enzyme with an apparent affinity (K m) of 3.6 mM, a value comparable to that of recombinant porcine DAO (Setoyama et al. 2002). Addition of excess benzoate completely inhibited the enzyme activity with an apparent K i of 7 μM. Based on these results, we prepared and crystallized a ternary complex comprised of the purified enzyme bound with FAD and benzoate (Fig. 1C).
Overall structure of the human DAO holoenzyme in complex with benzoate
The crystal structure of human DAO was determined by molecular replacement of the porcine enzyme (PDB code 1AN9; 2.5 Å; Miura et al. 1997). The asymmetric unit contained four molecules of human DAO in the form of two homodimers. Basically, each of the four molecules showed the same conformation, and the overall dimeric structure of human DAO (Fig. 2A) was identical to the “head-to-head” structure of porcine DAO (Mattevi et al. 1996; Mizutani et al. 1996), but different from the “head-to-tail” structure of the yeast enzyme (Pollegioni et al. 2002). The C terminus of the human DAO subunit (residues 341–347) was not clear in the electron density map, which is indicative of the flexibility of this region. The human DAO subunit (residues 1–347; 39 kDa) contained one molecule of noncovalently bound FAD as a cofactor and one molecule of benzoate as an inhibitory substrate analog (Fig. 2B). The Dali score (Holm and Sander 1993) between the human and porcine DAO subunits was 54.2 (RMSD of 0.6 Å for 340 Cα pairs; 85% sequence identity), while that between the human and yeast DAO subunits was 39.1 (RMSD of 1.9 Å for 319 Cα pairs; 28% sequence identity). As shown in Figure 2C, the human DAO subunit contains 11 α-helices and 14 β-strands, which fold into two domains, the FAD-binding domain and the interface domain.
Figure 2.

Overall structure of the human DAO holoenzyme in complex with benzoate. Structural models were prepared with PyMOL (http://www.pymol.org). FAD and benzoate are shown as ball-and-stick representations in A and B. (A) The DAO homodimer colored by secondary structure (helix in red, sheet in yellow, loop in green). (B) The DAO subunit colored spectrum in rainbow from the N terminus (blue) to the C terminus (red). Secondary structure elements are labeled. (C) Topology of the DAO subunit (helix in red, sheet in yellow). The cartoon was manually drawn based on the results of the TOPS algorithm (Michalopoulos et al. 2004). The DAO subunit consists of an FAD-binding domain (residues 1–88, 140–195, 286–340) and an interface domain (residues 89–139, 196–285). The first and the last residues are numbered for each secondary structure element.
Among the 30 residues located at the dimer interface of human DAO, 10 (33%) differ from the corresponding residue in porcine DAO, while 20 (67%) are conserved (Fig. 3). Thus, the frequency of substitution at the dimer interface is higher than the overall substitution frequency (53 residues; 15%). As a consequence, the electrostatic surface potential at the dimer interface of human DAO differs from that of porcine DAO (Fig. 4): The dimer interface of the human enzyme is negatively charged, while that of porcine enzyme is positively charged. We previously reported that the oligomerization state of human DAO significantly differs from that of porcine DAO (Molla et al. 2006). In solution, within a concentration range of 1–24 mg/mL, the human enzyme is always found as a dimeric holoenzyme. In contrast, the porcine enzyme exhibits an oligomerization state that is dependent on the protein concentration. Moreover, in contrast to other known DAO enzymes, the human DAO homodimer is stable even in the apoprotein form, presumably reflecting the different surface properties at the dimer interface.
Figure 3.
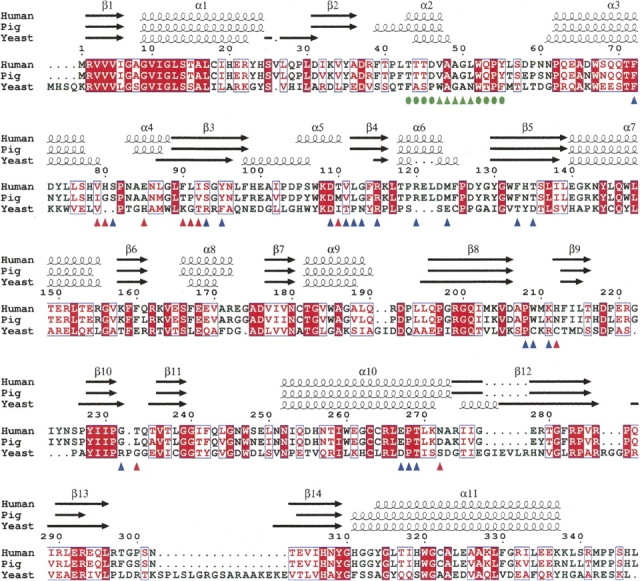
Structure-based sequence alignment of DAO. DAO sequences from human (Swiss-Prot no. P14920), pig (Swiss-Prot no. P00371), and yeast (Swiss-Prot no. P80324) were aligned using ClustalW 1.82 (Thompson et al. 1994) and colored using ESPript 2.2 (Gouet et al. 2003). White in a red box shows strict identity; red in a white box shows similarity within a group; and blue in a white box shows similarity across groups. The hydrophobic stretch is indicated (green triangles) with the residues locally conserved between the human and porcine enzymes (green circles). At the dimer interface of human DAO, 10 residues (red triangles) differ from the porcine enzyme, while 20 residues (blue triangles) are conserved.
Figure 4.

Electrostatic surface potential at the dimer interface. The dimer interface (shown within a dashed circle) of human DAO is negatively charged, while that of porcine DAO is positively charged. The corresponding surface area of the yeast DAO subunit is also shown in a dashed circle. The surface is colored from blue (positive) to red (negative). Only protein atoms were considered for surface calculation by GRASP (Nicholls et al. 1991). The electrostatic surface potential was scaled to a range between −10 kT and 10 kT e−1 (1 V = 38.94 kT e−1 at room temperature).
Context-dependent conformational variability of the VAAGL hydrophobic stretch
As expected from the 85% sequence identity, the active sites were conserved between the human and porcine enzymes at the re-face of the flavin ring (Fig. 5A). At the si-face of the flavin ring, however, we observed an important difference that was not expected, given the strict sequence identity at this region. In both enzymes, the si-face of the flavin ring is covered by a hydrophobic stretch (residues 47–51; VAAGL) (Mizutani et al. 1996), the conformation of which in human DAO differs significantly from both of the independently determined porcine DAO–benzoate complexes (Fig. 5B). The hydrophobic stretch is located inside the molecule, thus its conformation is considered to be rigid, as expected from the low average B-value of 44.6 Å2 for the 29 stretch atoms (the average B-value for the overall protein atoms is 52.2 Å2 as shown in Table 3, below). The human stretches were found to be in an identical conformation in all four molecules within the asymmetric unit (RMS of 0.23 ± 0.04 Å, among 29 atoms), which confirms the stability of the conformation, though a different conformation is favorable to the porcine DAO–benzoate structures. When compared with the overall structural similarity between the human and porcine enzymes (RMS of 0.40 Å for 215 Cα pairs comprising the all α-helices and β-sheets), the deviation of the hydrophobic stretch was evident from the RMS of 0.89 Å (Fig. 5C). This is surprising because the sequence of the hydrophobic stretch (VAAGL) is strictly conserved between the human and porcine enzymes. The stretch conformation in human DAO was confirmed by inspection of omit maps as described in the Materials and Methods section (Fig. 5D).
Figure 5.

Context-dependent conformational variability of the hydrophobic stretch. (A) At the re-face of the flavin ring, the active sites are conserved between the human (carbons in green) and porcine (carbons in magenta; PDB code 1VE9) enzymes. H-bonds are shown as dashed lines. (B) Context-dependent conformational variability of the hydrophobic stretch (residues 47–51) at the si-face of the flavin ring. The hydrophobic stretches of human (carbons in green) and porcine (carbons in magenta; PDB codes 1KIF, 1VE9) DAO showed differing conformations, despite strict sequence identity. The purple intermediate of porcine DAO or the structure in complex with imino tryptophan are also superimposed (carbons in cyan). For clarity, only the H-bond between the flavin N5 atom and the Ala49 backbone N atom in the porcine enzyme (PDB code 1KIF) is shown (dashed line). (C) The conformational difference of the hydrophobic stretch (colored in blue) is evident compared with the overall structural similarity between the human (green) and porcine (magenta) DAO structures. (D) The hydrophobic stretch of human DAO is shown with an F o–F c omit map contoured at 1.0 σ.
Table 3.
Refinement statistics

Because it lacks a side chain, glycine is a conformationally flexible residue. This means that the hydrophobic stretch, which includes a glycine at position 50 (Fig. 3), is capable of adopting a variety of conformations depending upon the environment in which it is located. When the backbone conformations were evaluated in φ/ψ plots of the stretch residues in the human and porcine enzymes (Fig. 6), it was found that despite the strict sequence identity, the φ/ψ combinations of Ala48, Ala49, and Gly50 are diversified among the three DAO–benzoate structures, while those of Val47 and Leu51 are conserved. This means that the VAAGL hydrophobic stretch can be considered a structurally ambivalent peptide (SAP) comprised of five residues (Kuznetsov and Rackovsky 2003). It has been shown that a significant difference between two distinct conformations of the same SAP can be the result of both the overall sequence and the structural properties of the protein harboring the SAP (global context) or the sequence and structural properties of the SAP's flanking regions (local context). In the case of DAO, the conformational variability of the hydrophobic stretch appears to reflect the global context, as the 13 local residues, i.e., the hydrophobic stretch and its flanking regions (residues 43–55; TTTD-VAAGL-WQPY in Fig. 3) are conserved between the two enzymes.
Figure 6.

φ/ψ plots of the hydrophobic stretch (residues 47–51) of human (green) and porcine (red; PDB codes 1KIF, 1VE9) DAO-benzoate structures. The φ/ψ combinations of Ala48, Ala49, and Gly50 are diversified among the three structures, while those of Val47 and Leu51 are conserved. [A, B, L] most favored regions; [a, b, l, p] additional allowed regions; [∼a, ∼b, ∼l, ∼p] generously allowed regions.
Structural implication of the weak FAD binding in human DAO
We previously reported that the K d of the FAD-apoenzyme complex is 40-fold higher for human DAO (8 ± 2 μM) than porcine DAO (0.2 μM) (Molla et al. 2006), and that the affinity for FAD is even stronger in yeast DAO (K d = 0.02 μM) (summarized in Molla et al. 2006). The different affinities of the porcine and yeast enzymes for FAD have been attributed to the environment at the flavin O2 position (Pollegioni et al. 2002). In porcine DAO, the partial positive charge of a dipole induced by helix α11 (residues 311–337 in Fig. 3) is presumed to contribute to the stabilization of the negative charge of the reduced flavin (Mattevi et al. 1996), a feature that is absent in yeast DAO (Pollegioni et al. 2002). The flavin O2 atom is H-bonded to Thr317 in both the human and porcine enzymes (Fig. 5A), whereas in yeast DAO, the atom is tightly H-bonded to Tyr338 and Gln339 (Pollegioni et al. 2002). To confirm the effect of Thr317 on FAD binding, Setoyama et al. (2002) designed and expressed a porcine DAO T317A substitution mutant and noted that the mutant enzyme had a lower affinity for FAD. On the other hand, the different affinities for FAD of the porcine and human enzymes cannot be explained by the Thr317 position, given the overall structural conservation at the re-face of the flavin ring (Fig. 5A).
In order to provide a structural basis for the observed kinetic difference between human and porcine DAO, we compared the FAD-binding patterns of the two enzymes. Aside from the conformation of the hydrophobic stretch, no remarkable differences were observed, at least within 6 Å of FAD, which indicates that the hydrophobic stretch plays an important role in determining the affinity for FAD. As compared with porcine DAO, the hydrophobic stretch in the human enzyme is shifted away from the FAD, resulting in the loss of the H-bond between the flavin N5 atom and the backbone N atom of Ala49 with the distance of 3.9 Å (Fig. 5B).
A noticeable shift in the hydrophobic stretch also occurs in the reduced state of porcine DAO when the enzyme is complexed with the reaction product imino tryptophan (PDB code 1DDO; 3.1 Å; Todone et al. 1997) and in the structure of the purple intermediate (a complex between the dehydrogenated product and the reduced form of DAO) (PDB code 1EVI; 2.5 Å; Mizutani et al. 2000) (Fig. 5B). In the porcine enzyme, the length of the H-bond between the flavin N5 atom and the Ala49 backbone N atom is 3.0 Å in the DAO–benzoate complex but is increased to 3.3 Å in the complex with imino tryptophan. Todone et al. (1997) suggested that upon reduction the flavin N5 atom most likely becomes protonated, causing the H-bond with the Ala49 backbone N atom to be weakened or even lost in the reduced enzyme.
Even when human DAO is in the oxidized state, the shift in the hydrophobic stretch is more apparent than that seen in the reduced porcine enzyme and raises the question as to how the conformation of the stretch affects the kinetic scheme of the reaction catalyzed by human DAO. Our kinetic data indicate that the rate of flavin reduction is slower in the human enzyme (180 ± 20 sec−1) than in the porcine enzyme (4000 sec−1 [Molla et al. 2006]; 1225 sec−1 [Pollegioni et al. 1994]), presumably reflecting the different conformations of the hydrophobic stretch. But further analysis of the structure of human DAO in the reduced state will be necessary to fully understand the effect of the stretch conformation on the enzyme's kinetics.
In summary, three-dimensional structural analysis of human DAO revealed that a SAP located at the si-face of the flavin ring exists in a significantly different conformation than in porcine DAO. The context-dependent difference in the conformation of the hydrophobic stretch is thought to be a key determinant of the enzyme's affinity for FAD as well as the rate of flavin reduction, thus highlighting the unique features of human DAO. Although purification and crystallization of human DAO is a very difficult task (in large part because of its low affinity for FAD), we are currently working on determining the structure of human DAO in the reduced state. In the present study, we provide the first structural evidence to explain the kinetic difference between the human and porcine enzymes, hopefully facilitating the understanding of this enigmatic enzyme, which may be pivotal for the treatment of disorders related to NMDA dysfunction, such as schizophrenia.
Protein Data Bank accession codes
The atomic coordinates and structure factors (code 2DU8) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Materials and methods
Subcloning
The cDNA encoding human kidney DAO (Momoi et al. 1988) was amplified using the N-terminal primer 5′-TCCGGCTGCTCATATGCGTGTGGTGGTGA-3′ and the C-terminal primer 5′-GCAGCAGTCACATATGTCTTCAGAGGTGG-3′. The PCR product was digested with NdeI and ligated into the similarly restricted pET-11b E. coli T7 expression vector (Novagen). The ligated product was introduced into E. coli nonexpression host DH5α supercompetent cells. The resultant construct was confirmed by DNA sequencing.
Production
The construct was introduced into an E. coli BL21(DE3) strain, after which a single colony of transformants was grown in cultures and stored in 50% (v/v) glycerol at −80°C. Transformants were cultured in terrific broth (1.2% tryptone, 2.4% yeast extract, and 0.4% [v/v] glycerol) with 0.5% (w/v) glucose and 50 mg/L ampicillin at 37°C to an optical density of 0.6 at 600 nm, and then induced with 0.1 mM IPTG. After an additional 24 h, the cells were harvested by centrifugation at 4°C. Usually 30 g of wet cell pellet were obtained from 4 L of culture and were kept frozen at −80°C until used.
Western blotting
Equal amounts of total cellular protein were fractionated on a 12.5% polyacrylamide gel and then transferred to a nitrocellulose membrane (Millipore) at room temperature. DAO was visualized using an ECL detection system (Amersham Biosciences) after incubation with rabbit polyclonal anti-pig kidney DAO primary antibody and a horseradish peroxidase-conjugated donkey anti-rabbit IgG secondary antibody (Promega KK).
Purification
The recombinant human DAO was purified using a modification of the procedure used to purify recombinant porcine DAO (Setoyama et al. 1996). The bacterial pellet was suspended (10 mL/g cell) in 17 mM Na pyrophosphate (pH 8.3) buffer containing 100 μM FAD, 1 mM Na benzoate, 0.3 mM EDTA, 0.5 mM DTT, and 4.5 μg/mL PMSF, after which the cells were disrupted by treatment with 1 mg/mL lysozyme for 1 h, followed by sonication for 30 sec, four times. The disrupted cells were treated with 1% (w/v) streptomycin sulfate, after which the cell debris was removed by centrifugation, and the soluble fraction was heated at 59°C for 3 min and then rapidly cooled to <10°C in an ice-water bath. The denatured proteins were removed by centrifugation, and the supernatant was precipitated with 70% ammonium sulfate. After dialysis overnight against buffer A (10 mM Tris-HCl at pH 8.0, 125 mM KCl, 10 μM FAD, 200 μM Na benzoate, and 4.5 μg/mL PMSF) followed by centrifugation, the supernatant was applied to an anion-exchange DEAE Sepharose CL-6B (Sigma) column (3 × 30 cm) equilibrated with buffer A without FAD, and the column eluate was fractionated. Yellow fractions, which contained the DAO holoenzyme, were detected based on the OD455/OD280 ratio and SDS-PAGE, then pooled and precipitated with 70% ammonium sulfate. After dialysis overnight against buffer B (50 mM Na phosphate at pH 6.8, 10 μM FAD, and 200 μM Na benzoate) followed by centrifugation, the supernatant was applied to a hydroxylapatite (nacalai) column (1 × 50 cm) equilibrated with buffer B without FAD, and the column eluate was fractionated. Again, the yellow fractions were detected based on the OD455/OD280 ratio and SDS-PAGE, pooled, and precipitated with 70% ammonium sulfate. The resultant purified protein was confirmed as a single band on SDS-PAGE (Fig. 1B). Protein concentrations were determined with a BCA Protein Assay Kit (Pierce) using BSA as a standard, or for the purified DAO, using an extinction coefficient previously obtained with porcine DAO (11.3 mM−1cm−1 at 455 nm). The N-terminal 10 residues of the purified enzyme were confirmed by protein sequencing.
Kinetic analyses
DAO activity was measured in oxygraphic assays using a modification of the method used to characterize recombinant porcine DAO (Miyano et al. 1991). A Clark oxygen electrode (Gilson, model 5/6 Oxygraph) was used for the assays. The standard reaction mixture contained DAO and 50 μM FAD in a total volume of 1.8 mL. The reactions were initiated by the addition of DAO and carried out in 50 mM Na pyrophosphate buffer (pH 8.3) at 25°C. The Michaelis constant (K m) and turnover number (k cat) were estimated from double reciprocal plots of the initial velocity versus the substrate concentration (Table 1B). The inhibition constant (K i) for benzoate was estimated from double reciprocal plots of the initial velocity versus the D-Pro concentration in the presence of benzoate (0–20 μM) (Table 1B).
Crystallization
The ammonium sulfate precipitant of the purified enzyme was dialyzed overnight against buffer containing 10 mM Na citrate (pH 8.0), 20 μM FAD, and 400 μM Na benzoate at 4°C, and then concentrated to 10 mg/mL. Crystallization conditions were screened using the hanging-drop vapor diffusion method. Yellow crystals were obtained from polyethylene glycol (PEG) 4000, ammonium acetate, and Na citrate (pH 8.0) at 20°C. Further screening resulted in single crystals after mixing 2 μL of protein sample (10 mg/mL) with the same volume of the reservoir solution (10% [w/v] PEG 4000, 0.2 M ammonium acetate, 0.1 M Na citrate at pH 8.0, and 12% [v/v] glycerol). The 12% (v/v) glycerol enhanced the crystal quality. Crystals grew to an average size of 0.1 × 0.1 × 0.05 mm in 10 d (Fig. 1C).
Data collection
The data collection statistics are summarized in Table 2. Using KEK Bl5a at the Photon Factory (Tsukuba, Japan), the native data were collected at 2.5 Å resolution using monochromatized radiation at λ = 1.0 Å and a ADSC Quantum 315 CCD detector. The distance between the crystal and detector was 350 mm, and the scan angle was 1.0°. Data were processed using the HKL2000 software package (Otwinowski and Minor 1997).
Table 2.
Data collection statistics

Structural determination and refinement
The refinement statistics are summarized in Table 3. Structural determination and refinement calculations were carried out using the CCP4 suite (Collaborative Computational Project, Number 4 1994). The structure was solved by molecular replacement using MOLREP with the porcine DAO dimer (Miura et al. 1997) serving as the search model. The rotation function and the translation function clearly identified the positions of two DAO dimers (Rfac = 0.543/Correlation coefficient = 0.551). Model building was carried out manually using XTALVIEW. FAD and benzoate were built into the difference electron density using strict noncrystallographic symmetry. Since the former data (2.4 Å) have a low degree of completeness (92.7% [85.0%]), we refined the model against other 2.5 Å data. However, both structures were basically the same and included the same conformation of the VAAGL hydrophobic stretch. Refinement was carried out using Refmac5 (Murshudov et al. 1997) and CNS (Brünger et al. 1998). SIGMAA-weighted maps calculated with coefficients 2F o–F c and F o–F c were used for the model rebuilding. The final model consists of residues 1–340 for each of the four DAO subunits. Water molecules with thermal factors >70 Å2 after refinement were excluded from the list. After the final round of refinement, the stereochemistry of the structure was assessed with PROCHECK, taking particular care of the conformation of the hydrophobic stretch. The conformation was confirmed by an omit map using a simulated annealing method after omitting the VAAGL hydrophobic stretch.
Acknowledgments
We thank Nobuhiko Katunuma and Kenji Aki for discussions; Hisaaki Taniguchi, Kazuko Fujiwara, and Kazuko Okamura-Ikeda for discussions on crystallization conditions; and Ritsu Chounan for help with the protein sequencing. H.T. thanks Masamichi Ikeguchi (Department of Bioinformatics, Faculty of Engineering, Soka University) for discussions on the context-dependent secondary structure formation. This work was supported in part by a Grant-in-Aid for Scientific Research and a Grant for the 21st Century COE Program from the Ministry of Education, Science, Sports, and Culture of Japan; a Grant for Scientific Research from the Japan Society for the Promotion of Science; and a Research Grant from the Ministry of Health, Labor and Welfare of Japan. This work also was supported in part by the National Project on Protein Structural and Functional Analyses from the MEXT of Japan.
Footnotes
Reprint requests to: Kiyoshi Fukui, The Institute for Enzyme Research, The University of Tokushima, 3-18-15 Kuramoto, Tokushima 770-8503, Japan; e-mail: kiyo@ier.tokushima-u.ac.jp; fax: 81-88-633-7431.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062421606.
References
- Brandish, P.E., Chiu, C.S., Schneeweis, J., Brandon, N.J., Leech, C.L., Kornienko, O., Scolnick, E.M., Strulovici, B., and Zheng, W.J. 2006. A cell-based ultra-high-throughput screening assay for identifying inhibitors of D-amino acid oxidase. J. Biomol. Screen. 11: 481–487. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., and Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Chumakov, I., Blumenfeld, M., Guerassimenko, O., Cavarec, L., Palicio, M., Abderrahim, H., Bougueleret, L., Barry, C., Tanaka, H., and La Rosa, P., et al. 2002. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc. Natl. Acad. Sci. 99: 13675–13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Fukui, K. and Miyake, Y. 1992. Molecular cloning and chromosomal localization of a human gene encoding D-amino-acid oxidase. J. Biol. Chem. 267: 18631–18638. [PubMed] [Google Scholar]
- Fukui, K., Watanabe, F., Shibata, T., and Miyake, Y. 1987. Molecular cloning and sequence analysis of cDNAs encoding pig kidney D-amino acid oxidase. Biochemistry 26: 3612–3618. [DOI] [PubMed] [Google Scholar]
- Fukui, K., Momoi, K., Watanabe, F., and Miyake, Y. 1988. In vivo and in vitro expression of pig D-amino acid oxidase: In vitro system for the synthesis of a functional enzyme. Biochemistry 27: 6693–6697. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Robert, X., and Courcelle, E. 2003. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31: 3320–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233: 123–138. [DOI] [PubMed] [Google Scholar]
- Krebs, H.A. 1935. Metabolism of amino-acids. III. Deamination of amino-acids. Biochem. J. 29: 1620–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov, I.B. and Rackovsky, S. 2003. On the properties and sequence context of structurally ambivalent fragments in proteins. Protein Sci. 12: 2420–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattevi, A., Vanoni, M.A., Todone, F., Rizzi, M., Teplyakov, A., Coda, A., Bolognesi, M., and Curti, B. 1996. Crystal structure of D-amino acid oxidase: A case of active site mirror-image convergent evolution with flavocytochrome b2. Proc. Natl. Acad. Sci. 93: 7496–7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalopoulos, I., Torrance, G.M., Gilbert, D.R., and Westhead, D.R. 2004. TOPS: An enhanced database of protein structural topology. Nucleic Acids Res. 32: D251–D254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, R., Setoyama, C., Nishina, Y., Shiga, K., Mizutani, H., Miyahara, I., and Hirotsu, K. 1997. Structural and mechanistic studies on D-amino acid oxidase·substrate complex: Implications of the crystal structure of enzyme·substrate analog complex. J. Biochem. 122: 825–833. [DOI] [PubMed] [Google Scholar]
- Miyano, M., Fukui, K., Watanabe, F., Takahashi, S., Tada, M., Kanashiro, M., and Miyake, Y. 1991. Studies on Phe-228 and Leu-307 recombinant mutants of pig kidney D-amino acid oxidase: Expression, purification, and characterization. J. Biochem. 109: 171–177. [DOI] [PubMed] [Google Scholar]
- Mizutani, H., Miyahara, I., Hirotsu, K., Nishina, Y., Shiga, K., Setoyama, C., and Miura, R. 1996. Three-dimensional structure of pig kidney D-amino acid oxidase at 3.0 Å resolution. J. Biochem. 120: 14–17. [DOI] [PubMed] [Google Scholar]
- Mizutani, H., Miyahara, I., Hirotsu, K., Nishina, Y., Shiga, K., Setoyama, C., and Miura, R. 2000. Three-dimensional structure of the purple intermediate of porcine kidney D-amino acid oxidase. Optimization of the oxidative half-reaction through alignment of the product with reduced flavin. J. Biochem. 128: 73–81. [DOI] [PubMed] [Google Scholar]
- Molla, G., Sacchi, S., Bernasconi, M., Pilone, M.S., Fukui, K., and Pollegioni, L. 2006. Characterization of human D-amino acid oxidase. FEBS Lett. 580: 2358–2364. [DOI] [PubMed] [Google Scholar]
- Momoi, K., Fukui, K., Watanabe, F., and Miyake, Y. 1988. Molecular cloning and sequence analysis of cDNA encoding human kidney D-amino acid oxidase. FEBS Lett. 238: 180–184. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Park, H.K., Shishido, Y., Ichise-Shishido, S., Kawazoe, T., Ono, K., Iwana, S., Tomita, Y., Yorita, K., Sakai, T., and Fukui, K. 2006. Potential role for astroglial D-amino acid oxidase in extracellular D-serine metabolism and cytotoxicity. J. Biochem. 139: 295–304. [DOI] [PubMed] [Google Scholar]
- Pilone, M.S. 2000. D-Amino acid oxidase: New findings. Cell. Mol. Life Sci. 57: 1732–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollegioni, L., Fukui, K., and Massey, V. 1994. Studies on the kinetic mechanism of pig kidney D-amino acid oxidase by site-directed mutagenesis of tyrosine 224 and tyrosine 228. J. Biol. Chem. 269: 31666–31673. [PubMed] [Google Scholar]
- Pollegioni, L., Diederichs, K., Molla, G., Umhau, S., Welte, W., Ghisla, S., and Pilone, M.S. 2002. Yeast D-amino acid oxidase: Structural basis of its catalytic properties. J. Mol. Biol. 324: 535–546. [DOI] [PubMed] [Google Scholar]
- Raibekas, A.A., Fukui, K., and Massey, V. 2000. Design and properties of human D-amino acid oxidase with covalently attached flavin. Proc. Natl. Acad. Sci. 97: 3089–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa, A. and Snyder, S.H. 2002. Schizophrenia: Diverse approaches to a complex disease. Science 296: 692–695. [DOI] [PubMed] [Google Scholar]
- Setoyama, C., Miura, R., Nishina, Y., Shiga, K., Mizutani, H., Miyahara, I., and Hirotsu, K. 1996. Crystallization of expressed pig kidney D-amino acid oxidase and preliminary X-ray crystallographic characterization. J. Biochem. 119: 1114–1117. [DOI] [PubMed] [Google Scholar]
- Setoyama, C., Nishina, Y., Tamaoki, H., Mizutani, H., Miyahara, I., Hirotsu, K., Shiga, K., and Miura, R. 2002. Effects of hydrogen bonds in association with flavin and substrate in flavoenzyme D-amino acid oxidase. The catalytic and structural roles of Gly313 and Thr317. J. Biochem. 131: 59–69. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todone, F., Vanoni, M.A., Mozzarelli, A., Bolognesi, M., Coda, A., Curti, B., and Mattevi, A. 1997. Active site plasticity in D-amino acid oxidase: A crystallographic analysis. Biochemistry 36: 5853–5860. [DOI] [PubMed] [Google Scholar]
- Umhau, S., Pollegioni, L., Molla, G., Diederichs, K., Welte, W., Pilone, M.S., and Ghisla, S. 2000. The X-ray structure of D-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. Proc. Natl. Acad. Sci. 97: 12463–12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi, K., Nagatsu, T., and Ozawa, T. 1956. Inhibitory action of chlorpromazine on the oxidation of D-amino-acid in the diencephalon part of the brain. Nature 177: 891–892. [DOI] [PubMed] [Google Scholar]