

Abstract
Photonic induced immobilization is a novel technology that results in spatially oriented and spatially localized covalent coupling of biomolecules onto thiol-reactive surfaces. Immobilization using this technology has been achieved for a wide selection of proteins, such as hydrolytic enzymes (lipases/esterases, lysozyme), proteases (human plasminogen), alkaline phosphatase, immunoglobulins’ Fab fragment (e.g., antibody against PSA [prostate specific antigen]), Major Histocompability Complex class I protein, pepsin, and trypsin. The reaction mechanism behind the reported new technology involves “photonic activation of disulfide bridges,” i.e., light-induced breakage of disulfide bridges in proteins upon UV illumination of nearby aromatic amino acids, resulting in the formation of free, reactive thiol groups that will form covalent bonds with thiol-reactive surfaces (see Fig. 1). Interestingly, the spatial proximity of aromatic residues and disulfide bridges in proteins has been preserved throughout molecular evolution. The new photonic-induced method for immobilization of proteins preserves the native structural and functional properties of the immobilized protein, avoiding the use of one or more chemical/thermal steps. This technology allows for the creation of spatially oriented as well as spatially defined multiprotein/DNA high-density sensor arrays with spot size of 1 μm or less, and has clear potential for biomedical, bioelectronic, nanotechnology, and therapeutic applications.
Keywords: biosensors, protein sensor arrays, protein immobilization, spatially oriented arrays, light-induced protein immobilization
Molecules can be immobilized on a carrier or a solid surface either passively through hydrophobic or ionic interactions, or covalently by attachment to activated surface groups. In response to the enormous importance of immobilization for solid phase chemistry, biological screening, and nanotechnology applications, the analytical uses of the technology have been widely explored. Immobilization technology has found broad application in many different areas of biotechnology, e.g., diagnostics, biosensors, affinity chromatography, immobilization of molecules in ELISA assays, and immobilization of molecules onto nanoparticles for drug/gene delivery. The value of immobilization techniques is demonstrated by the recent boost in the development of DNA and protein microarrays’ technologies, and is needed for the development of drug/gene delivery technologies for therapeutical purposes (e.g., cancer therapy, gene therapy). Common for most of the described immobilization methods is their use of one or more thermochemical/chemical steps, sometimes involving reagents that are likely to have a degrading effect on the structure and/or function of the bound protein (Johnsson et al. 1991; Collioud et al. 1993; Veilleux 1996; Sweeney et al. 2000; Wilson and Nock 2002). Also, commonly used protein immobilization methods lead to a random orientation of the proteins immobilized on a carrier, with the significant risk of lower biological activity and raised detection limits. There is a clear need in the science of protein immobilization to improve the immobilization method, where the structural and the functional properties of the immobilized component are retained and the orientation of the biomolecule can be controlled.
We report here a new photonic-induced method for immobilization of proteins and other biomolecules onto a carrier via disulfide bonds, which results in precise knowledge of the proteins’ attachment point to the surface while preserving the native structural and functional properties of the immobilized protein, avoiding the use of one or more chemical/thermal steps. The reaction mechanism behind the reported new technology involves “photonic activation of disulfide bridges,” i.e., light-induced breakage of disulfide bridges in proteins upon UV illumination of nearby aromatic amino acids, resulting in the formation of free, reactive thiol groups that will form covalent bonds with thiol reactive surfaces. This new technology (patent pending; Petersen and Neves-Petersen 2005) makes use of the spatial proximity between aromatic residues and disulfide bridges in proteins (Petersen et al. 1999). This constellation is present in many protein families such as hydrolases, oxidoreductases, transferases, lyases, Major Histocompability Complex (MHC) class proteins, and membrane receptor proteins, and is present in all members of the immunoglobulin superfamily (Ioerger et al. 1999), making these proteins candidates for photonic-induced protein immobilization. Immunoglobulins have a great biomedical relevance of in disease prognostic. For example, levels of the PSA (prostate specific antigen) protein can be correlated with the development of prostate cancer. Also, PSA is a new prognostic indicator for women with breast cancer. The 3D structures available for domains of Fab fragments of IgG immunoglobulins show that all of them are rich in the closely spaced triad of residues Aromatic/Cys–Cys. Each Fab fragment contains several of these triads. The intradomain disulfide bridges present in the CH, VH, CL, and VL domains are not solvent-accessible. An additional disulfide bridge bridging the light (L) and heavy (H) chains is present located far from the antigen binding site (see Fig. 1). This disulfide bridge is solvent-accessible, and in many cases located nearby Trp/Tyr residues. It is likely to induce opening of this interdomain disulfide bridge upon UV illumination of the nearby aromatic residue. Once this interdomain disulfide bridge is broken and free thiol groups have been formed, these solvent accessible groups can react with a gold (Au) or thiol-rich surface creating a covalent bond to the surface, immobilizing the immunoglobulin in a sterically defined way. The antigen binding site located distal to the Fab fragment’s binding site on the surface will maintain its availability to antigen recognition.
Figure 1.

UV light-induced immobilization illustrated with an immunoglobulin Fab fragment onto a thiol-derivatized surface. The disulfide bridge (red) located near a Tryptophan residue (blue) is located far away from the antigen binding site.
Results
UV light-induced protein immobilization studies
TIRF studies
In order to verify that the proteins were immobilized, upon illumination, onto, e.g., quartz surfaces, a gentle flow of protein solution was guided onto a thiol-derivatized quartz surface, and Total Internal Reflection Fluorescence (TIRF) was monitored using 280 nm (excites both Trp and Tyr residues in proteins) or 296 nm (selectively excites Trp residues in proteins) excitation light and 330 nm for emission detection. Since TIRF is only sensitive to a very thin surface layer of the order of λexc/2 above the quartz surface, it is ideal to monitor protein immobilization. Three sets of experiments were performed here exemplified for cutinase, a typical member of the hydrolase family: one where the protein solution passed over a thiol-derivatized quartz slide in the presence of UV light, a second experiment where the protein solution passed over a thiol-derivatized quartz slide in the absence of UV light, and a third experiment with a slide that had not been thiolated. All three slides were subsequently carefully washed and TIRF emission registered. As can be seen in Figure 2, native cutinase shows a strong fluorescence signal when immobilized on the thiol-derivatized slide in the presence of UV light, whereas if left in the dark or when nonthiol-derivatized slides were used, only a weak fluorescence was detectable.
Figure 2.

TIRF data confirming UV (296 nm) induced immobilization of cutinase molecules on a thiol-derivatized quartz glass surface. When native cutinase was illuminated both with 296-nm and 280-nm UV light, data showed protein immobilization on thiol-derivatized surfaces (see Cutinase Native, SH/UV296 [first bar] and Cutinase Native, SH/UV280 [fourth bar]; 296 means that illumination was carried out with 296-nm light, which exclusively excites the single tryptophan; 280 means that illumination was carried out with 280-nm light, which excites all aromatic residues). Almost no protein immobilization was achieved when the protein was not being UV-illuminated while flowing on the thiolated (SH) slide surface (see Cutinase Native, SH/Dark/UV296 [second bar]; UV296 means that after flowing the protein on the slide surface, TIRF was monitored exciting the slide surface with 296-nm light). Also, almost no protein immobilization was achieved when using a bare (OH) quartz slide (non-thiolated slide) (see Cutinase Native, OH/UV296 [third bar]), despite the fact that the protein was being UV-illuminated with 296-nm light while flowing on the slide. UV illumination of native and W69A mutant cutinase (Trp-depleted mutant) with 280-nm light revealed UV-induced immobilization of the proteins to a different extent: Native cutinase showed an approximately two times larger extent of light-induced immobilization onto thiolated slides (cf. Cutinase Native, SH/UV280 [fourth bar] and Cutinase W69A, SH/UV280 [fifth bar]).
Investigating the role of the aromatic residue in the photo-induced mechanism
In solution, it has been shown that UV illumination of native cutinase is associated with the formation of free thiol groups (Prompers et al. 1999; Neves-Petersen et al. 2002; Vanhooren et al. 2002), consistent with breakage of SS bonds (see Fig. 3). In order to investigate the molecular mechanism of the photon-induced reaction, we also studied the Trp-depleted mutant W69A cutinase. As reported previously (Weisenborn et al. 1996; Prompers et al. 1999; Neves-Petersen et al. 2002), as a consequence of the disruption of the disulfide bridge adjacent to the Trp residue, the fluorescence emission intensity of Trp increases >10-fold with increasing illumination time in concert with the disappearance of intact disulfide bridges, known to be excellent fluorescence quenchers. Upon UV illumination of the Trp-depleted mutant cutinase we no longer observe a large fluorescence increase upon illumination as observed for native cutinase.
Figure 3.

Correlation between the UV illumination time of a 17 μM cutinase protein solution (excitation at 296 nm) and the detected concentration of free thiol groups formed upon UV illumination. Free thiols were detected with the Ellman assay, upon reaction with 5,5′-dithiobis-(2-nitrobenzoic acid) (Ellman 1959; Riddles et al. 1983). Free thiol concentration is proportional to the absorbance value at 412 nm, as previously described (Neves-Petersen et al. 2002).
Detection of thiol groups’ concentration formed upon UV illumination of cutinase
Data displayed in Table 1 report the concentration of SH groups formed upon UV illumination of cutinase molecules. It can be observed that the presence of the Trp residue dramatically increases the likelihood of SS bond opening in the protein. Table 1 also shows that 280-nm UV excitation of Tyr residues of the mutant cutinase also leads to disulfide bridge disruption (~ 50% when compared to native cutinase), and that the disulfide bridge itself, to a small extent, is labile toward UV light.
Table 1.
Concentration of thiol (SH) groups formed upon UV illumination per mole of molecule (native or mutant W69A) irradiated (with 280 nm and 296 nm light)
| [SH]/[protein] | 280 nm | 296 nm |
| Native | 58.2% | 65% |
| W69A mutant | 25.9% | 6.5% |
Light-induced immobilization and protein microarrays
Figure 4 shows the first six-spot microarray developed using UV light-induced immobilization of cutinase molecules (light has been focused down to a few micrometers). The six peaks reveal the fluorescence emission intensity of the product umbelliferone formed upon substrate (methyl-umbelliferyl butyrate) catalysis by cutinase.
Figure 4.

Esterase activity detected on six-spot microarray of cutinase molecules covalently immobilized on a thiol derivatized glass surface after exposure to 280-nm laser light focused down to 100 μm for 1 min (16.8 μJ/spot).
Light-induced immobilization of IgG1-Fab fragments and other proteins
UV-induced immobilization of function IgG1-Fab fragments (Mouse IgG1 Fab fragment and antibody against PSA) alkaline phosphatase, proteases, and MHC class I (Snabe et al. 2005b) were also confirmed (data not shown).
Immunoassay after IgG1-Fab fragments immobilization
Antigen binding activity of the immobilized IgG1 Fab fragments has been detected as described in Materials and Methods. Western blotting of the control (non-illuminated) and illuminated mouse Fab fragments revealed no difference in antibody recognition.
Bioinformatic studies
Additional bioinformatics studies revealed several medically relevant proteins as suitable candidates for UV light-induced immobilization. Among those is a long list of immunoglobulins (IgGs). Fab fragments from IgGs are structurally identical, the variation lying mainly in the epitope region (the antigen recognition site). Interesting examples of antibodies found that are good candidates for UV light-induced immobilization are antibody against insulin (IgG1 insulin antibodies, IgG2a insulin receptor antibodies, IgG1k proinsulin antibodies), antibodies against cytokines (interferons, e.g., IFN-γ; interleukines, IL), antibodies against viral proteins (influenza A, influenza B), antibodies against T-cells, and antibodies against other antibodies. Interesting is the fact that the presence of aromatic residues nearby disulfide bridges seems to be preserved by nature also in receptor-type proteins. According to SCOP (structural classification of proteins), an interferon-γ receptor α-chain from humans belongs to the fibronectic type III family. This family of proteins includes the growth hormone receptor, the prolactin receptor, and the EPO (Erythropoietin) receptor. Common to all these proteins, and most likely for other proteins in this family, is that they display conserved nearby aromatic-disulfide bridge triads, having this way the potential for UV light-induced immobilization. Other members of this family are neural cell adhesion molecules 1, interleukin-4 receptor α-chain, cytokine receptor gp130 cytokine-binding domains, interleukin-10 receptor, interleukin-4 receptor, and interferon-α/β receptor.
Discussion
We have presented a new photonic technology used to immobilize proteins in a spatially oriented way to sensor surfaces. We have shown that highly interesting proteins can be immobilized with our technology (immunoglobulins, alkaline phosphatase, hydrolases, proteases, MHC class I). Regarding the results obtained with our model protein cutinase, data displayed in Table 1 are correlated with the 280-nm TIRF data displayed in Figure 2, since the ratio between the TIRF emission intensity of mutant protein immobilized is ~ 50% of the TIRF emission intensity of immobilized native cutinase, strongly indicating that the mechanism for opening the disulfide bridges (SS) is highly dependent on the immediate neighborhood: SS-Trp>SS-Tyr>SS. Undergoing work on the reaction mechanism behind the UV light-induced reactions has revealed that electron ejection from the side chains of aromatic residues occurs upon UV illumination of aromatic amino acids (M.T. Neves-Petersen, S. Klitgaard, V. Sundström, T. Polivka, A. Yartsev, T. Pasche, and S.B. Petersen, in prep.)
This new technology provides spatially controlled molecular immobilization since the UV light-induced immobilization of each biomolecule to a support surface can be limited to the focal point of illumination with dimensions as small as a few micrometers, as confirmed by our first protein microarray displayed in Figure 4. Due to the biomedical relevance of immunoglobulins in disease prognostic, our lab will now proceed with the construction of multiple densely-packed Fab sensor surfaces. The Fab fragments from IgGs are structurally identical, the variation lying mainly in the epitope region (the antigen recognition site). The “binding site” for UV light-induced immobilization is fairly conserved, and we have already shown that we can induce with light the immobilization of Fab fragments. Once immobilized the different Fab fragments could detect a multitude of different proteins.
The presented new technology allows for an extremely dense packing of identifiable and different molecules on a support surface ideal for charging microarrays with molecules aiming at the creation of protein nanobiosensors. Since the density of immobilized sensor proteins in a small area is extremely high, the development of very advanced high-density multipotent biosensors for human diagnostic is within reach. Additionally, the use of plasmon-assisted immobilization on, e.g., gold surfaces will increase the sensitivity of the biosensor through surface plasmon coupled emission. Also, the area onto which the biosensor is immobilized could further be reduced, since we would not be limited by the optical resolution of the excitation light (approximately half wavelength) but by the resolution of the plasmon wave (almost an order of magnitude shorter). Interestingly, this technology may bypass the use of microdispensers broadly applied in microarray technology since the spatial resolution of this technique will be defined by the area of the sensor surface that is illuminated and not by the physical size of the dispensed droplets of sensor molecules. This new technology is ideal to couple drugs, proteins, peptides, and other molecules to nanoparticles such as gold nanospheres, which can subsequently be used as molecular carriers into cells for therapeutic purposes.
Materials and methods
Protein immobilization on thiol-derivatized quartz slides
Proteins were immobilized upon shinning UV light on SH-activated quartz slides while using a TIRF setup according to the procedures described below.
Proteins
Cutinase (native and W69A mutant) both produced in our laboratories (Petersen et al. 1998); lysozyme (Sigma L-7651); alkaline phosphatase (Sigma P-3895); human plasminogen (Sigma P5661), pepsin (Sigma P6887/lot 062k76506 porcine gastric mucosa), trypsin (Boehringer Mannheim 109827/84856023-95/31), a general F(ab) fragment, and a PSA-specific F(ab) fragment was prepared from whole IgG1 antibodies (Mouse IgG1 M7894 from Sigma-Aldrich and RDI-TRK-4P33-PS2 from Research Diagnostics Inc.) by papain digestion and Protein-Abased purification using ImmunoPure F(ab) Preparation Kit (no. 44885 from Pierce Biotechnology Inc.). MHC class I protein was kindly supplied by Prof. Søren Buus. Prior immobilization proteins were diluted in 25 mM Tris (Applichem A1086) (pH 8.0 or 8.5) to a final concentration of 0.5–2 μM.
Surface preparation of TIRF quartz slides
Chemical modification of glass, quartz, and metal oxide surfaces involves silanization. Silane chemistry allows fuctionalization of a quartz surface with different active groups, such as free SH groups, by covalently linking Si-groups to the quartz OH-groups. Prior to silanization, the slides were cleaned by immersing them into 70°–75°C chromosulphuric acid (Merck 1.02499/Z624399) for 1 h, and rinsed thoroughly in water at ambient room temperature. Then the slides were hydroxylated in order to increase the number of OH groups. Hydroxylation was performed by immersing the slides for 1 h into 99°–100°C 5% (w/v) potassium persulphate (K2S2O8 99%, Acros Organics 20201-000) in deionized water. After hydroxylation, the slides were flushed with deionized water and dried rapidly (using compressed air). In order to prepare the quartz surface for S-S immobilization, the quartz slides were “SH activated.” SH activation was performed by applying 150 μL 0.03% (v/v) 3-mercaptopropyl-trimethoxysilane (Merck 63800) in m-xylene (99+%, Acros Organics 1808600100) on a slide (12 cm2). The solvent (m-xylene) was allowed to evaporate completely (~ 15 min in a fume hood at ambient room temperature, 20°–25°C). Subsequently the surface was flushed with pure xylene before flushing thoroughly with ethanol and deionized water. Finally, the slides were dried quickly using compressed air and placed in an oven at 100°C for 1 h or at 20°–25°C for at least 16 h. The SH activation was based on the procedure described in the document “Chemical Modification of TIRF Sensor Surface,” (BioElectrospec Inc., supplier of the current TIRF system).
Light-induced immobilization using TIRF—total internal reflection fluorescence spectroscopy
TIRF employs the phenomenon of total internal reflection of an electromagnetic wave that occurs at the interface between two transparent media with different refractive indices when the incident angle is greater than the critical angle. In brief, when a beam of light propagating within a medium of higher refractive index (n1) (e.g., quartz) encounters an interface with a medium of lower refractive index (n2) (e.g., water), the light undergoes total internal reflection if the incident angles (θi) is greater than the critical angle (θc). The critical angle is defined as θc = sin−1 (n1/n2). Interference of the incident and reflected beams generates a standing electromagnetic wave with maximum amplitude of the electric vector at the interface. An exponentially decaying electromagnetic wave, the evanescent wave, penetrates into the less condensed medium (i.e., the aqueous solution), while the main part of the incident light is reflected back into optically more condensed medium of quartz. The penetration depth (dp) of the evanescent wave into the sample medium is a function of the incidence angle, the refractive index ratio, and the incident light wavelength (λi):
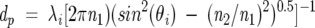 |
In the current system the penetration depth of the evanescent field is in the order of 112–118 nm (θi ≈ 72°; λi = 280–296 nm; nquartz = 1.46 and nwater = 1.33). Thus, the evanescent wave provides surface selectivity to TIRF and allows measurements of surface concentration of fluorescent adsorbed molecules, e.g., proteins.
A TIRF system (BioElectroSpec, Inc.) (Snabe et al. 2005a) coupled with a fluorescence spectrophotometer (PTI QM-2000 from Photon Technology Int.) with a 75-W Xenon arc source was used. The PTI instrument excitation and emission slits were set to 6 nm. The sample in the flow chamber was excited at 296 nm and the fluorescence emission intensity was monitored at 330 nm. The flow chamber was comprised of a “sandwich” consisting of a TIRF quartz prism, a TIRF quartz slide (with the sample surface), a 10-μm polyurethane gasket (comprising the chamber thickness), and a back block with holes to channel the solution through the flow chamber. In this setup, the excitation light travels through the prism and the slide, hits the interface between slide and solution, and totally internally reflects from the interface back into the slide and the prism. The reflected beam generates an evanescent electromagnetic field in the middle of the TIRF slide. This area of the TIRF slide serves thus as the sensor surface. Only fluorophores that are present at the surface and in close proximity of the surface (within the evanescent field) are excited and fluoresce. The dimension of the flow chamber is 16 mm × 24 mm × 0.01 mm (3.8 μL) and a standard flow rate of 0.25 mL min−1 was used.
The SH-activated slide was attached on the TIRF quartz prism using glycerol between them (glycerol and quartz have almost equal refractive indices and let the light pass with minimal interference). The TIRF flow chamber was assembled and mounted vertically in the instrument. Prior to each experiment buffer without protein was purged into the TIRF chamber for 5–10 min before adding protein (in the same buffer). After 10 or 20 min of protein exposure the system was rinsed in buffer without protein for 10 min. Then 2% (v/v) Helmanex II (Hellma GmbH & Co. KG) (pH 10–11), was applied for 5 min before again applying buffer. Experiments for analyzing UV-aided immobilization were performed in darkness (negative control) and by continuous excitation at 280 nm or 296 nm (with the resulting fluorescence intensity measurements from protein tyrosine and tryptophan residues emission at 330 nm). In order to acquire data from the experiments performed in darkness (i.e., without UV illumination), the sample was excited for a few seconds after the final rinse.
F(ab) fragment immobilization on SH-coated quartz slides was performed in the TIRF flow system at 25°C (± 0.5°C) in PBS at pH 7.4 (Fluka 79383). The surface was exposed to a solution of 0.5 μM F(ab) for 10 min in darkness (negative control) and with illumination at 295 nm. In order to rinse off nonspecific bound protein the sample surface (still placed in the TIRF flow system) was rinsed six times with 2.5 mL PBS alternately with and without 0.1% TWEEN20 (SigmaUltra P7949, Sigma-Aldrich). Illumination at 295 nm results in fluorescence emission at 350 nm from protein tryptophans. This fluorescence emission intensity was used as a measure of bound protein to the surface. In order to acquire data from the experiments performed in darkness, the sample was excited for 1–2 sec after the final rinse.
Functionality analyses of the light-induced immobilized proteins
After each experiment, the surface was cleaned carefully by flushing with deionized water before drying quickly with compressed air. A positive result is defined as when the SH-activated slide with UV-immobilized protein displays larger activity than (1) an SH-activated slide exposed to protein under same conditions but without UV-illumination and (2) a slide without SH activation (only Hydroxylated) exposed to protein with UV illumination. For cutinase activity detection, a droplet (typically 25–100 ML) of 10 μM 4-methyl-umbelliferyl butyrate (Fluka 19362) in 25 mM Tris (pH 8.0) was placed at the center of the horizontally orientated quartz slide (onto which cutinase was immobilized). Upon substrate hydrolyzes, the product fluoresces ~445 nm when excited at 365 nm. Cutinase activity was detected visually by observing the fluorescence intensity from the product formed (using a transilluminator, Unicon BTS20-LM, as excitation source). Product formation was monitored using a digital camera. For detection of lysozyme and alkaline phosphatase activity, the same principles were used as for the detection of cutinase activity, except that other substrates were used (EnzCheck Lysozyme, 22013, Molecular Probes Inc; 4-methylum-belliferyl phosphate, Sigma M-3168) and that the fluorescence signals were recorded spectrometrically. A drop of 125 μL of 50 μg/mL substrate (in 0.1 M phosphate/0.1 M NaCl at pH 7.5) was applied on the quartz slide. After incubation in darkness at ambient room temperature (20°–25°C) for 30 min (lysozyme) or 1–6 h (alkaline phosphatase), 100 μL of the solution was transferred to a well in a black multiplate (96 wells; Nunc 267742). In a fluorescence multiplate reader (SpectraMax Gemini XS) the samples were excited at 495 nm (lysozyme) or 360 nm (alkaline phosphatase), and the fluorescence emission intensity (FEI) at 525 nm (lysozyme) or 450 nm (alkaline phosphatase) was recorded.
Binding activity of the surfaces with immobilized F(ab) fragments was assayed using fluorescence-labeled antigens (anti-mouse IgG F[ab]-specific FITC conjugate F2653 from Sigma-Aldrich and FITC-conjugated PSA RDI-SCPCG117FT/13-142A from Research Diagnostics Inc., respectively). With the slide containing the immobilized F(ab) still mounted in the TIRF flow system, the surface was first blocked with 5% (w/v) nonfat skimmed milk in PBS (LABMMC27 from Bie-Berntsen) by 15-min incubation. The surface was then rinsed with PBS for 15 min in order to remove excess blocking agent before antigen solution (5 μM in PBS) was injected and allowed to incubate for 15 min. After a final rinse in PBS, the fluorescence emission at 525 nm (excitation at 495 nm) was observed and used as a measure of antibody/antigen recognition. Immobilization and binding analysis was performed at 25°C (± 0.5°C).
Preparation and illumination of IgG1-Fab fragment for Western blot and dot blot test
The purified Fab fragment from IgG digestion was diluted five times in deionized water (final concentration ~ 0.7 μM at pH 7.8) and illuminated for 1 h at 280 nm at 25°C. Illumination was performed in a 1-mL quartz cuvette in a fluorescence spectrophotometer (PTI QM-2000 from Photon Technology Int., with a 75-W Xenon arc source, and with the excitation slit set at 10 nm). A control sample was prepared as above without UV illumination.
Western blot analyses were carried out on the control and UV-illuminated samples. Twenty microliters of each sample was run on 12% SDS page under both native and denaturing conditions. Gels were blotted on to nitrocellulose membrane and blocked for 1 h, and the antibody (anti-mouse IgG Fab-specific fluoroscein isothyocyanate, dilution 1:500) was bound overnight with gentle rotation at 4°C. After sequential washing in TBE buffer with (0.1%) Triton X-100, the membrane was scanned on a typhoon PhosphoImager (Amersham Pharmacia), emission filter Cys 670 BP30, using a red laser. Experiments were repeated using dot blot methods where 20 μL of sample was placed directly onto nitrocellulose membrane hydrated in blotting buffer, blocked for 1 h with blocking buffer, and rinsed briefly with water, and then the antibody was bound at 1:500 dilution for 1 h.
Experimental setup used to obtain the fluorescence intensity vs. illumination time traces and detection of free thiol groups upon UV illumination of cutinase displayed in Figure 3
Steady-state fluorescence emission measurements were carried out using a 75-W Xenon arc lamp from a RTC 2000 PTI spectrometer coupled to a monochromator and equipped with a thermostated cuvette holder. Excitation was carried out at 295 nm and fluorescence emission intensity of cutinase at 350 nm was monitored throughout 6 h of illumination. The sample was continuously magnetically stirred at 700 rpm to maintain the homogeneity of the solution, and the temperature was kept constant at 25°C. Excitation and emission slits were set at 6 nm (Neves-Petersen et al. 2002).
Free thiols were detected with the Elman assay upon reaction with 5,5′-dithiobis-(2-nitrobenzoic acid). Detection and quantification of the protein thiol groups was carried out with the spectrophotometric assay based on the reaction of thiol groups with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB or Ellman’s reagent) (Ellman 1959). For this purpose, 3 mL of a 17.3 μM protein solution in 20 mM Tris–HCl (pH 8.5) was illuminated with 295-nm light, in a quartz macrocuvette (1-cm path length) for different time periods using an RTC 2000 PTI spectrometer. The time-dependent fluorescence emission intensity at 350 nm was acquired. All slits were set to 2-nm bandwidth. An excess of DTNB (100 μL of an 8.5-mM stock solution of DTNB in absolute methanol) was added to 900 μL of cutinase solution, prior to and after illumination of cutinase with 295-nm light. The DTNB stock in methanol is stable up to 2 wk at 4°C. The absorbance at 412 nm of the released NTB ion (nitrotehiobenzoate ion, ɛ412nm = 13,600 M−1cm−1) was measured with a UV/Visible Pharmacia spectrophotometer immediately after mixing the two components and after 20 min and 24 min reaction time. The readings were stable between 20 min and 24 min. Each data point is an average of three measurements after 24 min of mixing time at 25°C. The absorbance at 412 nm, proportional to the amount of thiol groups released by the photo-induced reaction, was measured as a function of the fluorescence intensity increase observed upon different illumination times, as described above. As a blank, 100 μL of an 8.5-mM stock solution of DTNB in absolute methanol was added to 900 μL of nonirradiated cutinase (17.3 μM cutinase solution in 20 mM Tris–HCl at pH 8.5). The concentration of illuminated cutinase sample had to be higher than in the previously described experiments to secure that enough free thiol group was formed upon illumination in order to be detected by the DTNB method (Neves-Petersen et al. 2002).
Microarray production—“six pack” array
The source of laser radiation used for the light-induced protein immobilization in order to create a protein microarray was a mode-locked titanium-Sapphire (Ti-Sapphire) laser (Tsunami 3960, Spectra Physics) pumped by a high power (5 W at 532 nm) solid-state laser (Millennia V, Spectra Physics). The tsunami output features were 840 nm, 80 MHz repetition rate, 50–60-fsec short pulse duration, 12-nm FWHM, and 900-mW average power at 840 nm. The pulsed laser radiation was sent into a pulse picker that changed its repetition rate to 8 MHz. The 840 nm radiation was further frequency tripled to 280 nm by using a frequency doubler/tripler (GWU; Spectra Physics). The average power at the GWU output was 460 μW at 280 nm. A filter was placed after the tripled output of the GWU unit in order to remove any 400 nm and 800 nm light. The average power output was 16.8 μJ/spot at 280 nm.
The 280-nm light was focused by an f100 lens, and at the same time the direction of the light beam was changed 90° using a mirror. The light beam reached its focal point after this mirror on the surface of an optically flat glass slide (average flatness of 2.0 nm, purchased from ArrayIt microarray technology, Telechem International Inc.) where 1-μL droplets of 2-μM solution (pH 8.5) (25 mM Tris–HCl buffer) were deposited by an automated dispenser. Each one of the six droplets was illuminated for 1 min. After illumination, the slide was washed with double-distilled water, PBS buffer, PBS plus Tween detergent, PBS buffer, and water. The slides were dried with a nitrogen stream.
Protein detection
From a 10-mM 4-methylum-belliferyl butyrate (Fluka 19362) in 25 mM Tris (pH 7.5) in 99.9% ethanol 1.5 μL was mixed with 1 mL 25mM Tris–HCl buffer (pH 7.5). From this stock 6 μL was deposited on each location where protein had been immobilized with UV light. The development of product (umbelliferone) formation was detected asa function of time, since upon excitation at 365 nm (provided by a transilluminator, Unicon BTS20-LM) it fluoresces in the blue (emission peak at 445 nm).
Control experiment
A control has been set up in order to show that protein immobilization was only achieved when UV light shined on the protein droplets and that all protein only adsorbed to the slide could be washed away. Two protein droplets have been placed at the same time on the optically flat glass slide and the laser beam has been used to illuminate only one such droplet (named “protein + UV”). The nonilluminated droplet (named “protein no UV”) was present on the slide upon illumination of the first droplet. After illumination of one droplet, the slide was washed as described above. Substrate has been added to the slide areas where the protein droplets have been previously present. A third substrate droplet was also placed on the slide in order to serve as an additional control.
Acknowledgments
M.T.N.-P. acknowledges the support from Novi Invest and Licfond and from the Danish Research Agency, Novo Nordisk A/S, Novozymes A/S. T.S. acknowledges the support from Novi Invest and Licfond. We thank Prof. Søren Buus for the MHC class I protein. We acknowledge Prof. Kristian Dalsgaard, Dr. Leonid Gurevich, and Dr. Zygmunt Gryczynski for fruitful discussions.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051885306.
References
- Collioud, A., Clemence, J.F., and Sanger, M. 1993. Oriented and covalent immobilization of target molecules to solid supports—Synthesis and application of a light-activatable and thiol-reactive cross-linking reagent. Bioconjugate Chem. 4: 528–536. [DOI] [PubMed] [Google Scholar]
- Ellman, G.G. 1959. Tissue sulphydryl groups. Arch. Biochem. Biophys. 82: 70–77. [DOI] [PubMed] [Google Scholar]
- Ioerger, T.R., Du, C., and Linthicum, D.S. 1999. Conservation of cys–cys trp structural triads and their geometry in the protein domains of immunoglobulin superfamily members. Mol. Immunol. 36: 373–386. [DOI] [PubMed] [Google Scholar]
- Johnsson, B., Lofas, S., and Lindquist, G. 1991. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface-plasmon resonance sensors. Anal. Biochem. 198: 268–277. [DOI] [PubMed] [Google Scholar]
- Neves-Petersen, M.T., Gryczynski, Z., Lakowicz, J., Fojan, P., Pedersen, S., Petersen, E., and Petersen, S.B. 2002. High probability of disrupting a disulphide bridge mediated by an endogenous excited tryptophan residue. Protein Sci. 11: 588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, S.B. and Neves-Petersen, M.T.D.C.A. 2005. Light induced immobilisation. WO2004065928/DK20030000081. PCT//US93/06388; WO 94/01773. BioNanoPhotonics A/S, Niels Jernes Vej 10, 9220 Aalborg Øst, Denmark. Aalborg University, Skjernvej 4A, DK-9220 Aalborg Ø, Denmark.
- Petersen, S.B., Jonson, P.H., Fojan, P., Petersen, E.I., Petersen, M.T.N., Hansen, S., Ishak, R.J., and Hough, E. 1998. Protein engineering the surface of enzymes. J. Biotechnol. 66: 11–26. [DOI] [PubMed] [Google Scholar]
- Petersen, M.T.N., Jonson, P.H., and Petersen, S.B. 1999. Amino acid neighbours and detailed conformational analysis of cysteines in proteins. Protein Eng. 12: 535–548. [DOI] [PubMed] [Google Scholar]
- Prompers, J.J., Hilbers, C.W., and Perpermans, H.A.M. 1999. Tryptophan mediated photoreduction of disulfide bonds causes unusual fluorescence behaviour of Fusarium solani pisi cutinase. FEBS Lett. 45: 409–416. [DOI] [PubMed] [Google Scholar]
- Riddles, P.W., Blakeley, R.L., and Zerner, B. 1983. Reassessment of Ellman’s reagent. Methods Enzymol. 91: 49–60. [DOI] [PubMed] [Google Scholar]
- Snabe, T., Neves-Petersen, M.T., and Petersen, S.B. 2005a. Enzymatic lipid removal from surfaces—Lipid desorption by a pH-induced “electrostatic explosion.” Chem. Phys. Lipids 133: 37–49. [DOI] [PubMed] [Google Scholar]
- Snabe, T., Røder, G.A., Neves-Petersen, M.T., Buus, S., and Petersen, S.B. 2005b. Oriented coupling of major histocompatibility complex (MHC) to sensor surfaces using light assisted immobilisation technology. Bio-sensors Bioelectronics (in press). [DOI] [PubMed]
- Sweeney, R.Y., Kelemen, B.R., Woycechowsky, K.J., and Raines, R.T. 2000. A highly active immobilized ribonuclease. Anal. Biochem. 286: 312–314. [DOI] [PubMed] [Google Scholar]
- Vanhooren, A., Devreese, B., Vanhee, K., Van Beeumen, J., and Hanssens, I.A. 2002. Photoexcitation of tryptophan groups induces reduction of two disulfide bridges in goat α-lactalbumin. Biochemistry 41: 11035–11043. [DOI] [PubMed] [Google Scholar]
- Veilleux, J.K. 1996. Covalent immobilization of biomolecules to preactivated surfaces. IVD Technol. 26–31.
- Weisenborn, P.C.M., Meder, H., Egmond, M.R., Visser, T.J.W.G., and van Hoek, A. 1996. Photophysics of the single tryptophan residue in Fusarium solani cutinase: Evidence for the occurrence of conformational substates with unusual fluorescence behaviour. Biophys. Chem. 58: 281–288. [DOI] [PubMed] [Google Scholar]
- Wilson, D.S. and Nock, S. 2002. Functional protein microarrays. Curr. Opin. Chem. Biol. 6: 81–85. [DOI] [PubMed] [Google Scholar]