

Abstract
A mutational analysis of the femtomolar-affinity anti-fluorescein antibody 4M5.3, compared to its wild-type progenitor, 4-4-20, indicates both context-dependent and -independent mutations are responsible for the 1800-fold affinity improvement. 4M5.3 was engineered from 4-4-20 by directed evolution and contains 14 mutations. The seven mutations identified as present in each of 10 final round affinity maturation clones were studied here. Affinities of the 4-4-20 single mutant addition and 4M5.3 single site reversion mutants were compared. These experiments identified four mutations, of these seven, that were context-dependent in their contribution to higher affinity. A simplified mutant containing only these seven mutations was created to analyze complete double mutant cycles of selected sets of mutations. Specific mutational sets studied included the ligand contact mutations, the heavy chain CDR3 mutations, the heavy chain CDR3 mutations plus the neighboring residue at site H108, and the early and late acquired mutations on the directed evolution pathway. The heavy chain CDR3 mutational set and the ligand-contacting mutations were shown to provide −1.4 and −2.0 kcal/mol, respectively, of the total −3.5 kcal/mol change in free energy of binding of the seven-site consensus mutant. The mutations acquired late in the directed evolution rounds provided much of the change in free energy without the earlier acquired mutations (−3.1 kcal/mol of the total −3.5 kcal/mol). Prior structural data and electrostatic calculations presented several hypotheses for the higher affinity contributions, some of which are supported by these mutational data.
Keywords: high affinity, antibody, antigen recognition, context-dependent mutations, directed evolution, double mutant cycles
High-affinity antibody binding interactions are important for both pharmaceutical and biotechnological uses. Antibodies identified from in vivo affinity maturation bind with an equilibrium binding constant ranging from 106 to 1010 M−1 (micromolar to 0.1 nm dissociation binding constant range) (Foote and Eisen 1995). Through protein engineering techniques, antibodies have been identified that bind from micromolar through femtomolar affinities (Boder et al. 2000; Maynard and Georgiou 2000). Nonetheless, designing high-affinity interactions remains difficult (Dixon et al. 2002).
Both high-affinity interactions from nature and the results from directed evolution may yield clues about the important biophysical contributions to tight noncovalent association. Structural, thermodynamic, and kinetic studies of the differences between the germ line or wild-type (lower affinity) and affinity-matured (higher affinity) interactions have been performed. Although no single common path for improvement in binding has been identified, a variety of important contributions have been noted including increased burial of hydrophobic area (Li et al. 2003), improved electrostatic complementarity balanced with losing favorable solvent interactions (Chong et al. 1999), an increase in a lock-and-key type fit (Yin et al. 2001) and a rearrangement of the heavy chain CDR3 loop (Terzyan et al. 2004). However, these prior studies of proteins with binding in the micromolar to picomolar range might differ qualitatively from the phenomena involved in femtomolar affinity binding.
Studies of natural high-affinity protein interactions have provided insight into very high-affinity mechanisms. The best studied higher affinity interactions are the barnase/barstar and avidin or streptavidin/biotin interactions. Computational studies of barnase/barstar have shown optimized electrostatics as well as strong hydrophobic interactions at the core of its binding mechanism (Lee and Tidor 2001). Studies of alanine scanning in streptavidin have shown the importance of the tryptophan residues in the binding pocket and also implicated hydrophobic binding contributions as significant for high-affinity interaction (Freitag et al. 1998; Dixon and Kollman 1999). However, these studies do not allow for easy identification of the most instrumental residues to be mutated to reach a higher binding affinity in designed protein interactions.
Characterization of individual mutations and their reversion mutant counterparts allows a precise dissection of structure–function relationships and also provides unique insights into the evolutionary pathways utilized in affinity maturation, including the extent of additivity and cooperativity that occurs. Few studies of this type for affinity-matured antibodies have been performed. The 48G7 catalytic antibody affinity maturation against p-nitrophenyl phosphonate hapten has been the most comprehensively studied by both structural and mutational analyses (Yang and Schultz 1999). Each of the nine somatic mutations was assessed for gain or loss of function in both the germ line and affinity-matured antibodies. Individual mutations were found to be either positive or neutral in their contribution to the 16-nM affinity, with context dependence for some sites.
4M5.3 is presently the highest engineered affinity antibody known (Boder et al. 2000). It was affinity matured from the single chain Fv antibody fragment (scFv) 4-4-20 by directed evolution against the hapten fluorescein–biotin. The 4-4-20 antibody has a 1.2-nM affinity for fluorescein–biotin, and has been extensively biophysically characterized through structural, thermodynamic, kinetic, computational, and mutational methods (Kranz et al. 1982; Herron et al. 1986; Denzin et al. 1993; Herron et al. 1994; Lim and Herron 1995; Lim etal. 1995; Whitlow et al. 1995). Through four rounds of random mutagenesis, off-rate screening on the surface of yeast, and selection with fluorescence activated cell sorting (FACS), the 4M5.3 clone was identified. This clone contains 14 mutations and has a 1800-fold higher affinity, with Kd = 300 fM. The structure of both the 4-4-20 and 4M5.3 binding domains were determined, and little difference could be observed (Midelfort et al. 2004). Here we study seven mutations present in all 10 final round high-affinity clones sequenced. These mutations were either added to the wild type or reverted back to wild type in the 4M5.3 clone. The affinity and ΔGbind were compared between these mutants. Additionally, clones with specific sets of mutations were also studied. Analyses of these results with respect to the previous structural data and electrostatic calculations are discussed.
Results
Individual addition and reversion mutants
The 14 mutations between 4-4-20 and 4M5.3 anti-fluorescein scFv antibody fragments conferred a 1800-fold affinity enhancement. Seven mutation sites were identified that appeared in all 10 of the sequenced fourth-round high-affinity clones from the affinity maturation study (L60, H31, H51, H101, H102, H106, and H108; Table 1) (Boder et al. 2000). These mutational sites include all the CDR mutations in 4M5.3 and are all located within three contact shells of the fluorescein–biotin binding site (Fig. 1A). These seven mutations were individually added into the wild-type 4-4-20 by site-directed mutagenesis and the affinity was determined by direct titration on the surface of yeast (Fig. 2). These mutations were also individually reverted back to wild type by site-directed mutagenesis in the high-affinity 4M5.3 construct. A competition equilibrium binding assay was performed to determine their Kd values (Fig. 3A). Competition assays were used, rather than a direct titration, due to the large volumes of extremely dilute (fM concentration) fluorescein–biotin required to ensure excess bulk ligand over antibody. This approach was previously validated as producing data quantitatively equivalent to in vitro determinations of binding equilibria and kinetics (Midelfort et al. 2004). These titration binding assay data were fit to determine the equilibrium dissociation binding constants (Kd) for the mutants and the ΔGbind was calculated for each. These results are shown in Table 2.
Table 1.
The 14 changes between 4-4-20 and 4M5.3
| Residue (4-4-20) | Chain number | Residue (4M5.3) | Location |
| Phe | L60 | Val | CDR2 |
| Ser | L81 | Asn | FR3 |
| Asp | H1 | Gly | FR1 |
| Arg | H16 | Gly | FR1 |
| Pro | H17 | Ala | FR1 |
| Ala | H24 | Thr | FR1 |
| Ser | H30 | Gly | FR1 |
| Asp | H31 | His | CDR1 |
| Ile | H51 | Phe | CDR2 |
| Met | H93 | Thr | FR3 |
| Ser | H101 | Ala | CDR3 |
| Tyr | H102 | Ser | CDR3 |
| Asp | H106 | Glu | CDR3 |
| Trp | H108 | Leu | FR4 |
The “Min7” mutations are bold; CDR: Complementarity determining region; FR: Framework region.
Figure 1.


Comparison of 4-4-20 and 4M5.3 Fv domain crystal structures. (A) Alignment of 4-4-20 and 4M5.3 structures. 4-4-20 is light teal and 4M5.3 is blue. Mutations are indicated, and mutations in Min7 are in bold type. (B) Hydrogen-bonding network of mutations sites H31, H101, H102, H106, and H108 in the 4-4-20 structure. (C) Hydrogen bonding network of mutations sites H31, H101, H102, H106, and H108 in the 4M5.3 structure.
Figure 2.

Direct fluorescein–biotin binding titration assay with yeast surface displayed 4-4-20 mutants. Representative titrations for each mutant are shown.
Figure 3.
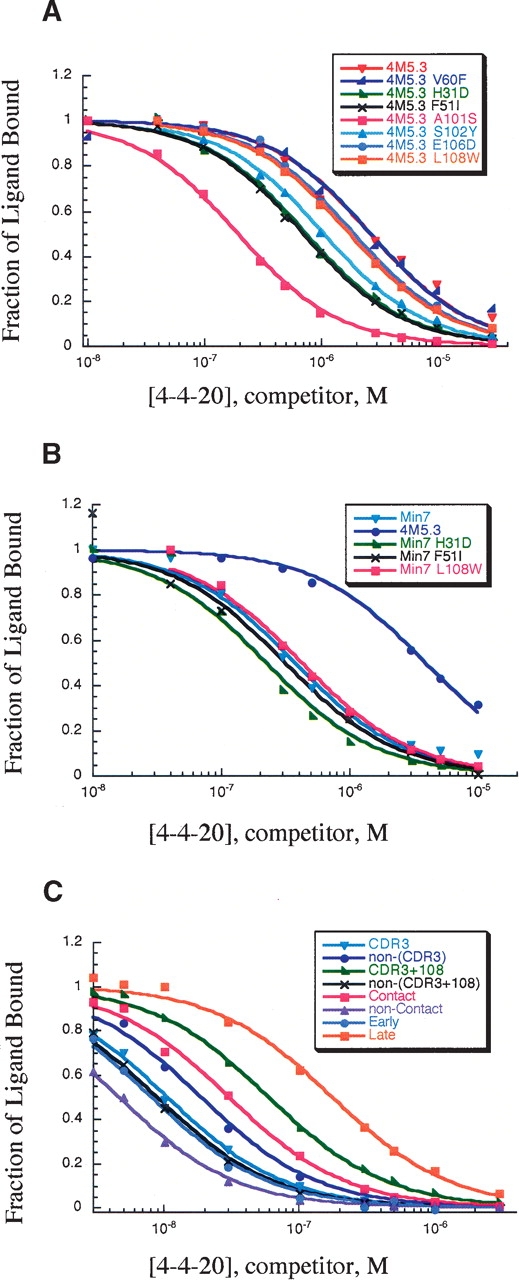
Competition equilibrium binding titration assay with yeast surface displayed. Representative titrations for each mutant are shown. (A) Titration assay with yeast surface displayed 4M5.3 reversion mutants. (B) Titration assay with yeast surface displayed Min7 reversion mutants. (C) Titration assay with yeast surface displayed partial mutants of Min7.
Table 2.
Dissociation binding constants and ΔG of binding for yeast surface displayed anti-fluorescein biotin binding scFv antibodies
| Mutant | No. of mutations, from 4-4-20 | Kd (± SD), pM | ΔGbind (± SD), kcal/mol | Δ ΔGbind from 4-4-20 |
| 4-4-20 | 0 | 1200 (± 300) | −12.16 (± 0.15) | 0 |
| 4M5.3 | 14 | 0.35 (± 0.08) | −17.0 (± 0.1) | −4.8 (± 0.2) |
| 4-4-20 F(L60)V | 1 | 540 (± 30) | −12.64 (± 0.03) | −0.5 (± 0.1) |
| 4-4-20 D(H31)H | 1 | 350 (± 40) | −12.89 (± 0.07) | −0.7 (± 0.1) |
| 4-4-20 I(H51)F | 1 | 2500 (± 300) | −11.73 (± 0.07) | 0.4 (± 0.1) |
| 4-4-20 S(H101)A | 1 | 720 (± 20) | −12.47 (± 0.02) | −0.3 (± 0.1) |
| 4-4-20 Y(H102)S | 1 | 320 (± 40) | −12.95 (± 0.07) | −0.8 (± 0.1) |
| 4-4-20 D(H106)E | 1 | 10,000 (± 5000) | −10.9 (± 0.3) | 1.3 (± 0.3) |
| 4-4-20 W(H108)L | 1 | 240 (± 40) | −13.1 (± 0.1) | −0.9 (± 0.1) |
| 4M5.3 V(L60)F | 13 | 0.7 (± 0.3) | −16.6 (± 0.3) | −4.4 (± 0.3) |
| 4M5.3 H(H31)D | 13 | 1.6 (± 0.2) | −16.08 (± 0.07) | −3.9 (± 0.1) |
| 4M5.3 F(H51)I | 13 | 2.0 (± 0.3) | −15.95 (± 0.09) | −3.8 (± 0.1) |
| 4M5.3 A(H101)S | 13 | 5.9 (± 0.4) | −15.31 (± 0.04) | −3.2 (± 0.1) |
| 4M5.3 S(H102)Y | 13 | 1.2 (± 0.1) | −16.27 (± 0.06) | −4.1 (± 0.1) |
| 4M5.3 E(H106)D | 13 | 0.7 (± 0.1) | −16.57 (± 0.08) | −4.4 (± 0.1) |
| 4M5.3 L(H108)W | 13 | 0.7 (± 0.1) | −16.6 (± 0.1) | −4.4 (± 0.1) |
| Min7 | 7 | 3.2 (± 0.3) | −15.67 (± 0.06) | −3.5 (± 0.1) |
| Min7 V(L60)F | 6 | N.D. | ||
| Min7 H(H31)D | 6 | 5.0 (± 0.4) | −15.41 (± 0.05) | −3.2 (± 0.1) |
| Min7 F(H51)I | 6 | 3.1 (± 0.8) | −15.7 (± 0.2) | −3.5 (± 0.2) |
| Min7 A(H101)S | 6 | N.D. | ||
| Min7 S(H102)Y | 6 | N.D. | ||
| Min7 E(H106)D | 6 | N.D. | ||
| Min7 L(H108)W | 6 | 2.3 (± 0.4) | −15.9 (0.1) | −3.7 (± 0.1) |
| CDR3 | 3 | 110 (± 10) | −13.57 (± 0.07) | −1.4 (± 0.1) |
| Non-CDR3 | 4 | 61 (± 7) | −13.93 (± 0.07) | −1.8 (± 0.1) |
| CDR3 + 108 | 4 | 21 (± 1) | −14.56 (± 0.03) | −2.4 (± 0.1) |
| Non-(CDR3 + 108) | 3 | 160 (± 20) | −13.36 (± 0.07) | −1.2 (± 0.1) |
| Contact | 3 | 38 (± 6) | −14.21 (± 0.09) | −2.0 (± 0.1) |
| Noncontact | 4 | 270 (± 60) | −13.1 (± 0.1) | −0.9 (± 0.2) |
| Early | 2 | 150 (± 20) | −13.38 (± 0.07) | −1.2 (± 0.1) |
| Late | 5 | 6.8 (± 1.4) | −15.2 (±.01) | −3.1 (± 0.2) |
N.D.: not determined. Error is indicated as standard deviation of replicate samples. The named mutants are defined as in Tables 1 and 3.
Five of the seven mutations [Phe(L60)Val, Asp(H31)His, Ser(H101)Ala, Tyr(H102)Ser, and Trp(H108)Leu] studied show an individual improvement in affinity when added into 4-4-20. In 4M5.3, all seven of the individual reversion mutants showed decreased affinity (higher Kd), indicating that all these mutations contribute favorably to 4M5.3’s affinity. The two single addition mutations in 4-4-20, Ile(H51)Phe and Asp(H106)Glu, that individually decreased affinity were, however, important in the high-affinity 4M5.3 because reversion of either site reduced affinity by at least twofold in 4M5.3.
This mutational study allowed for double mutant cycles to be analyzed between the addition of single mutations to the 4-4-20 and the reversion of the same mutational site from 4M5.3, as noted in Equation 1 below.
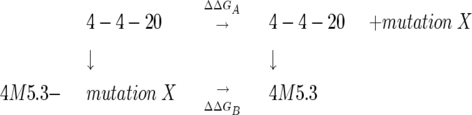 |
(1) |
Figure 4A shows the comparison in the change in the ΔΔG for the addition of a mutation into 4-4-20 (ΔΔGA) as well as the addition of this mutation to complete the 4M5.3 full mutant clone (ΔΔGB). The free energy of interaction with each individual mutation and the other 13 mutations in 4M5.3 can be quantified as ΔΔΔGint = ΔΔGB − ΔΔGA. For three of the mutations, at sites L60, H31, and H102, the same change in free energy was evident for both the addition of the mutations to 4-4-20 or the reversion in 4M5.3, indicating independence from the other mutations in the structure (|Δ Δ ΔGint | ≤ 0.2 kcal/mol). In contrast, the sites of H51, H101, H106, and H108 gave rise to significant differences between the addition and reversion of the same site, indicating a context-dependent effect in these mutational sites. I(H51)F or D(H106)E alone in 4-4-20 actually decrease the affinity for fluorescein–biotin, while reverting either mutation from 4M5.3 decreases affinity. Mutation at H101 provides a small gain in affinity for 4-4-20, but the reversion mutant in 4M5.3 loses 1.7 kcal/mol of binding affinity, indicating that the other mutations in 4M5.3 interact strongly with the S(H101)A mutation.
Figure 4.
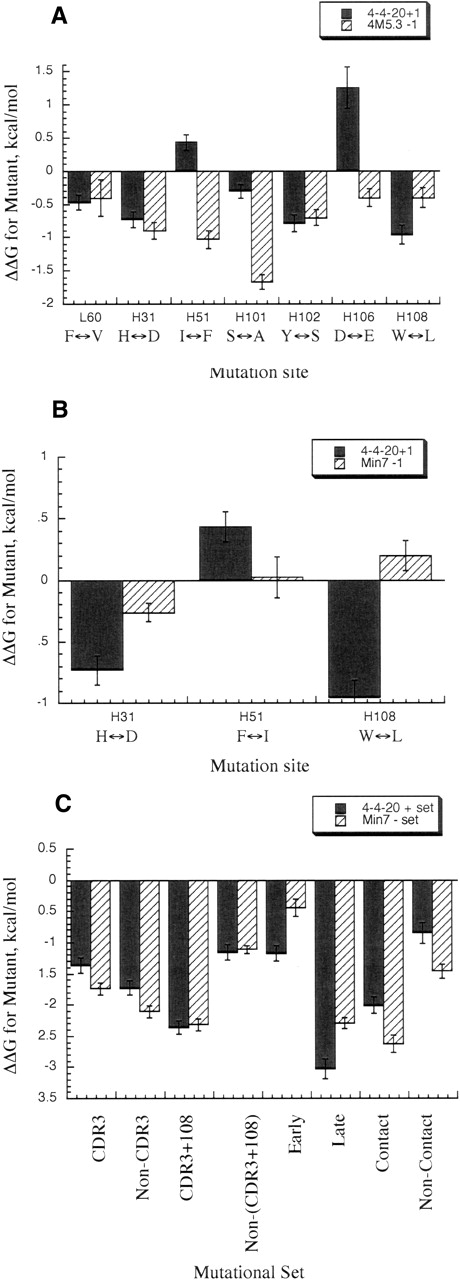
Change in ΔGbind for mutants in 4-4-20, Min7, and 4M5. Black bars represent Δ ΔGA, striped bars represent Δ ΔGB (see Equation 1 in text for A, and Equation 2 for B and C). Error bars are standard deviations of triplicate titrations. (A) Change in ΔGbind for addition mutants in 4-4-20 or reversion mutants in 4M5.3. In horizontal axis, amino acid noted on left of double arrow is wild type, right is 4M5.3 mutation. (B) Change in ΔGbind for addition mutants in 4-4-20 or reversion mutants in Min7. In horizontal axis, amino acid noted on left of double arrow is wild type, right is Min7 mutation. (C) Change in ΔGbind for addition of sets of mutations in 4-4-20 or reversion of sets of mutations in Min7.
Min7 partial mutant double mutant cycles
To search for the minimum set of mutations necessary for a majority of 4M5.3’s affinity improvement, a mutant with the seven mutations identified in all fourth-round affinity-maturation clones was created, and is referred to as “Min7” (Table 1). This mutant had a 10-fold lower affinity than 4M5.3 for fluorescein–biotin but still maintained a Δ ΔGbind of −3.5 kcal/mol, compared to 4-4-20 (Table 2). The seven individual Min7 reversion mutants were also created and displayed on the surface of yeast. Unfortunately, in four of the Min7 reversion mutants, the mutants were expressed poorly, and it was not possible to determine their affinity. There maining three mutants’ affinity on the surface of yeast were determined through the competition equilibrium binding assay (Fig. 3B), and the affinity and ΔGbind values are shown in Table 2. Double mutant cycles, as shown in Equation 2, can be constructed with this data.
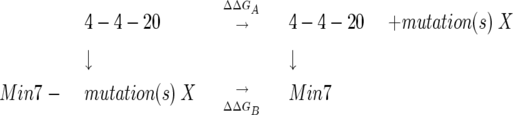 |
(2) |
The Δ ΔG of binding between the addition of the mutation to 4-4-20 (Δ ΔGA) and the addition of the same mutation to complete the Min7 mutant (Δ ΔGB) is shown in Figure 4B. These Min7 reversion mutants showed differences compared to their counterpart reversions in 4M5.3. Both the reversion mutations of H(H31)D and F(H51)I made less of a negative effect on the Min7 than on the 4M5.3 antibody affinity, with no loss in affinity in the Min7 for the F(H51)I mutation; this indicates that the significant Δ Δ ΔGint for I(H51)F results from energetic interactions with the six mutations not present in the Min7 mutant. In the case of L(H108)W, the mutation made no difference when removed from the Min7 mutant, while it caused a slight loss in affinity when removed from the 4M5.3 scFv.
Partial mutant dissection
A subset of mutations is often responsible for most of the binding free energy improvement in directed evolution or in vivo affinity maturation. The ligand-contacting residues or heavy chain CDR3 residues are often implicated as the predominant regions for mutations to improve affinity (Sundberg and Mariuzza 2002). To test these hypotheses, we made specific combinations of the Min7 mutations (Table 3) and determined their affinity for fluorescein–biotin by competition equilibrium binding assays (Fig. 3C). The Δ ΔGbind for addition to 4-4-20 or reversion from Min7 is shown in Figure 4C.
Table 3.
Partial mutant combinations
| Mutant set | Mutations in 4-4-20 |
| Contact | D(H31)H, S(H101)A, Y(H102)S |
| Noncontact | V(L60)F, I(H51)F, D(H106)E, W(H108)L |
| CDR3 | S(H101)A, Y(H102)S, D(H106)E |
| Non-CDR3 | V(L60)F, D(H31)H, I(H51)F, W(H108)L |
| CDR3+108 | S(H101)A, Y(H102)S, D(H106)E, W(H108)L |
| Non-(CDR3 + 108) | V(L60)F, D(H31)H, I(H51)F |
| Early | I(H51)F, W(H108)L |
| Late | V(L60)F,D(H31)H,S(H101)A,Y(H102)S,D(H106)E |
The heavy chain CDR3 residues are heavily mutated in vivo from junctional frameshifts during gene rearrangement to form a complete V-region exon and affinity maturation through somatic hypermutation that occurs in mature B cells in the complete V-region genes. These sites are therefore often studied for their importance to binding affinity (Maynard and Georgiou 2000; Xu and Davis 2000). We studied the three mutations in the heavy chain CDR3 (Table 2 and Fig. 4C, “CDR3” mutational set) versus the other four mutations (Table 2 and Fig. 4C, “Non-CDR3” mutational set) in the Min7 construct. These mutations showed a small difference between adding them to 4-4-20 versus reverting them from Min7, indicating very little coupling with other mutations.
However, we also noted that the mutation at heavy chain position 108 is directly C-terminal to the heavy chain CDR3 region. Grouping this mutation with the CDR3 mutations (Table 2 and Fig. 4C, “CDR3+108” mutational set) and comparing this to the remaining three Min7 mutations (Table 2 and Fig. 4C, “Non-(CDR3+108)” mutational set), Δ Δ ΔGint = −0.1 kcal/mol, or almost no detectable free energy of interaction between the {S(H101)A+Y(H102)S+D(H106)E+W(H108)L} and {V(L60)F+ D(H31)H+I( H51)F} mutations. Taken together with the single mutation double mutant cycles, the lack of a strong energetic interaction of the (CDR3 + 108) set with the other three mutations in Min7 implies that the W(H108)L, S(H101)A, and D(H106)E have a particularly strong interaction. Previous structural and computational results support this conclusion, as discussed further on.
The three mutations that contact the ligand (within 5 Å according to the crystal structure), are in positions H31, H101, and H102. These mutations added into 4-4-20 (Table 2 and Fig. 4C, “Contact” mutational set) and reverted in the Min7 construct (Table 2 and Fig. 4C, “Noncontact” mutational set) were compared. The contact mutations interact somewhat weakly with the non-contact mutations (Δ Δ ΔGint = 0.5 kcal/mol).
Finally, there is an element of sequential accumulation in the directed evolution studies. Through repeated rounds of mutatgenesis and screening, mutations accumulate (Boder et al. 2000). We studied the two mutations in Min7 that appeared in the first two rounds of the affinity maturation and remained through the successive rounds (H51 and H108; Table 2 and Fig. 4C, “Early” mutational set) versus the five in Min7 that appeared in the third and fourth rounds (L60, H31, H101, H102, and H106; Table 2 and Fig. 4C, “Late” mutational set) to deduce the effect of the later mutations without the support of the previous two mutations. The results show that the mutations are coupled; however, the Late mutational set do have a large effect (−3.0 kcal/mol of the total −3.5 kcal/mol for Min7) without the Early mutational set. There is negative coupling occurring since the addition of the ΔGbind of the Early mutational set plus the addition of the Late mutational set would add to a greater change than is seen when all seven mutations are placed in the construct.
The Early set with only the H51 and H108 mutations allows another double mutant cycle to be created with the single 4-4-20 mutations of H108 and H51. Here, the addition of the individual components (H51 and H108) to binding do not add up to the Early (H51/108) mutant (0.4 + (−0.9) ≠ −1.2, respectively), exhibiting a favorable interaction free energy between these two sites, Δ Δ ΔGint = −0.5 kcal/mol.
Mutant expression effects
Through these studies it was also possible to observe some evidence of mutations that helped the expression of the scFv antibody fragments (Table 4). 4M5.3 is expressed both on the surface of yeast and solubly about two- to fourfold better than 4-4-20. It was therefore interesting to note that Min7, the construct with the seven mutations that were present in all the fourth round affinity maturation clones, expressed not as well as 4-4-20. Furthermore, when individual mutations were reverted to wild type in this construct, expression dropped below a detectable level on the surface of yeast. When these same mutations were removed from 4M5.3, expression often dropped but never became undetectable. Also, when the individual mutations were added to the 4-4-20 antibody, these diminished expression of these antibodies.
Table 4.
Relative yeast surface display expression for antifluorescein scFv antibody fragments
| Yeast displayed mutant | No. of mutations, from 4-4-20 | Expression level (max. positive peak intensities) |
| Nondisplaying | 5 | |
| Yeast | ||
| 4-4-20 | 0 | 40–200 |
| 4M5.3 | 14 | 130–600 |
| 4-4-20 F(L60)V | 1 | 150 |
| 4-4-20 D(H31)H | 1 | 120 |
| 4-4-20 I(H51)F | 1 | 100 |
| 4-4-20 S(H101)A | 1 | 140 |
| 4-4-20 Y(H102)S | 1 | 200 |
| 4-4-20 D(H106)E | 1 | 10 |
| 4-4-20 W(H108)L | 1 | 130–190 |
| 4M5.3 V(L60)F | 13 | 100 |
| 4M5.3 H(H31)D | 13 | 200 |
| 4M5.3 F(H51)I | 13 | 200 |
| 4M5.3 A(H101)S | 13 | 120 |
| 4M5.3 S(H102)Y | 13 | 260 |
| 4M5.3 E(H106)D | 13 | 160 |
| 4M5.3 L(H108)W | 13 | 200 |
| Min7 | 7 | 10 |
| Min7 V(L60)F | 6 | 5 |
| Min7 H(H31)D | 6 | 40 |
| Min7 F(H51)I | 6 | 120 |
| Min7 A(H101)S | 6 | 5 |
| Min7 S(H102)Y | 6 | 5 |
| Min7 E(H106)D | 6 | 5 |
| Min7 L(H108)W | 6 | 20 |
| CDR3 | 3 | 200 |
| Non-CDR3 | 4 | 20 |
| CDR3+108 | 4 | 220 |
| Non-(CDR3+108) | 3 | 60 |
| Contact | 3 | 190 |
| Noncontact | 4 | 70 |
| Early | 2 | 15 |
| Late | 5 | 310 |
| 4-4-20 A(H24)T | 1 | 50 |
| 4-4-20 S(H30)G | 1 | 40 |
| 4-4-20 M(H93)T | 1 | 20 |
| Min7 A(H24)T | 8 | 20 |
| Min7 S3(H0)G | 8 | 10 |
| Min7 M(H93)T | 8 | 20 |
Analysis of the table of mutations found in the fourth round of affinity maturation indicated that the mutant antibodies frequently had one or more of the mutations at positions H24, H30, or H93 (Boder et al. 2000). All of these sites are well outside of the three contact shells around the ligand. They are all solvent exposed, and could affect the solubility of the mutants. We added these mutations individually into both the 4-4-20 wild type and the Min7 construct to observe changes in expression levels (Table 4). A very small increase in expression was seen in each of these single mutation additions; however, none of them rescued the level of the expression to a significantly higher level or to the level of 4M5.3.
Discussion
Individual addition and reversion mutations show contextual effect on binding in agreement with spatial location of mutations
We use genetic analysis and double mutant cycles here to study a protein–antigen interaction with femtomolar Kd, and attempt to parse the overall binding free energy among the 14 mutations in 4M5.3, as well as investigate energetic interactions amongst the mutations. The 4M5.3 antibody contains 14 mutations compared to the wild-type 4-4-20 antibody. However, in observing all of the clones identified from the final round of affinity maturation, only seven of these mutational sites were present in all 10 clones sequenced. These seven sites are the primary focus for the studies presented here. These sites include all of the CDR mutations found in 4M5.3. The study of these seven mutations reveals that all but two individually improved the affinity of 4-4-20, four of them by greater than twofold improvements. Surprisingly, two of the mutations (I(H51)F and D(H106)E) decrease 4-4-20 affinity, although these mutations contribute beneficial binding free energy in 4M5.3; removal of any of these seven mutations from 4M5.3 reduces affinity compared to the complete 4M5.3.
These mutational studies confirm observations from the structures and electrostatic calculations of 4-4-20 and 4M5.3 (Midelfort et al. 2004). The mutational sites of L60, H31, and H102 all appear to be somewhat energetically independent, and all provided beneficial improvement in affinity in 4-4-20 and deleterious effects in 4M5.3 when mutated. The locations of these sites in the crystal structure are distant from the other mutational sites, in support of this result (Fig. 1A). The L60 site is the only mutation, of these seven, located in the light chain. It is located at the VL–VH interface but does not contact other mutational sites in the heavy chain. The H31 site is located near the top of the binding site and is solvent exposed. Upon mutation from Asp to His, the side chain is further exposed to solvent and the removal of the Asp charge may lessen the negative charge repulsion between fluorescein and Asp. H31 is, however, spatially close to H102. These two sites do not form a hydrogen bond interaction in 4-4-20 (H31Asp and H102Tyr); however, in 4M5.3, there is a hydrogen bond between the H31His carbonyl oxygen and the H102Ser hydroxyl group (Fig. 1B,C). Since this interaction is formed between the backbone of the H31 site and the side chain of the H102 site, the side chain mutations need not interact to create their individual effects on affinity, supporting the observed independent effects of these side chain mutations.
Four (H51, H101, H106, and H108) of the seven mutations are seen to provide differential changes to Δ ΔGbind based on their environment. The mutations at positions H101, H106, and H108 are all near each other, and show interactions in their effects on ligand affinity, in agreement with crystal structure observations. The 4M5.3 backbone atoms of these residues remain very close to their locations in the 4-4-20 structure. These mutational sites form a hydrogen bond network in 4-4-20 through connections of H108Trp nitrogen (NE1) to carboxyl oxygen of the H106Asp side chain and the other carboxyl oxygen on H106Asp to the gamma oxygen on the H101Ser side chain (Fig. 1B). This hydrogen-bonding network was eliminated in 4M5.3 (Fig. 1C). The 4-4-20 hydrogen bond between H101Ser and H106Asp is very strained in its angle, and is computationally shown to be unfavorable compared with the mutated residues in those positions (Midelfort et al. 2004).
The change of H106 Asp to Glu moves the carboxyl oxygen from 5.8 Å to 6.2 Å away from the fluorescein oxygen, and may eliminate some unfavorable charge interactions as well as reduce the desolvation penalty. This is in agreement with the previous electrostatic calculations (Midelfort et al. 2004). From the crystal structure, no large rearrangements are observed in this area, but a collection of small favorable adjustments are also suggested by the electrostatic calculations and may have provided the desired binding improvement. The electrostatic calculations suggested that the Asp(H106)Glu mutation would be important, since the Ser(H101)Ala mutation was favored by eliminating an unsatisfied hydroxyl oxygen thereby removing a small desolvation penalty.
The mutation at position H108 shows a −0.9 kcal/mol improvement in affinity when added to the 4-4-20 scFv; however, the effect was somewhat smaller in the context of the 4M5.3 mutants (−0.4 kcal/mol). The observed change in the crystal structure with this mutation is surprisingly small. Although this large tryptophan side chain is mutated to the relatively smaller leucine, no other backbone or side chain atoms within 5 Å are seen to move to fill in this space.
Although the mutation at residue H51 also shows a context-dependent effect for fluorescein–biotin affinity, it is removed in space from the other mutations and is fairly solvent exposed. The backbone atoms of this position in both the 4-4-20 and 4M5.3 structures are identical, and it shows no obvious structural interactions with mutational sites (Fig. 1B,C). It remains unclear why this mutation has a context-dependent effect with the other mutations.
The Min7 partial mutant has a 10-fold lower affinity for fluorescein–biotin than the 4M5.3 scFv (Δ ΔGbind of −3.5 and −4.5 kcal/mol, respectively, relative to 4-4-20 binding). This indicates that the seven other mutations that were not studied here, but are found in 4M5.3, support the high-affinity interaction in 4M5.3. Three of these mutations are located in the framework regions within the second or third contact shells of the binding site while another four are well outside of the three contact shells of the fluorescein binding pocket.
The removal of these additional seven mutations from 4M5.3 (leaving the mutant named Min7) has also affected the context with which some of the Min7 mutations act. The three mutational sites, H31, H51, and H108, individually studied in the Min7 and 4-4-20 mutants exhibit some energetic interactions. Both H51 and H108 also showed this interaction in the 4-4-20 versus 4M5.3 environment, while H31 has become more sensitive to the environment in the Min7 mutant. The addition of H31 in 4-4-20 caused a 0.7 kcal/mol improvement in binding; however, the reversion of the H31 site in Min7 had only a very small effect on binding affinity (0.26 kcal/mol).
Effects of combinations of mutations show contextual effects of binding subgroups
Mutational experiments provide the opportunity for analysis of combinations of partial mutants to address mechanistic hypotheses for affinity maturation. Eight mutational sets were specifically studied, including the heavy chain CDR3 set, the non-CDR3 set, the CDR3 plus H108 (a neighboring residue) set, the non-(CDR3 + H108) set, the ligand contacting set, the ligand non-contacting set, the earlier acquired set, and the late acquired set. The double mutant cycle analysis with these sets show, in Figure 4C, all the sets of mutations have interacting mutations except for the CDR3 + 108 subgroup, which acts independently from the non-(CDR3 + 108) mutational set.
As expected, the mutations in the heavy chain CDR3 did provide a significant portion of the binding free energy; however, they were much more effective with an additional mutation at a position in the framework region adjacent to this CDR (H108). This is interesting, because mutation H108 is separated from the H101 and H102 sites in all but this CDR3 + 108 mutational set. This agrees with the structural interpretation that H108 has contextual effects with H101 and H102 through hydrogen bond networks. The result of framework residues enhancing the effects of the CDR3 mutations also provides encouragement for broader mutational strategies in antibody engineering rather than focusing on the heavy chain CDR3 loop exclusively.
The element of time in successive rounds of affinity maturation led to an effect where the two earliest accumulated mutations were necessary for the later mutations to provide a full free-energy contribution to binding. However, the effect was not as pronounced as one might expect. Serial rounds of directed evolution allows for context-dependent mutations to collect. Although that is generally seen here, the primary two mutations identified may have set the stage for the effects of the additional five mutations studied, but were not so influential as to determine the effect of the other mutations without the first two.
Finally, it is clear that the affinity increase was not due only to changes in the contact residues, as these residues alone provided only slightly more than half of the change in free energy of binding for the Min7 partial clone compared to the 4-4-20 wild type. Again, much emphasis for affinity engineering has been placed on the residues in direct contact with the ligand. Although a study of the affinity improvement possible through only mutations in contact residues has not been completed, it is clear that mutations in a larger surrounding area are beneficial for the improvements seen here. The affinity maturation results were not clearly visible from the crystal structures alone, and these mutational studies provide both further comprehension of the mechanisms involved and help confirm the insights from the computational electrostatic analysis.
Distant mutations affect expression levels
The effect of mutations on expression is not well understood, and is often thought to relate to folding stability or solubility of the final mutant. Here we observed changes in expression levels of the mutants on the surface of yeast. These expression levels did not correlate with affinity for fluorescein–biotin. It is often said, with respect to directed evolution, that “you get what you screen for.” In the isolation of 4M5.3, an explicit criterion for selection was detectable expression levels on the yeast surface. It is therefore hypothesized that many of the mutations far from the binding site in 4M5.3 contribute to higher expression levels of 4M5.3. The partial mutant Min7, with mutations in only the first three contact shells of the binding site, had only a 10-folddrop inaffinity compared to 4M5.3; however, it had very little expression, much less than the lower affinity 4-4-20 wild type. It was predicted that some of the other mutations that were seen often in the final round of affinity maturation might be playing a role in the expression improvement of 4M5.3 over 4-4-20. Three of these mutations (H24, H31, and H93) were checked in both the 4-4-20 and Min7 backgrounds for improvements in affinity. Although very slight improvements were seen, none of these individual mutations raised the expression levels greatly. Nevertheless, the marked difference in expression of 4M5.3 versus Min7 is clearly due to the combined effects of some subset of these seven mutations.
Deleterious individual kinetic mutations may support a greater contextual effect
Finally, a review of the order of the mutational accumulation in 4M5.3 shows that the H51 mutation occurred in the second round and remains throughout the following two rounds. However, this mutation on its own actually decreases the affinity for fluorescein–biotin. It is therefore hypothesized that this mutation may have association and dissociation rate kinetic effects on binding affinity. Although it does not help in the overall affinity, it may slow the dissociation rate so that it could be recovered under the dissociation kinetic sort strategy used to identify 4M5.3. The overall 4M5.3 association rate is slowed ninefold compared to the 4-4-20 wild type. This H51 mutant clone may have provided part of this slowed association rate, while also conferring a slower dissociation rate, overall providing a negative effect on affinity. Notably, this clone was also identified from a single-mutant 4-4-20 library that was specifically selected for slower dissociation rates (data not shown).
Overall conclusions
The evidence of both context-dependent and -independent mutational effects on binding affinity indicate the complexity in higher affinity mechanisms attained through directed evolution affinity maturation processes. Interestingly, all of the minimum seven mutations studied contributed beneficially to the binding free energy of the final high-affinity clone. However, the seven mutations which appeared as the most prevalent sites in the affinity maturation did not provide all of the Δ ΔGbind contribution, providing −3.5 of the total −4.5 kcal/mol difference for 4M5.3. Some or all of the remaining seven mutations in 4M5.3 must also play a part in this high-affinity interaction.
Although these results were found in a system where the wild-type binding interaction was already high affinity, the method of affinity maturation was through several rounds of affinity maturation and gene shuffling, and this binding interaction was between an antibody and small molecule antigen, the findings here are in agreement with other protein affinity maturation results and wild-type high-affinity protein interactions. Engineered affinity maturation studies have often attempted to select a small subset of important residues to mutate. In agreement with the affinity maturation of the nanomolar affinity catalytic antibody 48G7 (Wedemayer et al. 1997), this study emphasizes the importance of a wide range of residues working in concert to create a very high-affinity clone. As with other naturally occurring high-affinity interactions, this interaction has been found to involve many mechanisms, with some mutations with context dependence while others provide relatively independent improvements (Freitag et al. 1998). Both rational design and directed evolution studies will need to allow for mutations in a spatially broad range around the binding site and involving many biophysical contributions to the binding free energy.
Materials and methods
Gene constructs
Single mutations and sets of mutations were constructed by site-directed mutagenesis in the pCT-302 or pCT-4M5.3 vector (Boder et al. 2000). Mutations were performed with either the Quik Change Mutation Kit (Stratagene) or the Multi-Site Quik Change Mutation kit (Stratagene) following the provided protocols. The 4-4-20 gene with the following individual mutations were prepared: 4-4-20 V60VLF, 4-4-20 D31VHH, 4-4-20 I51VHF, 4-4-20 S101VHA, 4-4-20 Y102VHS, 4-4-20 D106VHE, 4-4-20 W108VHL. The 4M5.3 gene with the following individual reversion mutations were prepared: 4M5.3F60VLV, 4M5.3 H31VHD, 4M5.3 F51VHI, 4M5.3 A101VHS, 4M5.3 S102VHY, 4M5.3 E106VHD, and 4M5.3 L108VHW. A mutant with the seven mutations found in all the fourth-round affinity maturation clones was created and called Min7. This mutant was: 4-4-20 V60VLF, D31VHH, I51VHF, S101VHA, Y102VHS, D106VHE, and W108VHL. Additionally the following eight partial mutants of 4-4-20 were created: VH-CDR3 (4-4-20 S101VHA, Y102VHS, D106VHE), Non-VH-CDR3 (4-4-20 V60VLF, D31VHH, I51VHF, W108VHL), Contact (4-4-20 D31VHH, S101VHA, Y102VHS), Noncontact (4-4-20 V60VLF, D31VHH, I51VHF, D106VHE, W108VHL), VH-CDR3 + 108 (4-4-20 S101VHA, Y102VHS, D106VHE, W108VHL), Non-VH-CDR3 + 108 (4-4-20 V60VLF, D31VHH, I51VHF), Early (4-4-20 I51VHF, W108VHL), and Late (4-4-20 V60VLF, D31VHH, S101VHA, Y102VHS, D106VHE). Reversion mutants from the Min7 construct were also created including: Min7 F60VLV, Min7 H31VHD, Min7 F51VHI, Min7 A101VHS, Min7 S102VHY, Min7 E106VHD, and Min7 L108VHW. Finally, to assess the ability of the A24VHT, S30VHG, and M93VHT mutations in 4-4-20 and Min7 to affect expression levels, the following constructs were created: 4-4-20 A24VHT, 4-4-20 S30VHG, 4-4-20 M93VHT, Min7 A24VHT, Min7 S30VHG, and Min7 M93VHT. All plasmids were sequenced to assure correct changes.
Yeast surface display
Plasmids were transformed into the EBY100 yeast strain by the EZ Yeast Transformation II kit (Zymo Research) according to kit protocols. Colonies were then grown in 5-mL tube cultures of SD-CAA (2% dextrose, 0.67% yeast nitrogen base, 0.5% casamino acids, 50 mM sodium phosphate buffered to pH 6.6) media at 30°C, 250 rpm shaker, for 1–2 d to an OD600 of >5. Then ~ 5 × 107 cells were transferred in 100 μL of spent SD-CAA media to 5 mL of SG-CAA media (same as SD-CAA, except the dextrose is replaced with galactose), and placed in a 20°C shaker (250 rpm) for 18–24 h.
Determination of affinity by direct titration
Yeast surface-displayed equilibrium binding constants were determined by direct titration with fluorescein–biotin for the 4-4-20 wild type and 4-4-20 addition mutants. The ~ 5 × 104 yeast were incubated at 25°C with fluorescein–biotin, in the concentrations ranges of 40 pM to 0.1 μM, in 1-mL volumes with at least a 10-fold excess of antigen to scFv. Fluorescein–biotin binding was detected by secondary labeling with 1 : 100 streptavidin–Phycoerythrin (Pharmingen, BD Biosciences) for 3–10 min on ice, followed by analysis with flow cytometry (XL cytometer; Beckman Coulter). All experiments were performed in triplicate.
Determination of affinity by competition
The yeast surface-displayed equilibrium Kd of 4M5.3, 4M5.3 reversion mutants, Min7, Min7 reversion mutants, and the Min7 partial mutants (construct definitions listed in the Materials and Methods above) were determined by a competition assay with 4-4-20. Soluble 4-4-20 was produced as in Midelfort et al. 2004. Two million 4M5.3, or mutant, displaying yeast in a total final volume of 1 mL (~ 0.2 nM 4M5.3 scFv), 1.7 nM fluorescein–biotin (Molecular Probes), and varying concentrations of soluble 4-4-20 (0.01–40 μM) were placed in tubes. The experiments were either mixed with the 4M5.3 displaying cells and fluorescein–biotin first, allowed to incubate for 1 h at 25°C, and then the 4-4-20 competitor was added, or the competitor and fluorescein were allowed to incubate together for 1 h at 25°C, and then the 4M5.3 displaying cells were added. All experiments contained a final concentration of 1× Pen-Strep. The tubes were then incubated at 25°C for up to 15 d, with daily mixing of the samples. Fluorescein–biotin binding to the cells was detected by secondary labeling with streptavidin–Phycoerythrin (Pharmingen, BD Biosciences), with analysis by flow cytometry (XL cytometer; Beckman Coulter). All experiments were performed in triplicate.
Expression analysis
Antibody expression levels were probed through noting the highest labeling level under the antigen saturating condition in the affinity experiments or by labeling 1 × 106 yeast surface-displaying cells (induced as above) with 100 μL of 1:50 9e10 (anti-c-myc epitope tag; Covance) for 30 min on ice, followed by labeling with 10 μL of 1:50 goat–anti-mouse IgG-Phycoerythrin (Sigma Aldrich) for 30 min on ice. Analysis was by flow cytometry, as above.
Acknowledgments
This work was supported in part by NIH CA96504. We are grateful for helpful discussions with B. Tidor and S. Lippow.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051842406.
References
- Boder, E.T., Midelfort, K.S., and Wittrup, K.D. 2000. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. 97: 10701–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, L.T., Duan, Y., Wang, L., Massova, I., and Kollman, P.A. 1999. Molecular dynamics and free-energy calculations applied to affinity maturation in antibody 48G7. Proc. Natl. Acad. Sci. 96: 14330–14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzin, L.K., Gulliver, G.A., and Voss Jr., E.W. 1993. Mutational analysis of active site contact residues in anti-fluorescein monoclonal antibody 4-4-20. Mol. Immunol. 30: 1331–1345. [DOI] [PubMed] [Google Scholar]
- Dixon, R.W. and Kollman, P. 1999. The free energies for mutating S27 and W79 to alanine in streptavidin and its biotin complex: The relative size of polar and nonpolar free energies on biotin binding. Proteins 36: 471–473. [PubMed] [Google Scholar]
- Dixon, R.W., Radmer, R.J., Kuhn, B., Kollman, P.A., Yang, J., Raposo, C., Wilcox, C.S., Klumb, L.A., Stayton, P.S., Behnke, C., et al. 2002. Theoretical and experimental studies of biotin analogues that bind almost as tightly to streptavidin as biotin. J. Org. Chem. 67: 1827–1837. [DOI] [PubMed] [Google Scholar]
- Foote, J. and Eisen, H.N. 1995. Kinetic and affinity limits on antibodies produced during immune responses. Proc. Natl. Acad. Sci. 92: 1254–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag, S., Le Trong, I., Chilkoti, A., Klumb, L.A., Stayton, P.S., and Stenkamp, R.E. 1998. Structural studies of binding site tryptophan mutants in the high-affinity streptavidin–biotin complex. J. Mol. Biol. 279: 211–221. [DOI] [PubMed] [Google Scholar]
- Herron, J.N., Kranz, D.M., Jameson, D.M., and Voss Jr., E.W. 1986. Thermodynamic properties of ligand binding by monoclonal anti-fluorescyl antibodies. Biochemistry 25: 4602–4609. [DOI] [PubMed] [Google Scholar]
- Herron, J.N., Terry, A.H., Johnston, S., He, X.M., Guddat, L.W., Voss Jr., E.W., and Edmundson, A.B. 1994. High resolution structures of the 4-4-20 Fab–fluorescein complex in two solvent systems: Effects of solvent on structure and antigen-binding affinity. Biophys. J. 67: 2167–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz, D.M., Herron, J.N., and Voss Jr., E.W. 1982. Mechanisms of ligand binding by monoclonal anti-fluorescyl antibodies. J. Biol. Chem. 257: 6987–6995. [PubMed] [Google Scholar]
- Lee, L.P. and Tidor, B. 2001. Optimization of binding electrostatics: Charge complementarity in the barnase–barstar protein complex. Protein Sci. 10: 362–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., Li, H., Yang, F., Smith-Gill, S.J., and Mariuzza, R.A. 2003. X-ray snapshots of the maturation of an antibody response to a protein antigen. Nat. Struct. Biol. 10: 482–488. [DOI] [PubMed] [Google Scholar]
- Lim, K. and Herron, J.N. 1995. Molecular dynamics of the anti-fluorescein 4-4-20 antigen-binding fragment. 1. Computer simulations. Biochemistry 34: 6962–6974. [DOI] [PubMed] [Google Scholar]
- Lim, K., Jameson, D.M., Gentry, C.A., and Herron, J.N. 1995. Molecular dynamics of the anti-fluorescein 4-4-20 antigen-binding fragment. 2. Time-resolved fluorescence spectroscopy. Biochemistry 34: 6975–6984. [DOI] [PubMed] [Google Scholar]
- Maynard, J. and Georgiou, G. 2000. Antibody engineering. Annu. Rev. Biomed. Eng. 2: 339–376. [DOI] [PubMed] [Google Scholar]
- Midelfort, K.S., Hernandez, H.H., Lippow, S.M., Tidor, B., Drennan, C.L., and Wittrup, K.D. 2004. Substantial energetic improvement with minimal structural perturbation in a high affinity mutant antibody. J. Mol. Biol. 343: 685–701. [DOI] [PubMed] [Google Scholar]
- Sundberg, E.J. and Mariuzza, R.A. 2002. Molecular recognition in antibody–antigen complexes. Adv. Protein Chem. 61: 119–160. [DOI] [PubMed] [Google Scholar]
- Terzyan, S., Ramsland, P.A., Voss Jr., E.W., Herron, J.N., and Edmundson, A.B. 2004. Three-dimensional structures of idiotypically related Fabs with intermediate and high affinity for fluorescein. J. Mol. Biol. 339: 1141–1151. [DOI] [PubMed] [Google Scholar]
- Wedemayer, G.J., Patten, P.A., Wang, L.H., Schultz, P.G., and Stevens, R.C. 1997. Structural insights into the evolution of an antibody combining site. Science 276: 1665–1669. [DOI] [PubMed] [Google Scholar]
- Whitlow, M., Howard, A.J., Wood, J.F., Voss Jr., E.W., and Hardman, K.D. 1995. 1.85 A structure of anti-fluorescein 4-4-20 Fab. Protein Eng. 8: 749–761. [DOI] [PubMed] [Google Scholar]
- Xu, J.L. and Davis, M.M. 2000. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity 13: 37–45. [DOI] [PubMed] [Google Scholar]
- Yang, P.L. and Schultz, P.G. 1999. Mutational analysis of the affinity maturation of antibody 48G7. J. Mol. Biol. 294: 1191–1201. [DOI] [PubMed] [Google Scholar]
- Yin, J., Mundorff, E.C., Yang, P.L., Wendt, K.U., Hanway, D., Stevens, R.C., and Schultz, P.G. 2001. A comparative analysis of the immunological evolution of antibody 28B4. Biochemistry 40: 10764–10773. [DOI] [PubMed] [Google Scholar]