

Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) poses a major threat to human health, particularly through hospital acquired infection. The spread of MRSA means that novel targets are required to develop potential inhibitors to combat infections caused by such drug-resistant bacteria. Thymidylate kinase (TMK) is attractive as an antibacterial target as it is essential for providing components for DNA synthesis. Here, we report crystal structures of unliganded and thymidylate-bound forms of S. aureus thymidylate kinase (SaTMK). His-tagged and untagged SaTMK crystallize with differing lattice packing and show variations in conformational states for unliganded and thymidylate (TMP) bound forms. In addition to open and closed forms of SaTMK, an intermediate conformation in TMP binding is observed, in which the site is partially closed. Analysis of these structures indicates a sequence of events upon TMP binding, with helix α3 shifting position initially, followed by movement of α2 to close the substrate site. In addition, we observe significant conformational differences in the TMP-binding site in SaTMK as compared to available TMK structures from other bacterial species, Escherichia coli and Mycobacterium tuberculosis as well as human TMK. In SaTMK, Arg 48 is situated at the base of the TMP-binding site, close to the thymine ring, whereas a cis-proline occupies the equivalent position in other TMKs. The observed TMK structural differences mean that design of compounds highly specific for the S. aureus enzyme looks possible; such inhibitors could minimize the transfer of drug resistance between different bacterial species.
Keywords: S. aureus, thymidylate kinase, X-ray crystallography, substrate-induced conformational change, drug design
Staphylococcus aureus, a member of the Micrococcaceae family, is a versatile pathogen of human and various animal species. Humans are a natural reservoir of S. aureus (Lowy 1998), which typically lives as a commensal of the nose in 30%–70% of the population (Peacock et al. 2001). S. aureus is a common source of infection, causing a wide variety of diseases in humans, ranging in severity from minor skin conditions to life-threatening systemic infections such as endocarditis and haemolytic pneumonia. S. aureus can also cause toxin-mediated diseases such as toxic shock syndrome (Lowy 1998) and in recent years has become the most common cause of hospital-acquired infections, which are increasingly resistant to antibiotics (Peacock et al. 2001). Between 40% and 60% of S. aureus isolates from hospitals in several industrialized nations are now resistant to methicillin (Fluit et al. 2001). Methicillin-resistant S. aureus (MRSA) infections are currently treated with vancomycin; however, the emergence of vancomycin-resistant S. aureus (VRSA) has been reported (Hiramatsu 2001; Menichetti 2005). Thus the search for novel protein targets, against which to develop potential anti-S. aureus drugs, has become a priority in antibacterial research. Development of narrow-spectrum rather than broad-spectrum antibacterial agents may have some advantages in reducing the transfer of drug resistance between different bacterial species (Lee et al. 1999).
Nucleoside monophosphate (NMP) kinases are of special interest in a number of areas of drug discovery as they catalyze the production of vital precursors for the synthesis of DNA and RNA. Thymidylate kinase (TMK) (E.C. 2.7.4.9) is a member of the NMP kinase family and catalyzes the phosphoryl transfer from the preferred phosphoryl donor, ATP, to thymidine monophosphate (TMP), yielding thymidine diphosphate (TDP). The TMK reaction is positioned at the junction of the de novo and salvage pathway of thymidine triphosphate (dTTP) synthesis, with TMK being the last specific enzyme for dTTP synthesis.
NMP kinases exhibit a protein fold featuring a central five-stranded β-sheet surrounded by helices (Yan and Tsai 1999). The protein can be divided into three parts, namely, the CORE region, the NMP-binding region, and the LID region. The CORE region is the most conserved among NMP kinases, comprising mainly β-sheets with surrounding α-helices, and contains the P-loop, which is involved in the binding of ATP. The NMP-binding domain is largely helical among all NMP kinases except guanylate monophosphate kinases. The LID region is a flexible stretch of residues covering part of the phosphate donor site. Substrate-induced conformational changes have been observed in various family members of NMP kinases with large domain movements upon binding of one or both substrates (Muller-Dieckmann and Schulz 1994, 1995; Scheffzek et al. 1996; Blaszczyk et al. 2001; Sekulic et al. 2002).
Lavie et al. (1998b) identified two classes of TMKs: Class I enzymes are mainly from eukaryotes and have an arginine residue in position x1 of the consensus sequence Gxxx1xGKx of the P-loop, which interacts with ATP; class II TMKs are of prokaryotic origin and can be distinguished by the presence of a glycine residue instead of an arginine in the x1 position of the consensus sequence, along with additional basic residues (mostly Arg) in the LID region that interact with ATP. Inhibitors may thus potentially be designed to specifically target prokaryotic TMKs without affecting the host (human) enzyme, with the expectation that toxicity for the host can be minimized.
While structural studies on the TMP-binding site have been reported for Mycobacterium tuberculosis TMK (MtTMK) (Li de la Sierra et al. 2001), this enzyme belongs to neither of the common classes of TMKs; rather, it appears to have a distinctive catalytic mechanism (Li de la Sierra et al. 2001; Munier-Lehmann et al. 2001; Fioravanti et al. 2003; Haouz et al. 2003). We therefore set out to study TMP binding to S. aureus TMK (SaTMK), a class II TMK, by X-ray crystallography. SaTMK exhibits typical characteristics of a class II enzyme, containing a Gly at position x1 of the P-loop and a series of basic residues (Arg 141, 147, and 151, and Lys 144) in the LID region. This report describes the crystal structures of SaTMK in its unliganded and TMP-bound states, including a form with an intermediate conformation, allowing dissection of associated sequential conformational changes of the enzyme upon TMP binding. Additionally, significant differences in the TMP site of SaTMK compared to other bacterial TMKs and human TMK have been identified. The structures will thus potentially allow the design of novel inhibitors specific to SaTMK, which have potential as new antimicrobial drugs to tackle the increasing problem of drug-resistant bacteria, such as MRSA.
Results and Discussion
Overall comparison of differently liganded and tagged/ untagged SaTMK with other TMK structures
Although the his-tagged and untagged SaTMKs crystallized under similar conditions and belong to the same space group P21, the crystals have significantly different cell dimensions and varying diffraction limits (Table 1). Comparison of the structures revealed a difference in molecular packing of the TMK in the two crystal forms (data not shown). The N terminus of the untagged SaTMK crystal form is close to a neighboring molecule, and thus introduction of a his-tag is presumed to be incompatible with such packing, thus producing a different form. The untagged SaTMK proved to be easier to crystallize, without the need for microseeding, and the resolution of the data is also better compared to that of the tagged-form crystals. Also, differences in conformational states of TMP bound forms were observed between the his-tagged and untagged forms of the enzyme (see Table 1). In all cases, the ATP analogs and Mg2+ were not bound.
Table 1.
Data collection, refinement statistics, quality indicators, and description of SaTMK structures

aHigest resolution shell is shown in parentheses.
bAs defined by PROCHECK (Laskowski et al. 1993).
As expected, the overall SaTMK structure has an α/β-fold consisting of nine α-helices that surround a five- stranded β-sheet core (Fig. 1A) similar to those of the previously determined bacterial TMK structures, Escherichia coli TMK (EcTMK) (Lavie et al. 1998b) and MtTMK (Li de la Sierra et al. 2001) (31.6% and 21.8% amino acid identity, respectively) (Fig. 1B). Overlaying the EcTMK and MtTMK models in turn with SaTMK resulted in an overlap of the β-sheet core with an RMSD of 0.84 Å for 34 Cαs (EcTMK) and 0.70 Å for 29 Cαs (MtTMK). The nine helices show more divergent positioning relative to the β-sheet (RMSD of 1.26 Å for 81 Cαs [EcTMK] and 1.15 Å for 64 Cαs [MtTMK]). Overlaying the SaTMK structure with the human TMK structure (19% amino acid identity) (Ostermann et al. 2000), which has a similar fold to the bacterial TMKs, resulted in an overlap of the β-sheet core (RMSD of 0.47 Å for 23 Cαs) and the helices (RMSD of 1.00 Å for 78 Cαs).
Figure 1.
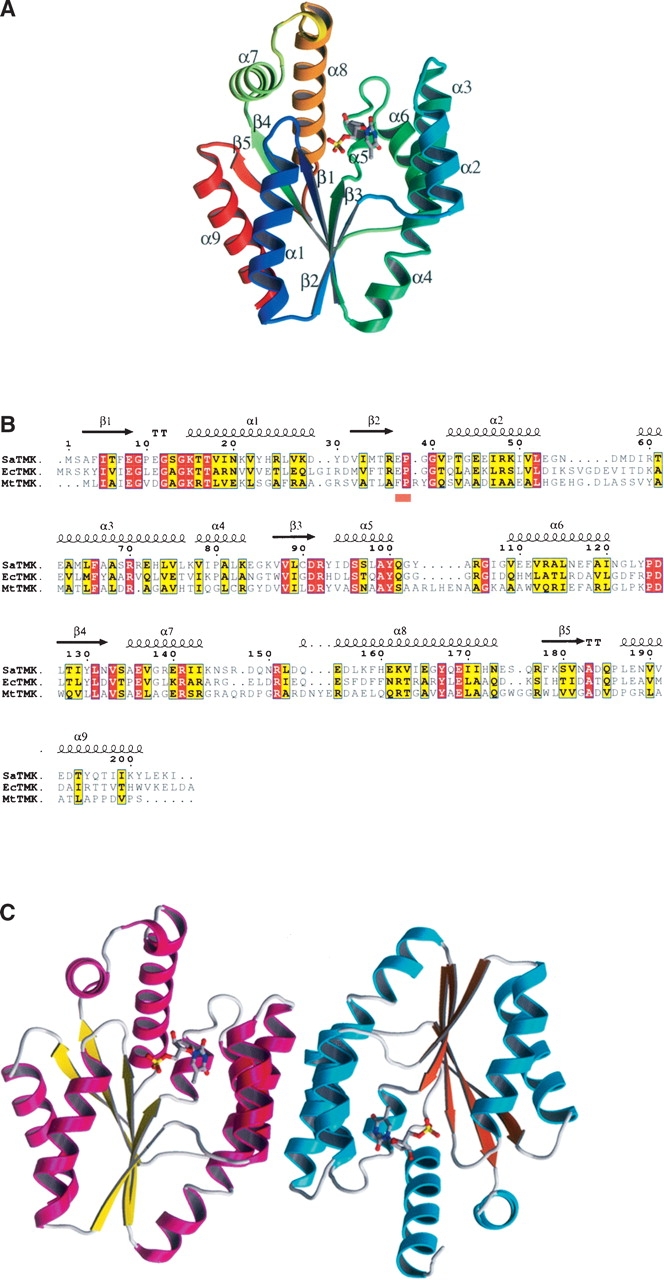
Structure of SaTMK. (A) Ribbon diagram of SaTMK with secondary structure marked and showing bound TMP. SaTMK is shown in rainbow colors from blue at the N terminus to red at the C terminus. TMP is shown in standard atom colors, with the carbon atoms in cyan. (B) Sequence alignment of SaTMK, EcTMK, and MtTMK. The secondary structure elements shown above the alignment are of SaTMK. The orange bar below the alignment indicates the conserved (E/F)P loop. (C) Ribbon diagram of the SaTMK dimer. The helices of the two molecules are shown in magenta and cyan, while the sheets are shown in yellow and orange. TMP is shown in standard atom colors, with the carbon atoms in gray.
In all the structures presented here, SaTMK crystallizes with a dimer per asymmetric unit. The two subunits of the dimer are related by noncrystallographic twofold symmetry with helices α2, α3, and α6 stacking to their antiparallel equivalents (Fig. 1C), similar to that seen in EcTMK (Lavie et al. 1997, 1998b). The dimer interface, although largely hydrophobic, also contains two pairs of hydrogen bonding interactions, Asp 58–Arg 71 and Arg 60–Asn 121, which are not observed in other TMK structures. Arg 92 of the conserved DRx loop is the only residue lying outside the allowed regions of the Ramachandran plot. Pro 38 of the conserved sequence (E/F)P is in a cis conformation, as in previously reported TMK structures (Lavie et al. 1997, 1998a, b; Ostermann et al. 2000, 2003; Li de la Sierra et al. 2001).
Comparing the three different SaTMK structures determined here shows conformational differences in the region of helices α2 and α3. Molecule A of the unliganded, untagged SaTMK superimposed on that of TMP-bound his-tagged SaTMK (Table 2), with the exception of the TMP-binding site and the LID region. Displacement of α2 and α3 at the TMP-binding domain was observed with unliganded SaTMK adopting a more open conformation than TMP-bound his-tagged TMK and the LID region being in the open conformation. We therefore designate the unliganded/untagged and histagged/TMP-bound SaTMK structures as the open and TMP site closed conformations, respectively (Table 1), and superposition of molecule A of the two SaTMK structures indicates that a rotation of 17.8° of molecule B has occurred. This rotation is a concerted movement of the dimer resulting from the movement of α2 and α3 as there are no significant changes in the interactions across the dimer interface. Comparison of the TMP-bound untagged SaTMK structure with the open and TMP site closed conformations of SaTMK revealed that molecule B has the open conformation while molecule A has an intermediate conformation (Table 2), which we designate as TMP site partially closed (Table 1). Such conformational changes are related to the presence or absence of TMP as well as the presence or absence of the his-tag and are discussed in more detail in the following sections.
Table 2.
Comparison of different SaTMK structures

aData set and molecule with reference to Table 1.
Comparison of the TMP-binding site in different TMKs
No ligands were seen to be bound in the untagged SaTMK structure despite the enzyme being crystallized in the presence of substrates, while for the his-tagged SaTMK structure, TMP was observed in both molecules with full occupancy (Table 1). It did, however, prove possible to bind TMP to untagged SaTMK by soaking in high concentrations of TMP, where it is observed in the expected site in molecule A with full occupancy. For molecule B, TMP is found in two positions, both with partial occupancy (Table 1), which appears to be in part due to the crystal packing of the untagged SaTMK molecules such that the substrate site in molecule B is held more in the open conformation by contacts with a neighboring molecule.
Analysis of TMP binding shows that, although some features are in common with other TMKs, a significant difference in one region of the binding site is apparent. The TMP-binding site of SaTMK and the associated network of hydrogen bonds are shown in Figure 2A. The features in common for TMP binding among different TMKs are (1) aromatic ring stacking interactions of the pyrimidine ring and a phenylalanine side chain (in this case Phe 66); (2) the positioning of Tyr 100, which allows selection of deoxynucleotides over ribonucleotides; and (3) an H-bond between O4 of the pyrimidine moiety of TMP and the side chain of Arg 70, which favors the selection of T/U over C. The importance of the interaction of Arg 70 to O4 of the thymine base of TMP is demonstrated in the equivalent residues of varicella zoster virus thymidine kinase (Bird et al. 2003) and Herpes simplex virus type 1 thymidine kinase (Hinds et al. 2000; Vogt et al. 2000). Other residues that are <3.9 Å away from TMP include Glu 11, Lys 15, Arg 36, Glu 37, Arg 92, Ser 97, and Gln 101. The interaction of the P-loop carboxylic residue Glu 11 with the ribose 3′-OH of TMP is conserved in all TMKs, an essential interaction for locating the sugar ring. O3 of the TMP phosphate group interacts with NE of Arg 92; the latter is the only residue to have a “disallowed” conformation in the enzyme structure. Arg 92 has been shown to be important for the phosphoryl transfer from the donor to TMP (Haouz et al. 2003).
Figure 2.

TMP-binding site of SaTMK. (A) Simulated annealing omit map of bound TMP in his-tagged SaTMK structure. Carbon atoms of TMP are drawn in cyan; those of residues interacting with TMP are drawn in gray. Potential hydrogen bonds are indicated by dotted lines; those that directly involve atoms of the substrate are drawn in gray, others in cyan. (B) Simulated annealing omit map of the (E/F)P loop and Arg 48 in his-tagged SaTMK structure. Carbon atoms of TMP are drawn in cyan; those of residues of SaTMK are drawn in gray. (C) Stereo view of the superposition of the TMP-binding site of SaTMK, EcTMK, MtTMK, and human TMK. The important residues defining the TMP-binding site and the positions of the conserved cis-proline are shown. The residues of SaTMK and TMP are drawn in atom colors, while the residues of EcTMK, MtTMK, and human TMK are drawn in green, yellow, and magenta, respectively. The residues of SaTMK are labeled in black, and the cis-Pro 43 of human TMK is labeled in magenta. Interaction of Arg 48 with the main-chain N atom of Glu 37 via water molecules and the interaction of Glu 37 with Arg 70 are indicated by cyan dotted lines. The interaction of Glu 40 with Arg 74 of EcTMK is indicated by black dotted lines.
Turning to the areas of significant difference in the TMP-binding site of SaTMK when compared to those in EcTMK, MtTMK, and human TMK: Besides the conserved interaction with the thymine base of TMP discussed above, Arg 70 of SaTMK also interacts with OD1 of Asn 116 via atom NH1. A similar interaction is observed in MtTMK and human TMK, where the equivalent residue, Arg 74 (Arg 76 in human TMK), forms an ion pair with Glu 124 (Asp 121 in human TMK), while in EcTMK it is replaced by interactions with Tyr 75 and Asp 102. Arg 70 NE and NH2 atoms form salt bridge/H-bonds with the carboxylate group of Glu 37, also observed in EcTMK; however, the interaction cannot be present with the structurally equivalent phenylalanine in both MtTMK and human TMK. While the thymidine N3 and O2 atoms H-bond with Gln 101 in SaTMK and EcTMK, for MtTMK the O2 of the TMP base does not form a contact with the protein and N3 interacts with an asparagine residue (Asn 100). In human TMK, the thymidine N3 and O3 atoms do not form direct contacts with the protein but interact with Thr 105 via water molecules. Apart from the interaction with Arg 70 discussed previously, the O4 of the TMP pyrimidine ring also H-bonds with Ser 97; this interaction is replaced by a threonine (Thr 105) in EcTMK and an arginine (Arg 74) in MtTMK, while in human TMK, this interaction is not observed since the equivalent residue is a glycine (Gly 102). Besides interaction with Arg 92, the O3 of the TMP base in SaTMK also forms an H- bond with the conserved Lys 15, an interaction not observed in other reported TMK structures. For example, in the EcTMK structure with the bisubstrate inhibitor TP5A, Lys 16 H-bonds with the β- and γ-phosphates of the phosphoryl donor (Lavie et al. 1998b), while in MtTMK, the equivalent lysine (Lys 13) interacts with the β-phosphate (Fioravanti et al. 2003). This conserved lysine is thought to have a catalytic role of stabilizing the pentavalent transition state involving the phosphoryl group to be transferred (Reinstein et al. 1990). The conserved lysine also plays an important role in maintaining the conformation of the P-loop, as suggested by site- directed mutagenesis in shikimate kinase (Krell et al. 2001) and adenylate kinase (Reinstein et al. 1990; Tian et al. 1990; Byeon et al. 1995). Arg 36 interacts with O1 of the TMP phosphate group, but this interaction is replaced by Tyr 39 in MtTMK and Arg 51 in EcTMK (equivalent of Arg 48 in SaTMK). Unlike Arg 51 of EcTMK, Arg 48 does not interact with TMP; instead, it takes the place of Pro 38 to form the bottom of the TMP-binding cavity.
The most striking difference in the substrate-binding site of SaTMK, compared with other TMKs, is at the base of the TMP-binding cavity. In all other reported TMK structures, the loop containing the conserved (E/F)P sequence, which has proline in a cis conformation and connects β2 to α2, forms the base of the TMP-binding cavity (Lavie et al. 1997, 1998a, b; Ostermann et al. 2000; Li de la Sierra et al. 2001). The main-chain N atom of the cis-proline is positioned ~4 Å from the pyrimidine group of TMP. In contrast, in SaTMK, Arg 36 forms part of the turn containing the conserved (E/F)P sequence (Fig. 2B) rather than being in at the end of β2, as is the case for EcTMK, MtTMK, and human TMK. Although the conserved Glu 37 of SaTMK still interacts with Arg 70, as is also observed in EcTMK, the cis-proline has turned away from the TMP-binding site and is located ~7.6 Å from the bound TMP, creating a space at the base of the TMP-binding site. The additional space is filled by the side chain of Arg 48 (Fig. 2C), which, in turn, is linked to the main-chain nitrogen atom of Glu 37 of the (E/F)P loop via water molecules.
TMP-induced conformational changes
As described in a previous section, a range of conformations of SaTMK have been identified that are related to TMP binding (Table 1). The structures appear to represent snapshots along the TMP-binding pathway of SaTMK (Fig. 3A). Combining SHP and HINGEFIND analyses from open and TMP site closed conformations revealed an overall domain movement of helices α2 and α3 (residues 43–75) toward the CORE region at the TMP-binding site with a rotation of ~14° (Fig. 3A). However, from analysis of torsion angle difference (TAD) plots, no hinge points relating to the domain movement were identified. In spite of this, the TAD plot showed significant torsion angle differences at the loop connecting α2 and α3 (residues 51–60) (Fig. 3B), which is flexible as indicated by the high B-factors.
Figure 3.
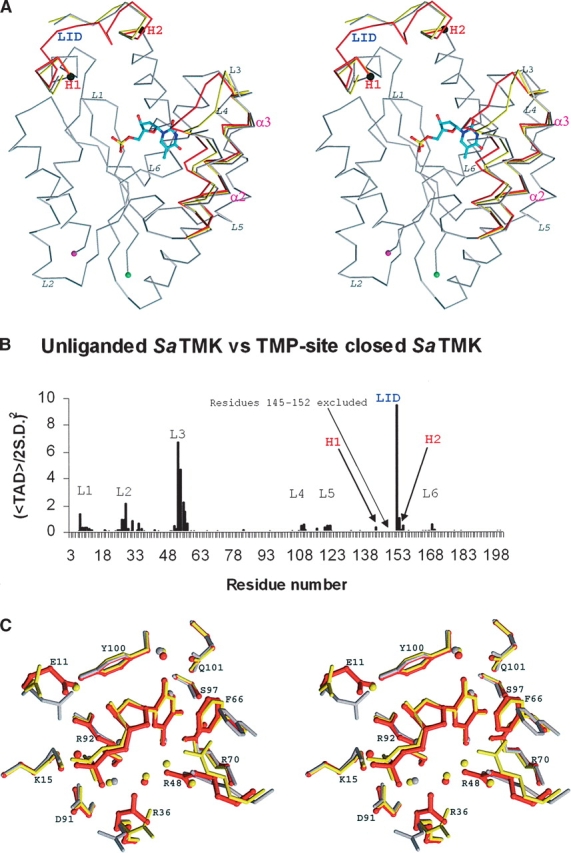
TMP-induced conformational change of SaTMK. (A) Stereo view of the alignmentof Cα traces of unliganded SaTMK (gray); TMP site partially closed SaTMK (yellow) and TMP site closed SaTMK (red). The N and C termini are indicated by green and by magenta spheres, respectively. The helices α2and α3 are labeled in magenta. Hinge points of the LID regions (labeled in blue) as identified with TAD analysis are drawn as black spheres and labeled in red. Flexible loops with torsion angle differences identified by TAD analysis are labeled in italics. (B) TAD plot of comparison of torsion angles between unliganded and TMP site closed SaTMK. The significant TAD peaks are labeled in black and correspond to the loops in A. The LID region is labeled in blue. The hinge points of the LID region are labeled as H1 and H2 in red. (C) Close-up stereo view of TMP-binding site. Residues are drawn in gray for unliganded SaTMK, in yellow for the SaTMK with partially closed TMP site, and in red for TMP site closed SaTMK.
Movement of the LID region toward the CORE region of the enzyme was also evident upon TMP binding, involving the rotation of the LID domain by 31°, with hinge points at residue 142 and residue 158 (Fig. 3A,B). This change results in the enzyme adopting a closed conformation with respect to the TMP site, as observed in other NMP kinases upon the binding of the non-ATP substrate (Vonrhein et al. 1995; Lavie et al. 1997; Briozzo et al. 1998; Fioravanti et al. 2003; Segura- Pena et al. 2004).
Closer analysis of the differences at the TMP-binding site from open to closed conformations revealed that the changes involved sequential movements of α3 and α2 toward the TMP-binding site. Upon TMP binding, helix α3 rotates by 4.3° toward the CORE region with little movement of α2, as seen in the transition from the open to TMP site partially closed conformation (Fig. 3A). Helix α2 then rotates toward the CORE region by 16.5°, closing the TMP-binding site. The extent of movement can also be expressed by the shift of the Cα positions of residues 53 and 58, which are located at the beginning and the end of the loop connecting the helices α2 and α3, respectively. The movement of the Cα of residue 58 is 0.67 Å between open and TMP site partially closed conformations and 0.92 Å between open and TMP site closed conformations, while residue 53 moves 1.4 Å between open and TMP site partially closed conformations and 4.7 Å between open and TMP site closed conformations.
The superposition of the open, partially closed, and closed conformations of the TMP-binding site of SaTMK is illustrated in Figure 3C. In the open conformation, a network of water molecules interacting with residues Gln 101 and Arg 70 occupies the thymine portion of the TMP site. Upon TMP binding, the rotation of helix α3 allows the stacking of Phe 66 to the pyrimidine ring of TMP, and although the site remains partially open, the essential features of TMP binding are established at this stage. The movement of α2 then brings Arg 48 to assume its position at the bottom of the TMP-binding cavity, thereby closing the TMP-binding site. Accompanying small induced-fit movements of helix α3 along with residues 11 and 36 also occur.
Substrate-induced conformational changes at the NMP-binding site have been observed in the other members of the NMP kinase family, e.g., human UMP/CMP kinase (Segura-Pena et al. 2004), yeast GMP kinase (Stehle and Schulz 1990; Blaszczyk et al. 2001), adenylate kinases (Vonrhein et al. 1995), and E. coli CMP kinase (Briozzo et al. 1998). However, the current report is the first description of substrate-induced conformational changes in TMK upon TMP binding in a class I or II TMK, as prior to this no unliganded structures had been solved. In MtTMK, which is distinctive to class I or II, only a small induced-fit mechanism at the level of the α-phosphate group and the ribose moiety upon TMP-binding has been reported (Fioravanti et al. 2003). This small difference can be explained, as the (E/F)P loop of MtTMK is already in position to form the base of the TMP-binding cavity. In this case, the TMP-binding site is already pre-tuned to accommodate the substrate, and therefore no major domain movement is required to close the binding site. However, as the base of the TMP-binding site is in a more open conformation than that of MtTMK and other bacterial TMKs (Fig. 2C), movement of helix α2 is required to bring Arg 48 into position to close the TMP-binding site. The TMP-induced conformational change observed in SaTMK therefore appears to be related to the position of the (E/F)P loop.
Conclusion
SaTMKs with differing conformational states were observed from crystal structure determination of his-tagged and untagged SaTMK together with the presence or absence of TMP. Differences in molecular packing between crystals of tagged and untagged forms of SaTMK appear to contribute to the observed range of conformational states. The crystal contacts at the TMP-binding site of untagged SaTMK crystal forms are more extensive than those of the his-tagged form, resulting in a hindrance of domain movement upon TMP binding. Analysis of the structures reveals sequential conformational changes from an open TMP-binding site to a closed state.
Significant differences in the conformation of the TMP-binding site in SaTMK compared to known bacterial and human TMK structures were identified that mainly relate to the positioning of the (E/F)P loop. Such differences are surprising considering the level of sequence identity particularly between SaTMK and EcTMK (~32%), typical class II TMKs, and the conserved role of substrate binding central to the biological function of these enzymes. The structural results appear unambiguous as the TMP site conformation was observed in both crystal forms of untagged and his-tagged SaTMK. It is clear that prediction of the TMP-binding site of SaTMK from homology modeling would be difficult, thus re-emphasising the need for caution in using such approaches.
The availability of crystal structures of TMP binding to SaTMK should stimulate the design of inhibitors that may have potential as novel antibacterial drugs using structure-based approaches. Two options to target the TMP-binding site of the enzyme are possible. First, to direct the design of inhibitors to regions of the TMP site conserved between the various bacterial TMKs but with structural differences to that of human TMK in order to obtain broad spectrum antibacterial agents. A second, more intriguing, option is to target the region with a conformational difference in the TMP-binding site of SaTMK with the aim of developing narrow-spectrum inhibitors against MRSA. One possibility would be to design inhibitors containing hydrogen-bonding groups targeting Arg 48 at the base of the TMP-binding cavity of SaTMK, an interaction not formed by other TMKs containing proline in this position of the active site. Such inhibitors would have the advantage of minimizing the transfer of drug resistance between different bacterial species as well as having the potential to minimize binding to the host (human) enzyme, hence minimizing toxicity.
Materials and Methods
Cloning, protein expression, and purification
The gene for SaTMK was amplified by PCR from S. aureus (strain SH1000) genomic DNA using primers incorporating NdeI/BamHI restriction sites, and the product was ligated into the expression plasmid pET15b (Novagen). The resulting plasmid, pMUT68 coding for N-terminal his-tagged SaTMK, was transformed into the E. coli expression strain Codon+. For untagged SaTMK, the PCR product was ligated into the expression plasmid pET3a (Novagen) via the NdeI and BamHI sites. The resulting plasmid, pMUT111, was transformed into the expression strain BL21(DE3). For both constructs, cells were grown at 37°C in LB with ampicillin selection and induced with IPTG for 5 h. Typically, 25 g of pelleted cells was sonicated in 500 mL of 50 mM K phosphate (pH 7.2), 1 mM DTT, and 1 mM benzamidine, and the lysate was clarified by centrifugation.
His-tagged SaTMK was purified using two column chromatography steps: (1) Zn2+-charged chelating Sepharose column eluted with a 0.0–0.3 M imidazole gradient in 50 mM K phosphate (pH 7.2) and 1 mM DTT, and (2) hydroxyapatite column eluted with a 50–400 mM gradient of K phosphate (pH 7.2) and 1 mM DTT. At each step in the purification procedure, fractions were analyzed by SDS-PAGE and appropriate fractions were pooled, giving a final yield of ~500 mg of SaTMK.
Untagged SaTMK was purified in a six-step protocol as follows: (1) DEAE Sephacel chromatography eluted with a 0.0–1.0 M NaCl gradient in 50 mM K phosphate (pH 7.2) and 1 mM DTT; (2) ammonium sulfate fractionation (35%–55% saturation); (3) phenyl-Sepharose eluted with a 1.0–0.0 M ammonium sulfate gradient in 50 mM K phosphate (pH 7.2) and 1 mM DTT; (4) hydroxyapatite chromatography in which the protein eluted following washes of 50 mM K phosphate (pH 7.2) and 1 mM DTT; (5) Mono Q FPLC eluted with a 0.0–1.0 M NaCl gradient in 50 mM K phosphate (pH 7.2) and 1 mM DTT; and (6) Sephacryl S300 gel-filtration chromatography run in 150 mM NaCl, 50 mM K phosphate (pH 7.2), and 1 mM DTT. At each step in the purification procedure, fractions were analyzed by SDS-PAGE, and appropriate fractions were pooled. The final yield was ~70 mg of purified untagged SaTMK. For both untagged and his-tagged SaTMK, protein was concentrated and buffer exchanged into 20 mM Tris-HCl (pH 7.4), 150 mM KCl using Vivaspin centrifugal concentrators (Vivascience) prior to crystallization.
Crystallization
For his-tagged SaTMK, an initial crystallization screen was performed using protein at 22 mg/mL complexed with 5 mM TMP, 5 mM ATPγS. Drops (100 nL) of protein solution were mixed with 100 nL of screening solution in a sitting drop vapor diffusion plate using a Cartesian robot (Walter et al. 2005). Crystals were obtained at 291 K from Hampton Grid Screen PEG/LiCl condition D4 (1 M LiCl, 0.1M HEPES at pH 7.0, 30% PEG 6000). Optimization of the condition gave crystals from 1 M LiCl, 0.1 M Na Cacodylate (pH 6.2–6.8), and 16%–22% PEG 6000; the best crystals were obtained by micro-seeding. Screening conditions for the crystallization of untagged SaTMK yielded crystals from 1 M LiCl, 0.1M HEPES (pH 6.8–7.2), and 16%–20% PEG 3350. For TMP bound untagged SaTMK structure, crystals were soaked in mother liquor containing 25 mM TMP, 25 mM AMPPcP, and 25 mM MgCl2 for 1 h prior to data collection.
Data collection and structure determination
The his-tagged and untagged SaTMK crystals were flash-cooled to 100 K after the addition of 10% MPD and 10% ethylene glycol to the mother liquor. For the his-tagged SaTMK crystals, data were collected on beamline ID 14.1 at the ESRF (Grenoble, France), while data for the untagged crystals were collected in-house using Cu Kα radiation from a Rigaku MicroMax 007 generator equipped with a MAR345 image plate. Data were processed with the HKL suite of programs (Otwinowski and Minor 1997). Crystals all belonged to space group P21 but with two distinct sets of cell dimensions dependent on the presence or absence of the his-tag. Data collection and processing statistics are shown in Table 1. Molecular replacement used a combination of EcTMK (PDB accession no. 4TMK) (Lavie et al. 1998b) and MtTMK structures (PDB accession no. 1GSI) (Li de la Sierra et al. 2001). The two TMK dimers were superimposed with the program SHP (Stuart et al. 1979), and structure factors were calculated from the overlapped models, which were used to solve the his-tagged SaTMK structure by molecular replacement with CNS (Brünger et al. 1998). O (Jones et al. 1991) was used for model building with EcTMK as the starting model. The untagged SaTMK structures were solved by molecular replacement with coordinates of the refined his-tagged SaTMK model using CNS (Brünger et al. 1998). The structures were refined in CNS using simulated annealing, positional, and B-factor refinement to give the final statistics shown in Table 1. O was used for model rebuilding (Jones et al. 1991).
Structure and sequence alignments
For comparison of the SaTMK structure with MtTMK, EcTMK, and human TMK (PDB accession no. 1E2F), SHP was used to overlap pairs of protein structures (Stuart et al. 1979). Different TMK amino acid sequences were retrieved via the ExPASy proteomics server Web site (http://www.expasy.org) and aligned with ClustalW.
Model analysis and identification of conformational changes
The SaTMK models were evaluated using PROCHECK (Laskowski et al. 1993). Different SaTMK structures were compared by overlapping coordinates sets using SHP (Stuart et al. 1979). Residues of SaTMK <3.9 Å away from TMP were determined by using the program CONTACT (Collaborative Computational Project, Number 4 1994). Cα torsion angles for each residue i, defined as the dihedral angle Cα(i – 1) – Cα(i) – Cα(i + 1) – Cα(i + 2), were examined for each SaTMK structure according to methods described by Flocco and Mowbray (1995). Torsion angle differences (TAD) between the structures were plotted using EXCEL with the Y-axis as (〈TAD〉/2 × SD)2, which allows significant TAD peaks to be observed above noise (Nichols et al. 2004). The locations of the hinges were found using the TCL script HINGEFIND (Wriggers and Schulten 1997) (partition value = 1.1 and maximum domains = 10), run, and visualized with the graphics package VMD (Humphrey et al. 1996).
Preparation of figures
The figures were prepared using BOBSCRIPT (Kraulis 1991; Esnouf 1999) and rendered with RASTER3D (Merritt and Bacon 1997).
Coordinates
The coordinates and structure factors have been deposited in the Protein Data Bank (accession nos. 2CCK, 2CCJ, and 2CCG, for the unliganded untagged, TMP bound untagged, and TMP bound his-tagged SaTMKs, respectively).
Acknowledgments
We thank the staff at beamline ID14.1 at the ESRF, Grenoble for help with the data collection; Dr. R. Esnouf, Ms. J. Dong, and Mr. A. Tamer for computer support; and Dr. K. Harlos for assistance with the in-house data collection. We thank Arrow Therapeutics for funding the research at Newcastle and at Oxford.
Footnotes
Reprint requests to: David K. Stammers, Division of Structural Biology, The Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, Oxford OX3 7BN, UK; e-mail: daves@strubi.ox.ac.uk; fax: +44-1865-287-547.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.052002406.
References
- Bird L.E., Ren J., Wright A., Leslie K.D., Degreve B., Balzarini J., Stammers D.K. 2003. Crystal structure of varicella zoster virus thymidine kinase J. Biol. Chem. 278: 24680–24687. [DOI] [PubMed] [Google Scholar]
- Blaszczyk J., Li Y., Yan H., Ji X. 2001. Crystal structure of unligated guanylate kinase from yeast reveals GMP-induced conformational changes J. Mol. Biol. 307: 247–257. [DOI] [PubMed] [Google Scholar]
- Briozzo P., Golinelli-Pimpaneau B., Gilles A.M., Gaucher J.F., Burlacu-Miron S., Sakamoto H., Janin J., Barzu O. 1998. Structures of Escherichia coli CMP kinase alone and in complex with CDP: A new fold of the nucleoside monophosphate binding domain and insights into cytosine nucleotide specificity Structure 6: 1517–1527. [DOI] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S.et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Byeon L., Shi Z., Tsai M.D. 1995. Mechanism of adenylate kinase. The “essential lysine” helps to orient the phosphates and the active site residues to proper conformations Biochemistry 34: 3172–3182. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: Programs for protein crystallography Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. 1999. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps Acta Crystallogr. D Biol. Crystallogr. 55: 938–940. [DOI] [PubMed] [Google Scholar]
- Fioravanti E., Haouz A., Ursby T., Munier-Lehmann H., Delarue M., Bourgeois D. 2003. Mycobacterium tuberculosis thymidylate kinase: Structural studies of intermediates along the reaction pathway J. Mol. Biol. 327: 1077–1092. [DOI] [PubMed] [Google Scholar]
- Flocco M.M. and Mowbray S.L. 1995. Cα-based torsion angles: A simple tool to analyze protein conformational changes Protein Sci. 4: 2118–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluit A.C., Verhoef J., Schmitz F.J. 2001. Frequency of isolation and antimicrobial resistance of Gram-negative and Gram-positive bacteria from patients in intensive care units of 25 European university hospitals participating in the European arm of the SENTRY Antimicrobial Surveillance Program 1997–1998 Eur. J. Clin. Microbiol. Infect. Dis. 20: 617–625. [DOI] [PubMed] [Google Scholar]
- Haouz A., Vanheusden V., Munier-Lehmann H., Froeyen M., Herdewijn P., Van Calenbergh S., Delarue M. 2003. Enzymatic and structural analysis of inhibitors designed against Mycobacterium tuberculosis thymidylate kinase. New insights into the phosphoryl transfer mechanism J. Biol. Chem. 278: 4963–4971. [DOI] [PubMed] [Google Scholar]
- Hinds T.A., Compadre C., Hurlburt B.K., Drake R.R. 2000. Conservative mutations of glutamine-125 in herpes simplex virus type 1 thymidine kinase result in a ganciclovir kinase with minimal deoxypyrimidine kinase activities Biochemistry 39: 4105–4111. [DOI] [PubMed] [Google Scholar]
- Hiramatsu K. 2001. Vancomycin-resistant Staphylococcus aureus: A new model of antibiotic resistance Lancet Infect. Dis. 1: 147–155. [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. 1996. VMD: Visual molecular dynamics J. Mol. Graph. 14: 33–38. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures J. Appl. Crystallogr. 24: 946–950. [Google Scholar]
- Krell T., Maclean J., Boam D.J., Cooper A., Resmini M., Brocklehurst K., Kelly S.M., Price N.C., Lapthorn A.J., Coggins J.R. 2001. Biochemical and X-ray crystallographic studies on shikimate kinase: The important structural role of the P-loop lysine Protein Sci. 10: 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Lavie A., Vetter I.R., Konrad M., Goody R.S., Reinstein J., Schlichting I. 1997. Structure of thymidylate kinase reveals the cause behind the limiting step in AZT activation Nat. Struct. Biol. 4: 601– 604. [DOI] [PubMed] [Google Scholar]
- Lavie A., Konrad M., Brundiers R., Goody R.S., Schlichting I., Reinstein J. 1998a. Crystal structure of yeast thymidylate kinase complexed with the bisubstrate inhibitor P1-(5′-adenosyl) P5-(5′-thymidyl) pentaphosphate (TP5A) at 2.0 Å resolution: Implications for catalysis and AZT activation Biochemistry 37: 3677–3686. [DOI] [PubMed] [Google Scholar]
- Lavie A., Ostermann N., Brundiers R., Goody R.S., Reinstein J., Konrad M., Schlichting I. 1998b. Structural basis for efficient phosphorylation of 3′-azidothymidine monophosphate by Escherichia coli thymidylate kinase Proc. Natl. Acad. Sci. 95: 14045–14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee V.J., Miller G.H., Yagisawa M. 1999. What's new in the antibiotic pipeline Curr. Opin. Microbiol. 2: 475–482. [DOI] [PubMed] [Google Scholar]
- Li de la Sierra I., Munier-Lehmann H., Gilles A.M., Barzu O., Delarue M. 2001. X-ray structure of TMP kinase from Mycobacterium tuberculosis complexed with TMP at 1.95 Å resolution J. Mol. Biol. 311: 87–100. [DOI] [PubMed] [Google Scholar]
- Lowy F.D. 1998. Staphylococcus aureus infections N. Engl. J. Med. 339: 520–532. [DOI] [PubMed] [Google Scholar]
- Menichetti F. 2005. Current and emerging serious Gram-positive infections Clin. Microbiol. Infect. 11:Suppl. 3 22–28. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon D.J. 1997. Raster3D: Photorealistic molecular graphics Methods Enzymol. 277: 505–524. [DOI] [PubMed] [Google Scholar]
- Müller-Dieckmann H.J. and Schulz G.E. 1994. The structure of uridylate kinase with its substrates, showing the transition state geometry J. Mol. Biol. 236: 361–367. [DOI] [PubMed] [Google Scholar]
- 1995. Substrate specificity and assembly of the catalytic center derived from two structures of ligated uridylate kinase J. Mol. Biol. 246: 522–530 ———. [DOI] [PubMed] [Google Scholar]
- Munier-Lehmann H., Chaffotte A., Pochet S., Labesse G. 2001. Thymidylate kinase of Mycobacterium tuberculosis: A chimera sharing properties common to eukaryotic and bacterial enzymes Protein Sci. 10: 1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C.E., Dhaliwal B., Lockyer M., Hawkins A.R., Stammers D.K. 2004. High-resolution structures reveal details of domain closure and “half-of-sites-reactivity” in Escherichia coli aspartate β-semialde-hyde dehydrogenase J. Mol. Biol. 341: 797–806. [DOI] [PubMed] [Google Scholar]
- Ostermann N., Schlichting I., Brundiers R., Konrad M., Reinstein J., Veit T., Goody R.S., Lavie A. 2000. Insights into the phosphoryl-transfer mechanism of human thymidylate kinase gained from crystal structures of enzyme complexes along the reaction coordinate Struct. Fold. Des. 8: 629–642. [DOI] [PubMed] [Google Scholar]
- Ostermann N., Segura-Pena D., Meier C., Veit T., Monnerjahn C., Konrad M., Lavie A. 2003. Structures of human thymidylate kinase in complex with prodrugs: Implications for the structure-based design of novel compounds Biochemistry 42: 2568–2577. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Peacock S.J., de Silva I., Lowy F.D. 2001. What determines nasal carriage of Staphylococcus aureus? Trends Microbiol. 9: 605–610. [DOI] [PubMed] [Google Scholar]
- Reinstein J., Schlichting I., Wittinghofer A. 1990. Structurally and catalytically important residues in the phosphate binding loop of adenylate kinase of Escherichia coli Biochemistry 29: 7451–7459. [DOI] [PubMed] [Google Scholar]
- Scheffzek K., Kliche W., Wiesmuller L., Reinstein J. 1996. Crystal structure of the complex of UMP/CMP kinase from Dictyostelium discoideum and the bisubstrate inhibitor P1-(5′-adenosyl) P5-(5′-uridyl) pentaphosphate (UP5A) and Mg2+at 2.2 A: Implications for water-mediated specificity Biochemistry 35: 9716–9727. [DOI] [PubMed] [Google Scholar]
- Segura-Pena D., Sekulic N., Ort S., Konrad M., Lavie A. 2004. Substrate-induced conformational changes in human UMP/CMP kinase J. Biol. Chem. 279: 33882–33889. [DOI] [PubMed] [Google Scholar]
- Sekulic N., Shuvalova L., Spangenberg O., Konrad M., Lavie A. 2002. Structural characterization of the closed conformation of mouse guanylate kinase J. Biol. Chem. 277: 30236–30243. [DOI] [PubMed] [Google Scholar]
- Stehle T. and Schulz G.E. 1990. Three-dimensional structure of the complex of guanylate kinase from yeast with its substrate GMP J. Mol. Biol. 211: 249–254. [DOI] [PubMed] [Google Scholar]
- Stuart D.I., Levine M., Muirhead H., Stammers D.K. 1979. Crystal structure of cat muscle pyruvate kinase at a resolution of 2.6 Å J. Mol. Biol. 134: 109–142. [DOI] [PubMed] [Google Scholar]
- Tian G.C., Yan H.G., Jiang R.T., Kishi F., Nakazawa A., Tsai M.D. 1990. Mechanism of adenylate kinase. Are the essential lysines essential? Biochemistry 29: 4296–4304. [DOI] [PubMed] [Google Scholar]
- Vogt J., Perozzo R., Pautsch A., Prota A., Schelling P., Pilger B., Folkers G., Scapozza L., Schulz G.E. 2000. Nucleoside binding site of herpes simplex type 1 thymidine kinase analyzed by X-ray crystallography Proteins 41: 545–553. [DOI] [PubMed] [Google Scholar]
- Vonrhein C., Schlauderer G.J., Schulz G.E. 1995. Movie of the structural changes during a catalytic cycle of nucleoside monophosphate kinases Structure 3: 483–490. [DOI] [PubMed] [Google Scholar]
- Walter T.S., Diprose J.M., Mayo C.J., Siebold C., Pickford M.G., Carter L., Sutton G.C., Berrow N.S., Brown J., Berry I.M.et al. 2005. A procedure for setting up high-throughput nanolitre crystallization experiments. Crystallization workflow for initial screening, automated storage, imaging and optimization Acta Crystallogr. D Biol. Crystallogr. 61: 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wriggers W. and Schulten K. 1997. Protein domain movements: Detection of rigid domains and visualization of hinges in comparisons of atomic coordinates Proteins 29: 1–14. [PubMed] [Google Scholar]
- Yan H. and Tsai M.D. 1999. Nucleoside monophosphate kinases: Structure, mechanism, and substrate specificity Adv. Enzymol. Relat. Areas Mol. Biol. 73: 103–134. [DOI] [PubMed] [Google Scholar]