

Abstract
The structure of the C-terminal antifreeze-like (AFL) domain of human sialic acid synthase was determined by NMR spectroscopy. The structure comprises one α- and two single-turn 310-helices and two β-strands, and is similar to those of the type III antifreeze proteins. Evolutionary trace analyses of the type III antifreeze protein family suggested that the class-specific residues in the human and bacterial AFL domains are important for their substrate binding, while the class-specific residues of the fish antifreeze proteins are gathered on the ice-binding surface.
Keywords: NMR, protein structure, antifreeze-like domain, sialic acid synthase, structural genomics
Sialic acids (acylated neuraminic acids) are widely found in nature and reside at the terminal ends of cell-surface glycoconjugates. Sialic acids play important roles in ligand-receptor binding and cell–cell and cell–extracellular matrix interactions during fertilization, embryogenesis, differentiation, and other phenomena (Schauer et al. 1995; Schauer 2004). The biological significance of sialic acids underscores the importance of characterizing their biosynthetic pathways. A variety of pathways involved in the biosynthesis of sialic acids in bacterial and mammalian cells have been identified (Angata and Varki 2002), although few structural and mechanistic studies on these enzymes have been performed. The biosynthesis of sialic acids begins with the formation of N-acetylmannosamine (ManNAc) from uridinediphospho-N-acetylglucosamine (UDP-GlcNAc). In bacteria, the ManNAc is then directly involved in a condensation reaction with phosphoenolpyruvate (PEP) to generate N-acetylneuramic acid (NeuNAc). In contrast, a three-step pathway converts ManNAc to NeuNAc in mammalian cells: phosphorylation of ManNAc to give ManNAc 6-phosphate (ManNAc-6P), condensation of ManNAc-6P with PEP to yield NeuNAc 9-phosphate (NeuNAc-9P), and then dephosphorylation of NeuAc-9P to generate NeuNAc.
Sialic acid synthase (SAS) catalyzes the condensation of PEP with ManNAc (bacteria) and ManNAc-6P (mammals) to yield NeuNAc and NeuNAc-9P, respectively. The bacterial and mammalian SASs share 30%–40% sequence identity (Lawrence et al. 2000), and both enzymes are composed of two distinct domains that are joined by an extended linker region. The N-terminal domain (NeuB domain) consists of ~250 amino acid residues, and is considered to bind the sugar substrates. The C-terminal domain consists of ~75 amino acid residues, and is called the antifreeze-like (AFL) domain, since it is similar to those observed in a variety of functional type III antifreeze proteins (AFPs). It has been proposed that the AFL domain is also involved in sugar binding, but the details of its function have remained elusive.
Recently, Gunawan et al. (2005) determined the crystal structure of an SAS from the bacterium Neisseria meningitidis, in complex with Mn2+, PEP, and ManNAc, which revealed a domain-swapped homodimeric architecture (PDB code 1XUZ). In the homodimer, the substrates are located in a cavity at the C-terminal end of a (β/α)8 barrel (TIM barrel) of the NeuB domain, and are capped by the AFL domain of the opposite monomer with an important hydrogen bond between the side chain of Arg314 and the acetyl oxygen of rManNAc. In addition, Ko and colleagues (Ko et al. 2003) proposed that the deletion of the AFL domain in the human SAS would abolish the SAS activity. Therefore, the AFL domain seems to play an important role in substrate binding by the SAS and to hold the key toward characterizing the sialic acid metabolic pathways.
To gain further insight into the structural and mechanistic properties of the AFL domain in sialic acid synthase, we solved the first three-dimensional structure of the C-terminal AFL domain of human sialic acid synthase (N-acetylneuraminate 9-phosphate synthase). Moreover, we have classified the type III antifreeze protein family into several subfamilies, and have compared each of them through tertiary structure-based sequence analyses, using the evolutionary trace (ET) method (Lichtarge et al. 1996).
Results
NMR resonance assignments and structure determination
Samples of the 13C/15N-labeled AFL domain of the human SAS, composed of 79 residues, were prepared for structure determination by a cell-free protein expression system. The protein sample has tag sequences (13 residues in total) at the N terminus (GSSGSSG) and the C terminus (SGPSSG), which are both derived from the expression vector. The amino acid numbering in the article is consistent with that of the recombinant AFL domain.
NMR resonances were assigned using conventional heteronuclear methods with the 13C/15N-labeled protein. The well-dispersed cross peaks in the 1H-15N HSQC spectrum showed that the domain has a well-folded structure (Fig. 1). The backbone resonance assignments are complete, with the exception of the amide groups of His67, Gly68, Ser73, and the 11 residues in the tag sequence regions at both termini. For each of the Xxx-Pro amide bonds, the trans conformation was confirmed independently by the presence of intense Xxx (Hα)-Pro (Hδ) sequential NOESY cross-peaks (Wüthrich 1986) and by the 13Cβ and 13Cγ chemical shift differences of the prolines (Schubert et al. 2002).
Figure 1.

[1H, 15N]-HSQC spectrum of the human AFL domain, recorded at 600 MHz 1H resonance frequency. The protein concentration was 1.1 mM, the pH was 7.0, and the temperature was 25°C.
The statistics of the structures are summarized in Table 1. Tertiary structures were calculated by the use of a total of 1652 NOE-derived distance restraints, including 336 intraresidual, 403 sequential, 247 medium-range (1 < |i − j| < 5), and 666 long-range (|i − j| > 4) ones. The final structures were refined by applying the calculation program CYANA (Herrmann et al. 2002; Güntert 2003) and were energy-minimized by the program OPALp (Koradi et al. 2000). No violation of the NOE distance constraints larger than 0.13 Å was observed in the final 20 conformers. Figure 2A shows a stereo view of the ensemble of the 20 lowest-energy calculated structures of the AFL domain. The root-mean-square deviations (RMSDs) from the mean structure were 0.33 Å for the backbone (N, Cα, C′) atoms and 0.74 Å for all non–hydrogen atoms in the well-ordered region (residues 7–66). The quality of the structures is also reflected by the fact that 85.1% of the (φ,ψ) backbone torsion angle pairs are in the most favored regions and 14.4% within the additionally allowed regions of the Ramachandran plot, according to the program PROCHECK-NMR (Laskowski et al. 1996).
Table 1.
Summary of conformational constraints and statistics of the final 20 best structures of the AFL domain in the human sialic acid synthase

aAverage values for the 20 energy-minimized conformers with the lowest CYANA target function values are given.
bBefore energy minimization.
cFrom PROCHECK-NMR (Laskowski et al. 1996).
Figure 2.

(A) Stereo view illustrating a trace of the backbone atoms for the ensemble of the 20 lowest energy structures of the human AFL domain (residues 5–67). (B) Ribbon diagrams of the AFL domains in the human sialic acid synthase (residues 5–67), the N. meningitidis sialic acid synthase (residues 289–349; PDB code 1XUZ), and the eel pout antifreeze protein (whole sequence; PDB code 1MSI) in the same view. The α- and 310-helices and β-strands are colored orange, pink, and green, respectively.
The molecular architecture of the AFL domain consists of a beta-clip fold with three short helices (helix1, Met23–Met25; helix2, Ile41–Leu44; and helix3, Glu61–Leu63), a two-stranded antiparallel β-sheet (β1, Ser8–Ala11; β2, Leu26–Lys29), and well-defined loops. Helices 1 and 3 are 310 helices, stabilized by O–HN hydrogen bonds between Met23 and Leu26, and Glu61 and Val64, respectively. Helix 2 is a single-turn α-helix involving a hydrogen bond between Ile41CO and Val45NH. In the antiparallel β-sheet, Ser8 and Val10 are involved in backbone hydrogen bonds with Lys29 and Thr27, respectively. These secondary structures are very similar to those of an Atlantic Ocean eel pout AFP (Jia et al. 1996; PDB code 1MSI), which shares high-sequence homology with the human AFL domain. On the other hand, the second helix of this domain was different from that of the bacterial AFL domain in the N. meningitidis SAS (Gunawan et al. 2005; PDB code 1XUZ). These are the only other SAS family members for which three-dimensional structures have been solved thus far.
Evolutionary trace analysis of the AFP family
In light of the new information provided by the structure of the AFL domain in the human SAS, we conducted a phylogenetic and sequence-structure analysis for this type of antifreeze protein family (34 sequences), using the evolutionary trace (ET) method (Lichtarge et al. 1996; http://www-cryst.bioc.cam.ac.uk/~jiye/evoltrace/evoltrace.html). The sequence identities between the AFL domain in the human SAS (the query sequence) and the other family proteins ranged from 33% to 90%, indicating that the family members share similar tertiary structures. The phylogenetic tree (Fig. 3) could be divided into two major branches: eukaryotic and prokaryotic proteins. The former branch could be subdivided into two branches so that the proteins were clustered into three subfamilies. Subfamily 1 consists of the fish type III AFP, while subfamilies 2 and 3 consist of the AFL domains in the vertebrate and the bacterial SAS proteins, respectively. The eel pout AFP (PDB code 1MSI) and the AFL domain in the N. meningitidis SAS (PDB code 1XUZ) were selected as the representative sequences for subfamilies 1 and 3, respectively. In addition, the three-dimensional structure of the human AFL domain (PDB code 1WVO), which was solved in this study, was also used as the representative for subfamily 2. An alignment of these three protein sequences is provided in Figure 4.
Figure 3.

Sequence identity dendrogram of 34 members of the type III antifreeze protein family, as produced by the ET server (Lichtarge et al. 1996). The sequence branches belonging to the three subfamilies are highlighted with bold lines and identified with brackets.
Figure 4.

Multiple sequence alignment of representative sequences from three subfamilies of the type III antifreeze protein family, as defined in Figure 3. Invariant residues are marked with asterisks, and nearly invariant residues are marked with colons and periods. The conserved and the class-specific residues from the first ET analysis are highlighted in orange and in green, respectively. The class-specific residues from the second ET analysis are colored in violet. The residues composing the ice-binding surface of the AFP are highlighted in red, and the Arg314 residue, which interacts with substrates, is boxed.
Discussion
The human AFL domain shares significant structural similarity with the two representatives of the other type III AFP subfamilies, the fish AFP (PDB code 1MSI) and the bacterial AFL domain (PDB code 1XUZ), with backbone RMSD values of 0.74 Å and 0.90 Å, respectively, for residues 7–66 of human AFL. To examine the structural context of the conserved and class-specific regions of the AFP family, we performed two distinct ET analyses between the eukaryotic and the bacterial proteins (subfamilies 1–2 vs. 3), and between the fish AFP and the vertebrate AFL domain (subfamilies 1 vs. 2).
The first ET analysis between the eukaryotic and bacterial proteins revealed only two conserved residues (Ser8 and Gly35 in the human AFL) in the family (colored orange in Figs. 4, 5C). Meanwhile, seven class-specific residues for the bacterial protein group were identified at Gly302, Lys313, Arg314, Pro315, Gly316, Gly330, and Lys331 (referring to N. meningitidis SAS), which are highlighted in green in Figures 4 and 5C. Interestingly, four of the seven residues are clustered around the Arg314 residue, which makes a key hydrogen bond to the substrates. Therefore, in the bacterial AFL domain family, the evolutionary tracing identified a single, functionally important surface that matches the ligand binding site. Although seven class-specific residues in the eukaryotic protein subfamily (subfamilies 1 and 2) are widely dispersed throughout the whole surface, most of the class-specific residues of the vertebrate AFL domain (subfamily 2) (Val28, Lys29, Val30, Glu32, Glu39, Ile41, Glu54, and Asp56; colored violet in Figs. 4 and 5C) are clustered on the d face, which is the substrate-binding surface for the bacterial SAS. In the vertebrate AFL domains, the Arg residue is missing from the ligand-binding site, but some class-specific residues (Lys12 and Lys29) with positively charged side chains lie near the subsite (Fig. 5B). Hence, we propose that both the vertebrate and bacterial SAS enzymes activate similar PEP condensation reactions, with ManNAc-6P and ManNAc, respectively. Considering the structural information from N. meningitidis SAS (Gunawan et al. 2005), the AFL domains seem to be used to cap the reaction chamber and to have been borrowed to perform this reaction. However, it became clear that the AFL domain has no substrate binding activity from the 15N HSQC perturbation experiments of the AFL domain with substrates (ManNAc-6P, PEP, and MnCl2) (data not shown). This means the AFL domain does not have the SAS function by itself. Further structural and mutational studies will clarify the details of the vertebrate SAS reaction mechanism.
Figure 5.
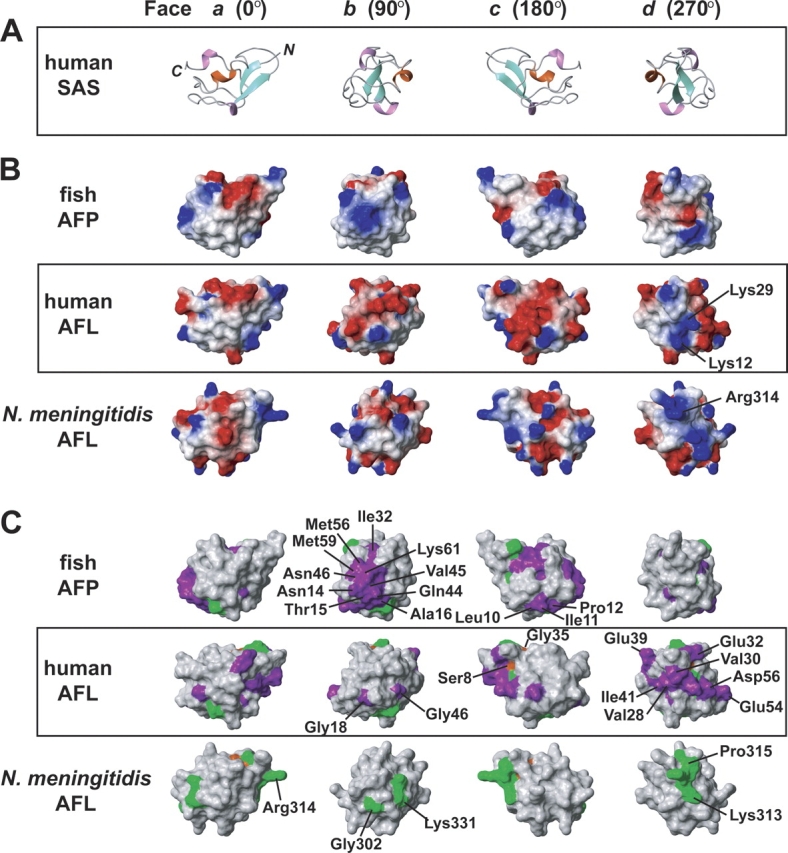
Structures of the representatives from the three subgroups of the type III antifreeze protein family. (A) Ribbon diagrams of the human AFL domain. Each row is depicted with sequential 90° rotations about the Y-axis. (B) The electrostatic surface potential of each protein: positive charges are blue, and negative charges are red. (C) Results of two ET analyses performed with the type III antifreeze protein family, as defined in Figure 3. Conserved and class-specific residues identified by the ET analysis are highlighted in the same color code as in Figure 4.
Next, the ET analysis between the fish AFP and the vertebrate AFL domain (subfamilies 1 vs. 2) was carried out. The results revealed seven conserved residues–Val9, Val10, Ala11, Leu21, Thr22, Met25, and Pro33 (referring to human AFL)—which are the class-specific residues identified by the previous ET analysis (colored green in Figs. 4 and 5C). Although Pro33 is a conserved residue in both subfamilies, they have opposite configurations of the amide bonds of the proline residues; that is, the peptide bond between Glu32 and Pro33 of the human AFL domain forms the trans-configuration, while the same peptide bond adopts the cis-configuration in all fish type III AFP structures. In all cases, this proline residue is located on a surface loop, but there is no obvious reason for this special configuration.
In the 19 class-specific residues for the fish AFPs (subfamily 1), 10 residues (Asn14, Thr15, Ala16, Ile32, Gln44, Val45, Asn46, Met56, Met59, and Lys61) were gathered on the b face. The bottom surface of the b face is located on the ice-binding surface of this family, indicating that the AFP has the critically preserved region for the antifreeze activity. On the other hand, there are no clusters of class-specific residues for the vertebrate AFL domain on the ice-binding regions, in spite of the high sequence identity (36.4%; four of 11 residues) with the residues on the ice-binding surface of the fish AFP (Fig. 4). In addition, the hydrophobicity of the AFL domain (subfamily 2) is significantly lower than those of the AFPs (Fig. 5B). These observations suggest that the vertebrate AFL domains have lost their antifreeze function.
Concluding remarks
The present NMR study revealed the structure of the AFL domain in the human SAS. To the best of our knowledge, this is the first three-dimensional structure of a eukaryotic AFL domain. The structure comprises a beta-clip fold, and is similar to the structures of other type III antifreeze proteins.
Our ET analyses highlighted three crucial findings: (1) The prokaryotic SAS (subfamily 3) shares the Arg314 residue, which plays an important role in binding substrates; (2) the class-specific residues of the human AFL domain are localized on one side (the d face in Fig. 5C), which might interact with substrates; and (3) the residues for the ice-binding by the fish AFP were conserved specifically in the subfamily. Interestingly, the ET analysis also demonstrated that the NeuB domains in all of the SAS proteins share the conserved residues around the substrate-binding site (data not shown).
The question might arise as to which structure represents the prototype: the sialic acid synthase or the fish type III AFP. According to the dendrogram in Figure 3, the vertebrate AFL domain is apparently more similar to the fish AFP than the bacterial AFL domain. This suggests that the SAS may be the prototype, which evolved to the type III AFP from the vertebrate SAS for polar fishes to survive in a severely cold environment. Unfortunately, we could not clarify whether there are any SASs in the polar fish, because the genome database of the polar fishes has not been published yet. Further genome research will be needed to define the evolutional relationship in the type III AFP family. This work provides not only a basis for future functional studies of the type III antifreeze protein family members but also a critical clue for solving the mechanism of the eukaryotic SAS.
Materials and methods
Protein expression and purification
The DNA encoding the AFL domain of the human sialic acid synthase (Ser294–Ser359) was subcloned by PCR from the human full-length cDNA clone, with the GenBank database code AF257466.1 and the NCBI protein database code gi:8453155 (Lawrence et al. 2000). This DNA fragment was cloned into the expression vector pCR2.1 (Invitrogen) as a fusion with an N-terminal histidine-affinity tag and a TEV protease cleavage site. The 13C/15N-labeled fusion protein was synthesized by a cell-free protein expression system, as described (Kigawa et al. 1999, 2004). The synthetic mixture was first adsorbed to a HisTrap column (Amersham Biosciences), which was washed with buffer A (20 mM Tris-HCl buffer at pH 8.0, containing 1 M sodium chloride and 12 mM imidazole) and was eluted with buffer B (20 mM Tris-HCl buffer at pH 8.0, containing 500 mM sodium chloride and 500 mM imidazole). After desalting on a HiPrep 26/10 column with buffer C (20 mM Tris-HCl buffer at pH 8.0, containing 1 M sodium chloride and 15 mM imidazole), the eluted protein was incubated at 30°C for 1 h with the TEV protease to cleave the His-tag. The sample was passed through a HisTrap column and then was desalted by a HiPrep desalting 26/10 column with buffer D (20 mM Tris-HCl buffer at pH 8.5, containing 1 mM EDTA). The sample was applied to a HiTrap SP column, and the flow-through fraction was fractionated on a HiTrap Q column by a concentration gradient of buffer D and buffer E (20 mM Tris-HCl buffer at pH 8.5, containing 1 M sodium chloride and 1 mM EDTA). Fractions containing the AFL domain were collected.
For NMR measurements, the purified protein was concentrated to 1.1 mM in 1H2O/2H2O (9:1) 20 mM Tris-d11-HCl buffer (pH 7.0), containing 100 mM NaCl, 1 mM 1,4-DL-dithiothreitol-d10 (d-DTT), and 0.02% NaN3. It was stable for at least 6 mo when stored at 4°C.
NMR spectroscopy, structure determination, and analysis
All NMR measurements were performed at 25°C on Varian Inova600 and Inova800 spectrometers. Backbone and Cβ resonances were assigned, using two-dimensional (2D) 1H-15N HSQC, constant-time-1H-13C HSQC, and 3D HNCA, HN(CO)CA, HNCO, (HCA)CO(CA)NH, CBCA(CO)NH, and HNCACB spectra. Side-chain and Hα proton assignments were obtained from HBHA(CO)NH, C(CO)NH, HC(CO)NH, HCCH-COSY, and HCCH-TOCSY spectra. 15N- and 13C-edited NOESY spectra with 75-msec mixing times were used to determine the distance restraints. The spectra were processed with the program NMRpipe (Delaglio et al. 1995). The program KUJIRA (N. Kobayashi, pers. comm.), created on the basis of NMRView (Johnson and Blevins 1994), was employed for optimal visualization and spectral analysis.
Automated NOE cross-peak assignments (Herrmann et al. 2002) and structure calculations with torsion angle dynamics (Güntert 2003) were performed using the software package CYANA1.0.7 (Güntert 2003). Peak lists of the two NOESY spectra were generated as input with the program NMRView (Johnson and Blevins 1994). No dihedral angle or hydrogen bond constraints were used.
A total of 100 conformers were calculated independently. The 20 conformers from CYANA cycle 7, with the lowest final CYANA target function values, were energy-minimized in a water shell with the program OPALp (Koradi et al. 2000), using the AMBER force field (Cornell et al. 1995). The structures were validated using PROCHECK-NMR (Laskowski et al. 1996). The program MOLMOL (Koradi et al. 1996) was used to analyze the resulting 20 conformers and to prepare drawings of the structures, unless noted otherwise in the legends.
The atomic coordinates of the 20 energy-minimized CYANA conformers of the AFL domain in the human SAS have been deposited at the RCSB Protein Data Bank (http://www.rcsb.org), under the PDB accession code 1WVO.
Evolutionary trace
Thirty-four sequences, identified nonredundantly by a BLAST query of the NCBI database code, were aligned with CLUSTALX (Thompson et al. 1997; http://workbench.sdsc.edu/) and were used as input for the TraceSuite server (Lichtarge et al. 1996; http://www-cryst.bioc.cam.ac.uk/~jiye/evoltrace/evoltrace.html). In the phylogenetic tree, these proteins were clustered into three subfamilies: fish type III AFP (subfamily 1), and the AFL domains in the vertebrate and bacterial SASs (subfamilies 2 and 3, respectively). The Atlantic Ocean eel pout AFP (gi:2781005, PDB code 1MSI) and the human and N. meningitidis sialic acid synthases (gi:8453155 and PDB code 1WVO, and gi:56554736 and PDB code 1XUZ, respectively) were chosen as the representatives for subfamilies 1, 2, and 3, respectively. Two distinct ET analyses between subfamilies 1–2 versus 3, and between subfamilies 1 versus 2 were performed (Fig. 5).
Electronic supplemental material
The supplemental material contains one figure showing the sequence alignment of the type III antifreeze protein family members.
Acknowledgments
We thank Yuichiro Higuchi and Yuki Kamewari for technical support. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), and the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
Footnotes
Supplemental material see www.proteinscience.org
Reprint requests to: Hiroshi Hirota, RIKEN Genomic Sciences Center, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan; e-mail: hirota@gsc.riken.jp; fax: (+81)-45-503-9210.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051700406.
References
- Angata T. and Varki A. 2002. Chemical diversity in the sialic acids and related α-keto acids: An evolutionary perspective Chem. Rev. 102: 439–470. [DOI] [PubMed] [Google Scholar]
- Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules J. Am. Chem. Soc. 117: 5179–5197. [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Gunawan J., Simard D., Gilbert M., Lovering A.L., Wakarchuk W.W., Tanner M.E., Strynadka N.C.J. 2005. Structural and mechanistic analysis of sialic acid synthase NeuB from Neisseria meningitidis in complex with Mn2+, phosphoenolpyruvate, and N-acetylmannosaminitol J. Biol. Chem. 280: 3555–3563. [DOI] [PubMed] [Google Scholar]
- Güntert P. 2003. Automated NMR protein structure calculation Prog. NMR Spectrosc. 43: 105–125. [Google Scholar]
- Herrmann T., Güntert P., Wüthrich K. 2002. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA J. Mol. Biol. 319: 209–227. [DOI] [PubMed] [Google Scholar]
- Jia Z., DeLuca C.I., Chao H., Davies P.L. 1996. Structural basis for the binding of a globular antifreeze protein to ice Nature 384: 285–288. [DOI] [PubMed] [Google Scholar]
- Johnson B.A. and Blevins R.A. 1994. NMRView: A computer program for the visualization and analysis of NMR data J. Biomol. NMR 4: 603–614. [DOI] [PubMed] [Google Scholar]
- Kigawa T., Yabuki T., Yoshida Y., Tsutsui M., Ito Y., Shibata T., Yokoyama S. 1999. Cell-free production and stable-isotope labeling of milligram quantities of proteins FEBS Lett. 442: 15–19. [DOI] [PubMed] [Google Scholar]
- Kigawa T., Yabuki T., Matsuda N., Matsuda T., Nakajima R., Tanaka A., Yokoyama S. 2004. Preparation of Escherichia coli cell extract for highly productive cell-free protein expression J. Struct. Funct. Genomics 5: 63–68. [DOI] [PubMed] [Google Scholar]
- Ko T.P., Robinson H., Gao Y.G., Cheng C.H.C., DeVries A.L., Wang A.H.-J. 2003. The refined crystal structure of an eel pout type III antifreeze protein RD1 at 0.62-Å resolution reveals structural microheterogeneity of protein and solvation Biophys. J. 84: 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R., Billeter M., Wüthrich K. 1996. MOLMOL: A program for display and analysis of macromolecular structures J. Mol. Graph. 14: 51–55. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter M., Güntert P. 2000. Point-centered domain decomposition for parallel molecular dynamics simulation Comput. Phys. Commun. 124: 139–147. [Google Scholar]
- Laskowski R.A., Rullmann J.A., MacArthur M.W., Kaptein R., Thornton J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR J. Biomol. NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- Lawrence S.M., Huddleston K.A., Pitts L.R., Nguyen N., Lee Y.C., Vann W.F., Coleman T.A., Betenbaugh M.J. 2000. Cloning and expression of the human N-acetylneuraminic acid phosphate synthase gene with 2-keto-3-deoxy-D-glycero-D-galacto-nononic acid biosynthetic ability J. Biol. Chem. 275: 17869–17877. [DOI] [PubMed] [Google Scholar]
- Lichtarge O., Bourne H.R., Cohen F.E. 1996. An evolutionary trace method defines binding surfaces common to protein families J. Mol. Biol. 257: 342–358. [DOI] [PubMed] [Google Scholar]
- Schauer R. 2004. Sialic acids: Fascinating sugars in higher animals and man Zoology 107: 49–64. [DOI] [PubMed] [Google Scholar]
- Schauer R., Kelm S., Reuter G., Roggentin P., Shaw L. 1995. Biochemistry and role of sialic acids In Biology of the sialic acids (ed. Rosenberg A.) . pp. 7–67. Plenum, New York.
- Schubert M., Labudde D., Oschkinat H., Schmieder P. 2002. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics J. Biomol. NMR 24: 149–154. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools Nucleic Acids Res. 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K. In NMR of proteins and nucleic acids . 1986. Wiley, New York.