

Abstract
Aldehyde dehydrogenases are general detoxifying enzymes, but there are also isoenzymes that are involved in specific metabolic pathways in different organisms. Two of these enzymes are Escherichia coli lactaldehyde (ALD) and phenylacetaldehyde dehydrogenases (PAD), which participate in the metabolism of fucose and phenylalanine, respectively. These isozymes share some properties with the better characterized mammalian enzymes but have kinetic properties that are unique. It was possible to thread the sequences into the known ones for the mammalian isozymes to better understand some structural differences. Both isozymes were homotetramers, but PAD used both NAD+ and NADP+ but with a clear preference for NAD, while ALD used only NAD+. The rate-limiting step for PAD was hydride transfer as indicated by the primary isotopic effect and the absence of a pre-steady-state burst, something not previously found for tetrameric enzymes from other organisms where the rate-limiting step is related to both deacylation and coenzyme dissociation. In contrast, ALD had a pre-steady-state burst indicating that the rate-limiting step was located after the NADH formation, but the rate-limiting step was a combination of deacylation and coenzyme dissociation. Both enzymes possessed esterase activity that was stimulated by NADH; NAD+ stimulated the esterase activity of PAD but not of ALD. Finding enzymes that structurally are similar to the well-characterized mammalian enzymes but have a different rate-limiting step might serve as models to allow us to determine what regulates the rate-limiting step.
Keywords: aldehyde dehydrogenase, ALDH, phenylacetaldehyde dehydrogenase, lactaldehyde dehydrogenase, pre-steady-state burst, esterase activity
During the past decade many proteins have been identified as belonging to a large family of aldehyde dehydrogenases (ALDH) (Yoshida et al. 1998; Perozich et al. 1999a,b; Vasiliou and Nebert 2005). The sequences of many and structures of a few have been determined (Liu et al. 1997; Steinmetz et al. 1997; Johansson et al. 1998; Moore et al. 1998). Based on the sequences, the enzymes were placed into different families or classes (Vasiliou et al. 1995; Yoshida et al. 1998). Though many of the enzymes have been characterized with respect to substrate specificity, mechanistic work has been done with just a few. Perhaps the best-studied ones are the mammalian enzymes from liver cytosol (class 1) (Hart and Dickinson 1982; Vallari and Pietruszko 1984; Dickinson and Haywood 1986), liver mitochondria (class 2) (Feldman and Weiner 1972a, b; Sidhu et al. 1975; Zheng et al. 1993; Ni et al. 1997; Sheikh et al. 1997), and dimeric liver and stomach cytosol (class 3) (Hsu et al. 1992; Mann and Weiner 1999; Perozich et al. 2000; Hempel et al. 2001;). Some generalizations about the enzymes have been presented based on properties of the enzymes found in these three classes. For example, the class 1 and class 2 members contain 500 amino acids, while those of class 3 have 450 residues. The class 3 enzymes are homodimers, while the class 1 and class 2 family members are homotetramers. When sequences were compared, it was found that the class 3 enzymes had some 17 residues extending beyond where the other two terminated and were missing the first 56 residues. Our laboratory reported that the reason why the class 3 enzyme was a dimer was not related to the extension at the tail-end as we once thought (Hurley et al. 1999), but was related to differences in the hydrophobicity of the exposed surfaces of the subunit when compared with the subunit from the tetrameric enzyme (Rodríguez-Zavala and Weiner 2002). In spite of the tail not being responsible for dimer or tetramer formation, few, if any, exceptions to this generalization have been documented.
The subunit structures of members of the three major ALDH classes are very similar and the sequence of the class 1 and class 3 enzymes are 70% and 40% identical to that of class 2 ALDH (Perozich et al. 1999a). The rate-limiting step, however, for each isozyme is different. Based on the mechanistic scheme shown in Figure 1, it is coenzyme dissociation (k9) for class 1 (MacGibbon et al. 1977), deacylation (k7) for class 2 (Wang and Weiner 1995), and hydride transfer (k5) for class 3 (Mann and Weiner 1999). Addition of Mg2+ ions affected each isozyme differently. The ions enhance the rate of deacylation so class 2 enzymes are activated by the ions (Takahashi and Weiner 1980). Their presence decreases the rate of NADH dissociation so the class 1 enzymes are inhibited (Vallari and Pietruszko 1984; Ho et al. 2005). No magnesium ion effect was found with the class 3 enzyme, so it was concluded that the rate of hydride transfer is not affected.
Figure 1.

Scheme of the general reaction mechanism of ALDH. The rate-limiting step for the three main mammalian isozymes is indicated. For ALD1, it is coenzyme dissociation, or k9; for ALDH2, deacylation, or k7; and for ALDH3, hydride transfer, or k5.
While using Escherichia coli for mammalian protein expressions, a new NADP+-dependent ALDH was found (Ho and Weiner 2005). Its properties were that of a typical class 2 enzyme. The E. coli literature described two other ALDHs that were involved in specific metabolic pathways rather than being a general detoxifying enzyme like so many others (Caballero et al. 1983; Hanlon et al. 1997). These two enzymes were of interest as one was reported to be a dimer, yet its sequence was more characteristic of a tetrameric enzyme. Since little was known about these two isozymes, we undertook a detailed characterization to determine if some of the generalization made using mammalian enzyme could be extended into the prokaryotic families. In this paper we report on properties of an enzyme involved in the catabolism of phenylalanine (Hanlon et al. 1997) and one involved in fucose metabolism (Caballero et al. 1983).
Results
Amino acid sequences
The amino acid sequences for the two E. coli ALDHs have been published (Hidalgo et al. 1991; Ferrandez et al. 1997). Phenylacetaldehyde dehydrogenase (PAD) was 39% identical to the mammalian tetrameric class 1 and 2 isozymes and 35% identical to dimeric class 3 ALDH, while lactaldehyde dehydrogenase (ALD) was 34% identical to the three mammalian enzymes. However, PAD possessed a 6–amino acid portion at position 147 that was not present in the other sequences. To see where in the structure these might be located, we threaded the sequence data for the PAD bacterial enzymes into the known structure for the class 2 mammalian ALDH and that of ALD into the E. coli medium-chain aldehyde dehydrogenase (1WNB), reported to be a member of the betaine aldehyde dehydrogenase family according to its sequence identity (Gruez et al. 2004). All the published structures of ALDH show that they are virtually identical, so it can be expected that the models represent what would be found had the structure been determined. Shown in Figure 2 is one subunit from human aldehyde dehydrogenase class 2 (ALDH2) and human aldehyde dehydrogenase class 3 (ALDH3) and the deduced three-dimensional models for the bacterial enzymes. The six extra amino acids found in PAD are located on what can be expected to constitute the oligomerization domain forming the cross-interaction between subunits from different dimer pairs in the tetramer.
Figure 2.
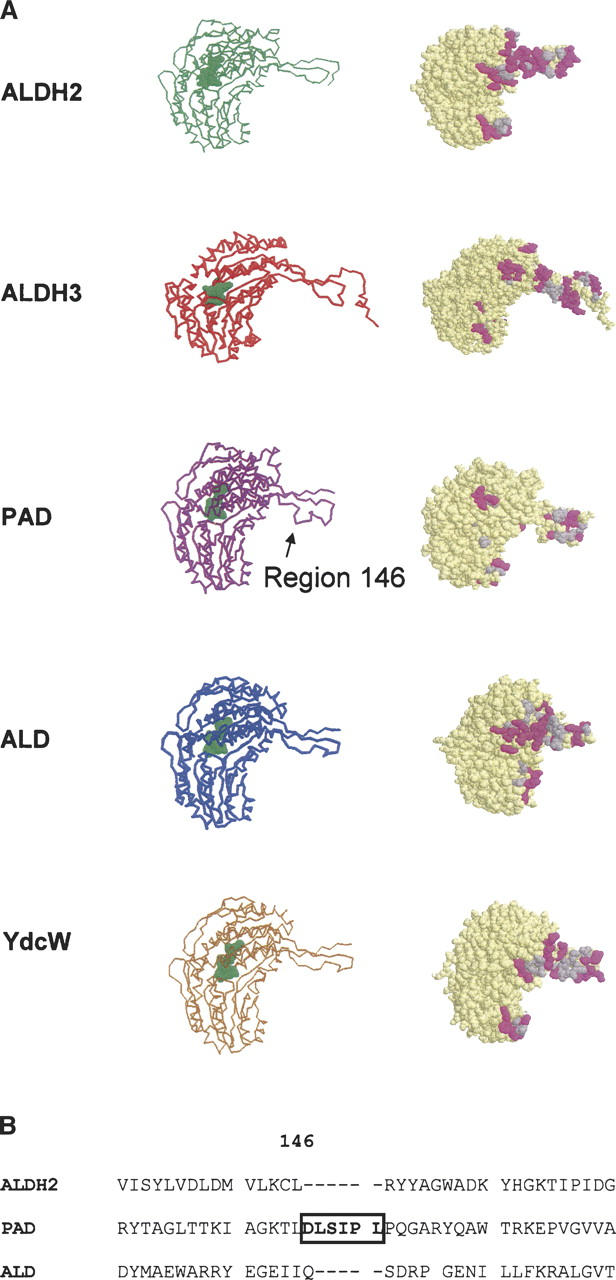
(A) Models of the tertiary structure of PAD and ALD. (Left) Backbone representation of the subunits obtained by modeling PAD and ALD as indicated in Materials and Methods, and subunit “A” from the tetramers of ALDH2 (used as the template to generate the model for PAD) and E. coli medium-chain ALDH (YDCW) (used as the template to generate the model for ALD), and the dimer of ALDH3. (Right) Spacefill representation of the same subunits, showing the dimer–dimer interface area if these subunits were tetramers. Hydrophobic and hydrophilic residues are shown in gray and in purple, respectively. (B) Alignment of the sequence of the region of residue 146 of PAD with the corresponding region of the sequence of human ALDH2 and ALD. PAD possesses a region of six extra amino acids shown by a rectangle in the sequence alignment and indicated by an arrow in the model; that region is not present in the other enzymes aligned.
Since there is a correlation between C-terminal domain and the quaternary structure, these residues were compared. Presented in Figure 3 are the residues of the known mammalian class 1, 2, and 3 isoforms as well as sequences from yeast and E. coli. These E. coli enzymes are typical of what one would expect for a tetrameric form of the enzyme.
Figure 3.

C-terminal region of ALDHs from mammalian, yeast, and E. coli. Residues 487 and 475, which are important for the stabilization of the coenzyme binding domain, are shown in rectangles. The residue numbers are based on human class 2 numbering.
The one known variant of the human class 2 enzyme is the inactive Asian variant, where the residue at position 487 is a lysine rather than a glutamate (Hempel et al. 1984; Yoshida et al. 1984). It was noted that all active tetrameric forms of the enzyme possessed either a glutamate or a glutamine at position 487 and had an arginine at position 475. Structural studies revealed that these two residues were in contact and that disrupting the salt or hydrogen bone caused the enzyme to have an altered NAD+-binding domain (Larson et al. 2005). Class 3 ALDH had histidine–serine rather than glutamatearginine interaction. ALD was also found to have a noninteracting pair of amino acids, glutamine, and glycine. This point will be discussed below.
Molecular weight determination
Addition of a 6× His-tag to the N terminus permitted a one-step purification of the recombinantly expressed E. coli isoenzymes with a high yield of pure, active enzymes. Gel filtration analysis of these enzymes demonstrated that PAD was a tetramer with a mass of 210.7 ± 10 kDa as expected for an enzyme terminating near residue 500. ALD eluted in a fraction corresponding to a lower mass of 178.3 ± 20 kDa. Finding a mass of ∼200 kDa for PAD was not unexpected based on the sequence prediction that it should be a tetramer, but it differed from the literature report that the enzyme was a dimer. That experiment was performed with crude samples (Halton et al. 1997). To verify that the enzymes might be tetramers, they were subjected to ultracentrifugation analysis.
The calculated masses for the enzymes were essentially identical: 219 kDa for PAD and 211 kDa for ALD, consistent with both enzymes being tetrameric. The expected masses based upon the amino acid sequence were 218 and 213 kDa for PAD and ALD, respectively. Thus, consistent with the prediction based upon the C-terminal domain, each formed a stable tetramer.
Substrate specificity
Even though it has been shown that the two bacterial enzymes were involved with the oxidation of specific aldehydes, a substrate specificity screening was performed to identify a more convenient aldehyde that could be used in routine assays. The physiological substrate for PAD was reported to be phenylacetaldehyde (Ferrandez et al. 1997). It proved to be the best substrate of those tested, as the kcat/Km value was double that found with medium-chain-length aliphatic aldehydes. Benzaldehyde proved to be a poor substrate but it had a very low Km value. These results are tabulated in Table 1. Though the kcat value for propionaldehyde was similar to those of the longer-chain aldehydes, its Km was 1 order of magnitude higher. Acetaldehyde proved to be a very poor substrate, consistent with the need for a hydrophobic substrate.
Table 1.
Specificity of PAD and ALD for different substrates

The natural substrate for ALD might be lactaldehyde that would be formed during the metabolism of fucose (Baldoma and Aguilar 1988). A variety of aldehydes were assayed, and ALD showed a higher preference for glycolaldehyde over other substrates consistent with its purported role in fucose metabolism (Table 1). The values for Km between glycoaldehdye and acetaldehyde differed by a factor of 100, but the Km for the hydroxyaldehyde was essentially identical to the value found when propionaldehyde was the substrate. It was not convenient to assay with the physiological substrate, but the need for the hydroxyl group is apparent as reflected in the kcat term.
Coenzyme specificity
PAD can use either NAD+ or NADP+ as the coenzyme but there was a distinct preference for NAD+ as shown by the data presented in Table 2. In contrast, no detectable activity for ALD could be found in the presence of NADP+. Dimeric class 3 ALDHs can use both coenzymes, but it shows a preference for NADP+ (Yin et al. 1989; Perozich et al. 2000).
Table 2.
Activity of PAD and ALD with NAD+ or NADP+, using propionaldehyde as the substrate

Cooperativity
Analysis of many sequences of ALDHs showed that if the residue at position 487 (using class 1 or 2 numbering) was either a glutamate or a glutamine, then the residue at position 475 was an arginine (Fig. 3). Mutation of R475 in ALDH2 caused the coenzyme domain to become disorganized, resulting in NAD+ now binding in a cooperative manner; i.e., sigmodial kinetics were observed (Wei et al. 2000). ALD possessed a glycine at position 475 (Fig. 3), but a plot of activity measured against increasing concentrations of NAD+ did not fit a sigmoidal curve (data not shown).
Rate-limiting step
A variety of assays were used to determine the possible rate-limiting step for the various isozymes of human ALDH (Ni et al. 1997). Some of these were employed to investigate the rate-limiting step for PAD and ALD. Pre-steady-state burst analysis was performed with both enzymes to determine if a burst of NADH would be found, showing that the rate-limiting step occurred after the hydride transfer step. ALD exhibited a pre-steady-state burst with a magnitude of 2 nmols of NADH/nmol of enzyme, indicating that the rate-limiting step was located after the NADH formation. A burst magnitude of 2 for a tetrameric enzyme is indicative of half-of-the-site reactivity; this was found to occur for most other tetrameric ALDHs (Weiner et al. 1976). In contrast to what was found with ALD, no burst was detected when PAD was investigated, suggesting that the rate-limiting step occurred prior to NADH formation.
Our studies with the human enzymes revealed that magnesium ions increased the rate of hydrolysis of the acyl intermediate (k7 in Fig. 1; Wang and Weiner 1995), a step that occurred after NADH formation. Assays run in the presence of Mg2+ ions showed that the ALD enzyme's activity was inhibited by the addition of Mg2+ ions just as it did for the human cytosolic class 1 isozyme (Takahashi and Weiner 1980; Ho et al. 2005). This finding is consistent with NADH dissociation being partly rate-limiting.
Assays with deuterium-containing substrates and/or substrates with electron-withdrawing groups were performed in order to provide an alternative way to assess the rate-limiting step of the E. coli enzymes. PAD exhibited a twofold decrease in activity when (2H)-acetaldehyde was the substrate. Only a 10% isotope effect was found for ALD when (2H)-benzaldehyde was the substrate. These results, shown in Table 3, lead us to conclude that hydride transfer (k5) is rate-limiting for PAD, but a different step, perhaps related to coenzyme dissociation or deacylation, could be rate-limiting for ALD.
Table 3.
Determination of the rate-limiting step of the E. coli PAD and ALD enzymes

If hydride transfer were rate-limiting, a substrate with an electron-withdrawing group such as chloroacetaldehyde should be a poorer substrate than an aldehyde such as acetaldehyde. The electron-withdrawing effect of the chlorine atom would make it more difficult to remove a hydride during the oxidation of the aldehyde. Indeed, chloro-acetaldehyde was oxidized slower than acetaldehyde under Vmax conditions with PAD. In contrast, the electron-withdrawing p-nitrobenzaldehyde was oxidized more rapidly than was benzaldehyde or p-methoxybenzaldehyde by ALD. The fact that the nitro derivative was oxidized more rapidly is consistent with a nucleophilic attack contributing to the rate-limiting step. Thus, for ALD both coenzyme dissociation and deacylation could contribute to the rate-limiting step.
To complete the kinetic analysis of the two enzymes, the inhibition by NADH was determined and reported as Kiq data in Table 4. The other kinetic constants for the two substrate reactions were calculated by two substrate kinetic analyses.
Table 4.
Comparison of the kinetic properties of ALD and PAD, with those of the E. coli AldB and human ALDHs, when propionaldehyde was the substrate

Esterase reaction
It is not readily convenient to measure the attack of the active site nucleophilic residue (C302) on the aldehyde to form the hemiacetal covalent intermediate between substrate and enzyme. The hydrolysis of p-nitrophenyl acetate has often been used to mimic this step (Takahashi and Weiner 1981). It is known that ALDHs can hydrolyze the ester in the absence of coenzyme so that the ester can bind to the apo-enzyme but it is not known with certainty if aldehydes can bind in the absence of NAD+.
Both PAD and ALD were found to possess esterase activity. NAD+ and NADH each enhanced the esterase hydrolysis rate when PAD was tested. In contrast, the addition of NAD+ had no effect on the reaction when ALD was investigated. NADH, though, activated the esterase reaction of ALD. These results are tabulated in Table 5. The addition of Mg2+ induced a small inhibition effect on the rate of ester hydrolysis in the absence of coenzyme for ALD. A more appreciable effect on activity was found in the presence of NADH (Table 6). Mg2+ ions did not affect the dehydrogenase or esterase activity for PAD.
Table 5.
Effect of the coenzyme on the esterase activity of the E. coli PAD, ALD, and the human ALDH2

Table 6.
Inhibition of E. coli ALD by Mg2+ ion

Discussion
Aldehyde dehydrogenases are found in virtually all species. The physiological role of the enzyme varies between species and even organs within a eukaryote. Some isozymes appear to function as detoxifying enzymes converting various aldehydes into their corresponding carboxylic acids. Virtually every species tested possesses a form of the enzyme that can oxidize acetaldehyde to acetate. In addition to the general detoxifying enzymes, there appear to be forms of the enzyme that play a defined role. For example, the betaine-specific enzyme is involved in osmolarity regulation (Weretilnyk and Hanson 1990), while a retinaldehyde and nonphosphorylating glyceraldehyde dehydrogenase are necessary for the biotransformation of those substrates (Bhat et al. 1995; Brunner et al. 2001). Another example of an aldehyde dehydrogenase participating in a metabolic pathway is the ALDH from Euglena gracilis, which oxidizes acetaldehyde produced from pyruvate to generate acetate that is then used to form acetyl CoA that enters the Krebs cycle (Rodríguez-Zavala et al. 2006).
From both genomic analysis and observed activities, a number of E. coli ALDHs have been identified (Caballero et al. 1983; Hanlon et al. 1997). We recently reported that AldB was an NADP+-dependent general detoxifying enzyme (Ho and Weiner 2005). Here we characterized two previously identified isozymes that have properties that make them potentially interesting models for studying mechanistic structure/function relations. What makes these isoforms particularly interesting is that they have properties that do not conform to all the generalizations made about aldehyde dehydrogenases.
The activities for these two enzymes have been reported by others, and some partial characterizations of the enzymes were presented (Caballero et al. 1983; Hanlon et al. 1997). Both ALD and PAD are now shown to be tetrameric enzymes. This was an expected result since both enzymes had sequences that could align with the initiation and termination of the tetrameric class 1 and class 2 mammalian enzymes and not with dimeric class 3 enzyme. What was totally unexpected was to find that the rate-limiting step did not correspond to what would be expected for a tetrameric enzyme as will subsequently be discussed.
From a comparison of the structures of the tetrameric ALDH2 and dimeric ALDH3, it was concluded that the C-terminal tail might be involved in determining the oligomerization state of the enzyme (Hurley et al. 1999). While trying to change the quaternary structure of enzyme, it became apparent that exposed surface of a subunit played an important role in determining the oligomerization state (Rodríguez-Zavala and Weiner 2001, 2002). Analysis of the surface of the subunit of the E. coli enzymes reveal that by forming a tetramer large patches of hydrophobic surface become buried (Fig. 2). This appears to be a major driving force in causing the enzyme to become a tetramer (Fig. 2) and not the C-terminal tail that is often referred to as the oligomerization domain.
An important salt or hydrogen bond exists between residue 487 in one subunit of the mammalian tetrameric enzymes and residue 475 in the subunit that forms a dimer pair. In the human enzyme the residue at 487 is a glutamic acid and the one at 475 is an arginine (Steinmetz et al. 1997). Mutating the glutamate to a glutamine did not affect the activity of the enzyme, but if a lysine was introduced to mimic a natural variant of the enzyme found in people from Southeast Asia, then the enzyme had little activity (Farrés et al. 1994). The structure of the E487K enzyme revealed that the coenzyme-binding domain is destroyed, showing why NAD+ binds with such difficulty to the Asian variant (Larson et al. 2005). A similar situation occurs when arginine 475 is mutated (Wei et al. 2000). The structure of this mutant has not yet been published but NAD+ binding is severely affected in the R475Q mutations. Presumably, this is a result of a similar disruption of the NAD+-binding domain as found in the Asian variant of the enzyme.
Finding that PAD possessed both a 487E and a 475R was expected since those are found in most tetrameric ALDHs, though some have Q487. Finding that ALD had both a Q and a G was unexpected. This situation is more like that found in the class 3 dimeric enzymes that do not possess the stabilizing E–R bond between the monomers (Liu et al. 1997). Disruption of the salt bond in the tetrameric mammalian enzymes caused the Km and Kd for NAD+ to increase dramatically and to exhibit cooperativity in binding (Wei et al. 2000). This was not the case with ALD. The Km and Kd (measured as Kia in the two substrate kinetics analyses shown in Table 5) were essentially of the same magnitude as those for PAD, AldB, and the class 2 human mitochondrial isozyme. Further, kcat/Ka, related to k1, the binding of NAD+ to the enzyme, was of the same magnitude when compared among the various ALDHs. Apparently, the bacteria enzyme has evolved to not require this salt bond just as did the class 3 form of the enzyme. In the latter the residues corresponding to E487 and R475 are H423 and A411 (Liu et al. 1997). These are located in the monomer–monomer interface, just as are E487 and R475, but they are >6 Å apart. In the class 1 and class 2 enzymes E487 and R475 are 3.15 and 2.77 Å apart, respectively (Steinmetz et al. 1997; Moore et al. 1998).
Most NAD(P)+-dependent dehydrogenases show a strong preference for one coenzyme over the other. PAD, like class 3 ALDH, can use both NAD+ and NADP+ as the coenzyme, but there is a strong preference for NAD+, with both Km and kcat being more favorable when NAD+ is compared with NADP+. Based upon structure it appears that residues near position 197 might influence preference for one coenzyme over the other. In Figure 4 are shown residues near this position in a series of ALDHs. All the ALDHs that either exclusively use NAD+ or can use both coenzymes have a glutamate, while ALDB, the NADP+-dependent enzyme, has an arginine. It is possible that the phenylalanine ring at position 198 in ALD is contributing some steric effect that prevents the enzyme from using both coenzymes as does PAD. However, the coenzyme preference cannot be explained by a single residue difference as demonstrated in other reports. Changing the E197 equivalent residue in rat ALDH3 (E140) to a neutral or inserting a shorter acidic group shifted the preference for coenzyme favoring NAD+ or increasing the preference for NADP+, respectively, though in these mutants there was always a preference for NADP+ (Perozich et al. 2000). Whereas, when residue R197 in aldB was mutated to E, a residue conserved in most of the NAD-dependent ALDHs in an intent to modify the coenzyme preference, the resulting enzyme possessed only 10% of the original activity, and any detectable activity was found with NAD+ as the coenzyme (Ho and Weiner 2005).
Figure 4.

Alignment of the 197 region of the human and E. coli ALDHs. It has been proposed that this region is important for the coenzyme binding. Residue 197 and its two adjacent residues are shown by a rectangle. Numbers are according to E. coli aldB numbering.
The rate-limiting step for a number of human ALDHs has been determined. For the class 1 enzymes it is the dissociation of NADH (k9 in Fig. 1; MacGibbon et al. 1977), while for the class 2 enzyme it is deacylation (k7) (Wang and Weiner 1995). With class 3 it is hydride transfer governed by k5 (Mann and Weiner 1999). Each E. coli ALDH also possessed a different rate-limiting step. For AldB we reported it was k7 (Ho and Weiner 2005), while for PAD it was found to be k5. The data for ALD make it appear that k9 is involved with the rate-limiting step. This conclusion is based upon the finding that a pre-steady-state burst exists, showing that the rate-limiting step occurred after NADH was formed, but Mg2+ ions inhibited the reaction. For the mammalian enzymes it was shown that the ions inhibited the rate of dissociation of NADH (Takahashi et al. 1980; Ho et al. 2005). This step cannot be totally rate-limiting for the ALD isozyme since we found that the reaction rate also was a function of the electron-withdrawing ability of the substrate. The latter was found with the class 2 enzyme where Mg2+ ions activate the reaction by increasing the rate of hydrolysis of the thio-ester formed during the reaction.
It appears that for ALD there is no distinct rate-limiting step as found with the other forms of the enzyme. The pre-steady-state burst magnitude of 2 mol NADH/mol enzyme might not be indicative of half-of-the-site reactivity. The equation for pre-steady-state burst for a simple reaction such as that catalyzed by chymotrypsin is
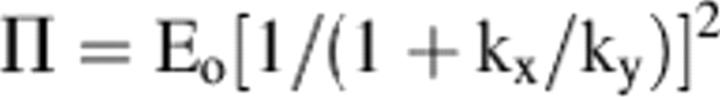 |
where Π is the burst magnitude (Fersht 1999) and kx and ky are the rate constants for the formation and disappearance of the chromophore (NADH in the case of a dehydrogenase). This equation should represent what is occurring in the ternary complex of ALDH where the rate constants represent the formation of NADH (k5) and either deacyl (k7) or NADH dissociation (k9) (see Fig. 1). With ALD, where there was a burst magnitude of 2, and Eo, the number of subunits, is 4, it can be calculated that a ratio of rate constants of just 0.4 will make it appear that the enzyme was functioning with half-of-the-site reactivity. Thus, it appears that the burst magnitude may not represent half-of-the site reactivity but was a consequence of two steps governed by both these rate constants and not just one as was found for the mammalian class 1 or 2 isozymes (MacGibbon et al. 1977; Wang and Weiner 1995).
It is usually difficult to study k3. Often we have used the esterase reaction since the initial step requires the active site nucleophile to attack nitrophenyl acetate forming the acylated enzyme. Coenzyme can affect this rate since the presence of NAD(H) can increase the nucleophilicity of the active site cysteine. Here we show that both ALD and PAD can hydrolyze the ester, a reaction not catalyzed by stomach class 3 ALDH but by the human liver isozyme (Mann and Weiner 1999). NADH dramatically enhanced the esterase rate for both enzymes. NAD+, though, only enhanced the velocity of PAD and had no effect on the velocity of ALD. This is in contrast with the class 1 and class 2 enzymes that are activated by both NAD+ and NADH (Feldman and Weiner 1972b; Sidhu and Blair 1975; Takahashi and Weiner 1981; Ho et al. 2005).
Recent studies with the human class 2 enzyme showed that the nicotinamide ring can sample two different populations, and that one favors acylation while the other favors deacylation (Hammen et al. 2002). That is, the nicotinamide ring binds in two different conformations with the enzyme with one favoring acylation and the other deacylation. NADH, though, favors only the deacylation conformation. This apparently is not occurring with the two E. coli enzymes. Ester hydrolysis is more enhanced by NADH as shown in Table 5. The structural basis for the large NADH enhancement or the lack of an effect of NAD+ on the ALD-catalyzed reaction is not understandable by comparison of the sequences of the E. coli enzymes. It will need structural comparison to determine if indeed the movement of the nicotinamide ring is responsible for these effects.
The properties of the two new E. coli enzymes were not predictable based on what was known with other members of the ALDH family. The only exception to this was the prediction that they should be tetramers based on their lengths and hydrophobic surface areas. The rate-limiting steps and ability to bind NAD+ tightly, in spite of not having a 487E–475R salt bond, were not expected for tetrameric enzymes. Neither were the coenzyme effects on the esterase reaction. These two isozymes perform an essential role in the bacteria with each being involved in the metabolism of a potential carbon source. Of perhaps greater interest is not their metabolic role, but having found enzyme forms with unique properties might aid us to design experiments to allow the understanding of what regulates the rate-limiting step in the reaction catalyzed by the various ALDHs.
Materials and methods
Expression and purification of the recombinant enzymes
The plasmids containing the DNA encoding the PAD and ALD proteins were kindly provided by Dr. J.L. García (Centro de Investigaciones Biológicas, Madrid, Spain) and Dr. J. Aguilar (University of Barcelona, Spain). These sequences were subcloned into a vector to add a His-Tag to the N terminus of the protein to facilitate the purification (Mann and Weiner 1999). E. coli BL21 strain was transformed with the vectors containing the His-Tag constructs. The proteins were overexpressed as reported elsewhere (Zheng et al. 1993). The cultures were started by the addition of 10 mL of an overnight grown culture to a 2 L XYT medium. After 3 h of incubation (or Abs600 nm = 0.6) the protein expression was induced by the addition of 0.4 mM IPTG; the incubation was continued overnight. The cells were harvested and washed twice with 100 mL saline solution. Cells were disrupted by the use of a French press cell at 4°C. Extracts of cells containing the recombinant protein were applied to a 20-mL Chelating-Sepharose column packed with NiCl2 and equilibrated with a buffer containing 50 mM H2NaPO4 (pH 7.5), 500 mM NaCl, and 20 mM 2-mercaptoethanol at 4°C. The column was washed with 50 mM imidazole in the same buffer, and the protein was eluted applying a 100-mL total volume of a 50–500 mM linear imidazole gradient. Fractions with activity were pooled and dialyzed overnight against 2 L of a buffer containing 100 mM H2NaPO4 (pH 7.5), 100 mM NaCl, and 0.025% 2-mercaptoethanol. The pure enzyme was then concentrated and stored at −20°C in the presence of 50% glycerol until use. The protein concentration was determined with the Bradford protein assay kit (Bio-Rad Laboratories), using bovine serum albumin as a standard. The enzyme purified this way had a purity of >95% as judged from SDS-PAGE gels (Laemmli 1970) and was stable for >6 mo.
Molecular weight analysis
Size exclusion chromatography was performed using a Bio-Sil Sec-250 HPLC gel filtration column (Bio-Rad Laboratories). A 200-μL sample (0.5–1 mg/mL) was loaded onto the column, and the elution of the protein was performed using a buffer containing 100 mM H2NaPO4 (pH 7.4), 100 mM NaCl, 20 mM 2-mercaptoethanol, and 10 mM NaN3, at room temperature. The peaks were detected measuring the absorbance at 280 nm, and the ALDH activity was determined as indicated below. Equilibrium centrifugations were performed on a Beckman XL-1 ultracentrifuge in a buffer containing 100 mM H2NaPO4 (pH 7.4), 100 mM NaCl. Samples were spun at 6500 rpm for 20 h at 4°C. Weight-average molecular weights were calculated using the software provided by National Analytical Biotechnology Center.
Activity assay
For the determination of the activity, 20 μg of protein were added to a buffer containing 100 mM H2NaPO4 (pH 7.5), 100 mM NaCl, 20 mM 2-mercaptoethanol, and 2 mM NAD+. The reaction was initiated by the addition of aldehyde. The ALDH activity was measured with the use of an Aminco filter spectrofluorometer, following the increase in fluorescence due to the formation of NADH, using 340 nm for the excitation and recording the emission at 460 nm. The Km and Vmax values for phenylacetaldehyde, glycolaldehyde, and other aldehydes were determined in the presence of 2 mM NAD+. The dissociation constants of NADH (Kiq) were determined as inhibition constants using NADH as a competitive inhibitor against NAD+, with propionaldehyde as the substrate.
Rate-limiting step
To determine the rate-limiting step of PAD, the purified enzyme was assayed with acetaldehyde and its analogs (2H)-acetaldehyde and chloro-acetaldehyde under apparent Vmax conditions (Table 1), while for ALD the activity was assayed with benzaldehyde and its analogs (2H)benzaldehyde, p-nitro-benzaldehyde, and p-methoxy-benzaldehyde.
Determination of the presteady-state burst
The pre-steady-state burst magnitude of NADH formation was determined by the use of a Fluorolog-3 spectrofluorometer (ISA JOBIN YVON-SPEX Instruments S.A., Inc.) as reported previously (Farrés et al. 1994). Ten micromolars of the protein were incubated in a buffer composed of 100 mM H2NaPO4 (pH 7.4), 100 mM NaCl, and 2 mM NAD+. The reaction was started by the addition of propionaldehyde for both enzymes. The magnitude of the burst of NADH formation was calculated extrapolating the linear portion of the steady-state rate of the reaction to the point of the addition of the substrate. This value was correlated with a calibration curve generated with NADH.
Spectrophotometric assay for the esterase activity
The esterase activity of the enzymes was determined by assaying the rate of p-nitrophenol formation at 400 nm, incubating the protein in 100 mM H2NaPO4 (pH 7.4), with 0.1 mM p-nitrophenylacetate as the substrate. The coenzyme effect on esterase activity was determined by adding increasing concentrations of NAD+ or NADH to the esterase assay.
Surface area and modeling of the tertiary structures
Models of the ALD and PAD subunits were obtained using the SWISS-MODEL software (available at http://swissmodel.expasy.org/) (Peitsch 1995; Guex and Peitsch 1997; Schwede et al. 2003). Determination of the amount of hydrophobic and hydrophilic residues in what would be the dimer–dimer contact area of the tetramers was performed by the use of the Protein Explorer program (Erick Martz).
Acknowledgments
This work was supported by the NIH grant no. AA05812. This is paper no. 2006-17908 from Purdue University Agriculture Experiment Station. We wish to acknowledge Craig Mann for initiating the project, and thank him for providing us with expression vectors. We also wish to thank John Burgner (Purdue University) for performing and analyzing the ultracentrifuge runs. The writing of the manuscript was initiated while H.W. was a Fellow at the Institute of the Advanced Study at La Trobe University, Melbourne, Australia.
Footnotes
Reprint requests to: Henry Weiner, Department of Biochemistry, Purdue University, 175 South University Street, West Lafayette, IN 47907-2063, USA; e-mail: hweiner@purdue.edu; fax: (765) 494-7897.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.052039606.
Abbreviations: PAD, phenylacetaldehyde dehydrogenase; ALD, lactaldehyde dehydrogenase; ALDH, aldehyde dehydrogenase; ALDH1, human aldehyde dehydrogenase class 1; ALDH2, human aldehyde dehydrogenase class 2; ALDH3, human aldehyde dehydrogenase class 3; yALDH1, yeast aldehyde dehydrogenase class 1; yALDH5, yeast aldehyde dehydrogenase class 5; yALDH2, yeast aldehyde dehydrogenase class 2.
References
- Baldoma L. and Aguilar J. 1988. Metabolism of L-fucose and L-rhamnose in Escherichia coli: Aerobic-anaerobic regulation of L-lactaldehyde dissimilation J. Bacteriol. 170 416–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat P.V., Labrecque J., Boutin J.M., Lacroix A., Yoshida A. 1995. Cloning of a cDNA encoding rat aldehyde dehydrogenase with high activity for retinal oxidation Gene 166 303–306. [DOI] [PubMed] [Google Scholar]
- Brunner N., Siebers B., Hense L.R. 2001. Role of two different glyceraldehyde-3-phosphate dehydrogenases in controlling the reversible Embden-Meyerhof-Parnas pathway in Thermoproteus tenax. Regulation on protein and transcript level Extremophiles 5 101–109. [DOI] [PubMed] [Google Scholar]
- Caballero A., Baldoma L., Ros J., Boronat A., Aguilar J. 1983. Identification of lactaldehyde dehydrogenase and glycolaldehyde dehydrogenase as functions of the same protein in Escherichia coli J. Biol. Chem. 258 7788–7792. [PubMed] [Google Scholar]
- Dickinson F.M. and Haywood G.W. 1986. The effects of Mg2+ on certain steps in the mechanisms of the dehydrogenase and esterase reactions catalysed by sheep liver aldehyde dehydrogenase. Support for the view that dehydrogenase and esterase activities occur at the same site on the enzyme Biochem. J. 233 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrés J., Wang X., Takahashi K., Cunningham S.J., Wang T.T., Weiner H. 1994. Effects of changing glutamate 487 to lysine in rat and human liver mitochondrial aldehyde dehydrogenase J. Biol. Chem. 269 13854–13860. [PubMed] [Google Scholar]
- Feldman R.I. and Weiner H. 1972a. Horse liver aldehyde dehydrogenase. I. Purification and characterization J. Biol. Chem. 247 260–266. [PubMed] [Google Scholar]
- Feldman R.I. and Weiner H. 1972b. Horse liver aldehyde dehydrogenase. II. Kinetics and mechanistic implications of the dehydrogenase and esterase activity J. Biol. Chem. 247 267–272. [PubMed] [Google Scholar]
- Ferrandez A., Prieto M.A., Garcia J.L., Diaz E. 1997. Molecular characterization of PadA, a phenylacetaldehyde dehydrogenase from Escherichia coli FEBS Lett. 406 23–27. [DOI] [PubMed] [Google Scholar]
- Fersht A. In Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding . 1999. W.H Freeman and Company, New York.
- Gruez A., Roig-Zamboni V., Grisel S., Salomoni A., Valencia C., Campanacci V., Tegoni M., Cambillau C. 2004. Crystal structure and kinetics identify Escherichia coli YdcW gene product as a medium-chain aldehyde dehydrogenase J. Mol. Biol. 343 29–41. [DOI] [PubMed] [Google Scholar]
- Guex N. and Peitsch M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modelling Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Hammen P.K., Allali-Hassani A., Hallenga K., Hurley T.D., Weiner H. 2002. Multiple conformations of NAD and NADH when bound to human cytosolic and mitochondrial aldehyde dehydrogenase Biochemistry 41 7156–7168. [DOI] [PubMed] [Google Scholar]
- Hanlon S.P., Hill T.K., Flavell M.A., Stringfellow J.M., Cooper R.A. 1997. 2-Phenylethylamina catabolism by Escherichia coli K-12: Gene organization and expression Microbiol. 143 513–518. [DOI] [PubMed] [Google Scholar]
- Hart G.J. and Dickinson F.M. 1982. Kinetic properties of highly purified preparations of sheep liver cytoplasmic aldehyde dehydrogenase Biochem. J. 203 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempel J., Kaiser R., Jornvall H. 1984. Human liver mitochondrial aldehyde dehydrogenase: A C-terminal segment positions and defines the structure corresponding to the one reported to differ in the Oriental enzyme variant FEBS Lett. 173 367–373. [DOI] [PubMed] [Google Scholar]
- Hempel J., Kuo I., Perozich J., Wang B.C., Lindahl R., Nicholas H. 2001. Aldehyde dehydrogenase. Maintaining critical active site geometry at motif 8 in the class 3 enzyme Eur. J. Biochem. 268 722–726. [DOI] [PubMed] [Google Scholar]
- Hidalgo E., Chen Y.M., Lin E.C., Aguilar J. 1991. Molecular cloning and DNA sequencing of the Escherichia coli K-12 ald gene encoding aldehyde dehydrogenase J. Bacteriol. 173 6118–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho K.K. and Weiner H. 2005. Isolation and characterization of an aldehyde dehydrogenase encoded by aldB gene of Escherichia coli J. Bacteriol. 187 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho K.K., Allali-Hassani A., Hurley T.D., Weiner H. 2005. Differential effects of Mg2+ ions on the individual kinetic steps of human cytosolic and mitochondrial aldehyde dehydrogenases Biochemistry 44 8022–8029. [DOI] [PubMed] [Google Scholar]
- Hsu L.C., Chang W.C., Shibuya A., Yoshida A. 1992. Human stomach aldehyde dehydrogenase cDNA and genomic cloning, primary structure, and expression in Escherichia coli J. Biol. Chem. 267 3030–3037. [PubMed] [Google Scholar]
- Hurley T.D., Steinmetz C.G., Weiner H. 1999. Three-dimensional structure of mitochondrial aldehyde dehydrogenase. Mechanistic implications Adv. Exp. Med. Biol. 463 15–25. [DOI] [PubMed] [Google Scholar]
- Johansson K., El-Ahmad M., Ramaswamy S., Hjelmqvist L., Jornvall H., Eklund H. 1998. Structure of betaine aldehyde dehydrogenase at 2.1 A resolution Protein Sci. 7 2106–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4 Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Larson H.N., Weiner H., Hurley T.D. 2005. Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant J. Biol. Chem. 280 30550–30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.J., Sun Y.J., Rose J., Chung Y.J., Hsiao C.D., Chang W.R., Kuo I., Perozich J., Lindahl R., Hempel J.et al. 1997. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold Nat. Struct. Biol. 4 317–326. [DOI] [PubMed] [Google Scholar]
- MacGibbon A.K., Buckey P.D., Blackwell L.F. 1977. Evidence for two-step binding of reduced nicotinamide-adenine dinucleotide to aldehyde dehydrogenase Biochem. J. 165 455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann C.J. and Weiner H. 1999. Differences in the roles of conserved glutamic acid residues in the active site of human class 3 and class 2 aldehyde dehydrogenases Protein Sci. 8 1922–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore S.A., Baker H.M., Blythe T.J., Kitson K.E., Kitson T.M., Baker E.N. 1998. Sheep liver cytosolic aldehyde dehydrogenase: The structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases Structure 6 1541–1551. [DOI] [PubMed] [Google Scholar]
- Ni L., Sheikh S., Weiner H. 1997. Involvement of glutamate 399 and lysine 192 in the mechanism of human liver mitochondrial aldehyde dehydrogenase J. Biol. Chem. 272 18823–18826. [DOI] [PubMed] [Google Scholar]
- Peitsch M.C. 1995. Protein modeling by E-mail Biotechnology (N. Y.) 13 658–660. [Google Scholar]
- Perozich J., Nicholas H., Lindahl R., Hempel J. 1999a. The big book of aldehyde dehydrogenase sequences. An overview of the extended family Adv. Exp. Med. Biol. 463 1–7. [DOI] [PubMed] [Google Scholar]
- Perozich J., Nicholas H., Wang B.C., Lindahl R., Hempel J. 1999b. Relationships within the aldehyde dehydrogenase extended family Protein Sci. 8 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozich J., Kuo I., Wang B.C., Boesch J.S., Lindahl R., Hempel J. 2000. Shifting the NAD/NADP preference in class 3 aldehyde dehydrogenase Eur. J. Biochem. 267 6197–6203. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Zavala J.S. and Weiner H. 2001. Role of the C-terminal tail on the quaternary structure of aldehyde dehydrogenases Chem. Biol. Interact. 130 151–160. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Zavala J.S. and Weiner H. 2002. Structural aspects of aldehyde dehydrogenase that influence dimer-tetramer formation Biochemistry 41 8229–8237. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Zavala J.S., Ortíz-Cruz M.A., Moreno-Sánchez R. 2006. Characterization of an aldehyde dehydrogenase from Euglena gracilis J. Eukaryot. Microbiol. 53 36–42. [DOI] [PubMed] [Google Scholar]
- Schwede T., Kopp J., Guex N., Peitsch M.C. 2003. SWISS-MODEL: An automated protein homology-modeling server Nucleic Acids Res. 31 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh S., Ni L., Hurley T.D., Weiner H. 1997. The potential roles of the conserved amino acids in human liver mitochondrial aldehyde dehydrogenase J. Biol. Chem. 272 18817–18822. [DOI] [PubMed] [Google Scholar]
- Sidhu R.S. and Blair A.H. 1975. Human liver aldehyde dehydrogenase. Esterase activity J. Biol. Chem. 250 7894–7898. [PubMed] [Google Scholar]
- Steinmetz C.G., Xie P., Weiner H., Hurley T.D. 1997. Structure of mitochondrial aldehyde dehydrogenase: The genetic component of ethanol aversion Structure 5 701–711. [DOI] [PubMed] [Google Scholar]
- Takahashi K. and Weiner H. 1980. Magnesium stimulation of catalytic activity of horse liver aldehyde dehydrogenase. Changes in molecular weight and catalytic sites J. Biol. Chem. 255 8206–8209. [PubMed] [Google Scholar]
- Takahashi K. and Weiner H. 1981. Nicotinamide adenine dinucleotide activation of the esterase reaction of horse liver aldehyde dehydrogenase Biochemistry 20 2720–2726. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Weiner H., Hu J.H. 1980. Increase in the stoichiometry of the functioning active sites of horse liver aldehyde dehydrogenase in the presence of magnesium ions Arch. Biochem. Biophys. 205 571–578. [DOI] [PubMed] [Google Scholar]
- Vallari R.C. and Pietruszko R. 1984. Interaction of Mg2+ with human liver aldehyde dehydrogenase. II. Mechanism and site of interaction J. Biol. Chem. 259 4927–4933. [PubMed] [Google Scholar]
- Vasiliou V. and Nebert D.W. 2005. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family Hum. Genomics 2 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V., Weiner H., Mareslos M., Nebert D.W. 1995. Aldehyde dehydrogenase genes: Classification based on evolution, structure and regulation Eur. J. Drug Metabol. Pharmacokinet. 20 53–64. [Google Scholar]
- Wang X.P. and Weiner H. 1995. Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis Biochemistry 34 237–243. [DOI] [PubMed] [Google Scholar]
- Wei B., Ni L., Hurley T.D., Weiner H. 2000. Cooperativity in nicotinamide adenine dinucleotide binding induced by mutations of arginine 475 located at the subunit interface in the human liver mitochondrial class 2 aldehyde dehydrogenase Biochemistry 39 5295–5302. [DOI] [PubMed] [Google Scholar]
- Weiner H., Hu J.H., Sanny C.G. 1976. Rate-limiting steps for the esterase and dehydrogenase reaction catalyzed by horse liver aldehyde dehydrogenase J. Biol. Chem. 251 3853–3855. [PubMed] [Google Scholar]
- Weretilnyk E.A. and Hanson A.D. 1990. Molecular cloning of a plant betaine-aldehyde dehydrogenase, an enzyme implicated in adaptation to salinity and drought Proc. Natl. Acad. Sci. 87 2745–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S.J., Liao C.S., Wang S.L., Chen Y.J., Wu C.W. 1989. Kinetic evidence for human liver and stomach aldehyde dehydrogenase-3 representing an unique class of isozymes Biochem. Genet. 27 321–331. [DOI] [PubMed] [Google Scholar]
- Yoshida A., Huang I.Y., Ikawa M. 1984. Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals Proc. Natl. Acad. Sci. 81 258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A., Rzhetsky A., Hsu L.C., Chang C. 1998. Human aldehyde dehydrogenase gene family Eur. J. Biochem. 251 549–557. [DOI] [PubMed] [Google Scholar]
- Zheng C.F., Wang T.T.Y., Weiner H. 1993. Cloning and expression of the full-length cDNAs encoding human liver class 1 and class 2 aldehyde dehydrogenases Alcohol. Clin. Exp. Res. 17 828–831. [DOI] [PubMed] [Google Scholar]