

Abstract
In an era of rapid genome sequencing and high-throughput technology, automatic function prediction for a novel sequence is of utter importance in bioinformatics. While automatic annotation methods based on local alignment searches can be simple and straightforward, they suffer from several drawbacks, including relatively low sensitivity and assignment of incorrect annotations that are not associated with the region of similarity. ProtoNet is a hierarchical organization of the protein sequences in the UniProt database. Although the hierarchy is constructed in an unsupervised automatic manner, it has been shown to be coherent with several biological data sources. We extend the ProtoNet system in order to assign functional annotations automatically. By leveraging on the scaffold of the hierarchical classification, the method is able to overcome some frequent annotation pitfalls.
Keywords: protein family, hierarchical classification, InterPro, clustering
Accurate automatic functional annotation holds the potential for enormous benefits in speeding up the annotation process of new biological data. This is particularly true for genome annotation. Genome information is rapidly accumulating for a multitude of species. At present, there are at least 500 genomes that are either completed or at final stages of draft phase. The genomes of additional 522 genomes (as of October 2005) are currently in the pipeline. This unprecedented number of genomes includes ∼200 eukaryotes that are in their final stage of assembly or in progress (http://www.ncbi.nlm.nih.gov/Genomes). These new genomes largely outnumber the 18 complete eukaryotic genomes currently available. Therefore, the need for automation in the painstaking task of functional annotation becomes critically important.
In addition to ongoing “whole genome” projects, other types of experimental data are becoming available from numerous high-throughput methodologies. In recent years, standardization in the technologies of SNP arrays, DNA micro-array, and DNA chips has increased the quality and reproducibility of the results. Overall, the volume of data that is collectively referred to as “nonsequence data” is rapidly growing. However, the quality of the data varies. While the quality of some data sources may be very high, other types may be of inherently poor quality. For example, structural genomics projects produce detailed and accurate three-dimensional information from crystallography and NMR spectroscopy. The function of many of these structures is still unknown (Skolnick et al. 2000). In contrast, data on protein–protein interactions originating from two-hybrid systems suffer from large amounts of false positives and low reproducibility. With the addition of proteomics data from LC MS/MS experiments, protein chips, and subcellular localization data, the data that emerges is protein rather than genomic centered (Bork et al. 2004).
The notion of protein function is elusive. To apply computational methods, we need to provide an unambiguous definition. We propose equating function to annotations. Annotations are simply categorical biological properties describing the protein's functionality. Annotations can describe various biological aspects of the protein such as its structure, enzymatic classification, taxonomy, cellular localization, and more. Local alignment search tools such as BLAST (Altschul et al. 1997) provide the most straightforward method for performing automatic function prediction on a new sequence (Jones and Swindells 2002), via function inference. With this method, a protein database is searched for high-scoring local alignments with the query protein. The annotations on the sequence that score the highest alignment are assigned to the query sequence, provided the alignment score passes a predetermined threshold. The underlying logic is simple: Proteins with similar sequences are conjectured to have evolved from a single ancestor gene and thus to have retained similar functionality. While this approach is simplistic, it performs fairly well in many cases. However, local alignment searches suffer from some important caveats:
Excessive transfer of annotations. In some cases, similarity is restricted to a local region in the sequence. While only annotations that are functionally linked to the region of similarity should be transferred, annotations that are not related to the local region of similarity will be transferred as well, even though they are not shared by both proteins. This difficulty arises even when using manual inference of the annotations, as it is not possible to conclusively determine what annotation is linked to the region of similarity. The reason is that the connection between specific segments of the protein to its function is often unknown. Excessive transfer of annotations occurs more frequently for annotations that describe a high-level functionality than for annotations that are motif-based and can be localized in sequence.
Annotation errors in the source database. Because many databases employ computational methods in the assignment of annotations, isolated cases of false annotation assignment occur. Studies have shown that once an erroneous annotation is introduced into a database, it tends to propagate via automatic annotation inference methods that are based on sequence similarity (Linial 2003). If the best matching sequence has been assigned a false annotation, the annotation will be transferred to the new protein sequence.
Threshold relativity. Various scoring methods exist for assessing the quality of an alignment. The score threshold used for annotation is usually arbitrary and fails to reflect the relativity that scoring methods tend to exhibit (different thresholds are suitable for different groups of proteins).
Low sensitivity/specificity. Depending on the annotation threshold that is used, simple local alignment methods are usually outperformed by advanced supervised methods in terms of sensitivity/specificity. This is due to the fact that advanced methods take into account features that are shared by the family of proteins to which the protein belongs, while a simple local alignment search does not consider these data.
Paralogs versus orthologs. Two different proteins in one species that resulted from a gene duplication event might possess significant sequence similarity but will often have different functions. In contrast, two proteins from different species that may have almost undetected similarity can still share the same function or a similar one. Sequence comparison methods frequently fail to distinguish between these two instances (Sonnhammer and Koonin 2002).
We hereby present a scheme for inference of functional annotations of protein sequences. The scheme consists of two parts: (1) ProtoNet, an automatic hierarchical organization of protein sequence databases representing functional and evolutionary relations amongst the proteins, and (2) an automatic method for predicting the function of a new protein based on its localization in the protein tree (Sasson et al. 2003; Kaplan and Linial 2005). We start by describing the ProtoNet classification hierarchy, proceed by discussing its biological validity, and conclude by explaining the annotation inference method and showing how it avoids the common annotation assignment pitfalls mentioned above.
Results
The ProtoNet method
Given a set of proteins (typically a protein from database such as UniProt) (Bairoch et al. 2005), ProtoNet aims at organizing the proteins into a hierarchy of trees, each tree representing a biologically related group of proteins and its division into functional subgroups. Much work was done in the field of protein classification and, in particular, hierarchical clustering (e.g., Systers [Krause et al. 2005], CLusTr [Kriventseva et al. 2001]).
In contrast to a nonhierarchical functional grouping, this hierarchical representation of proteins provides a much more accurate view on protein functional relations, because functionality encompasses several degrees of granularity, from very general effects at the organism level to very specific descriptions of biochemical function. To achieve this organization, we use the following three phases:
All-against-all BLAST. A matrix is constructed so that it holds the e-values resulting from NCBI-BLAST comparisons (McGinnis and Madden 2004) on all possible pairs of sequences. E-values >100 are set to be equal 100.
- Clustering. An initial hierarchy is constructed by progressively clustering the proteins according to their e-values. We use the well-known paradigm of hierarchical agglomerative clustering (Kaufman and Rousseeuw 1990) using group average linkage. We use arithmetic averaging and define the score between two clusters to be
At each step of the clustering method, the pair of clusters that has the lowest score is merged. The clustering process stops when a score of 100 is reached. The clustering process results in a set of binary tree hierarchies in which the proteins are arranged into clusters.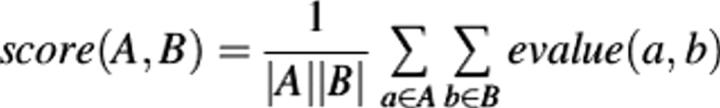
Pruning. The number of clusters generated in the hierarchical clustering is equal the number of proteins in the database minus one. Obviously, some of these clusters hold little information. Some are very large clusters that will be created inevitably as methodological artifacts, and others are intermediate partial clusters. Therefore, an automatic unsupervised method is needed to distinguish biologically valid clusters from clusters that are artifacts of the method. Following the method presented in Kaplan et al. (2004), the resulting hierarchy is automatically pruned according to an intrinsic measure, producing the final hierarchy. The pruning method has been shown to eliminate 88% of the clusters while keeping the validity of the measured its correspondence to external data sources.
In light of the explosive growth of sequence databases, scalability is an important issue. Although the presented method scales well in terms of result quality (tested on a database of 90,000 up to 200,000 proteins), the computation itself is more challenging. For large protein databases such as UniProt (containing >1,600,000 sequences), performing the hierarchical clustering requires very large memory. To avoid this problem, we divide the clustering problem into several clustering steps, each of which considers a subset of the similarity graph. Preliminary results indicate that the biological validity of the hierarchy produced by this method is not reduced significantly (Sasson 2005). ProtoNet is available at http://www.protonet.cs.huji.ac.il.
Biological validity of ProtoNet
The validity of clusters can be determined in comparison to other classifications, e.g., InterPro (Mulder et al. 2002). At present, the InterPro classifier uses a combination of 12 supervised detection methods based on state-of-the-art methods such as hidden Markov models (HMMs), position-specific scoring matrices (PSSMs), and profiles (Mulder et al. 2005). To determine if ProtoNet is able to detect the weak functional relationships that are detected by InterPro, we perform the following test: For each InterPro annotation (each InterPro entry can be thought of as an annotation), we consider the set of all proteins that were assigned that annotation (S). Next, we define the following score between a cluster C and the set S (this score is also known as the Jaccard coefficient):
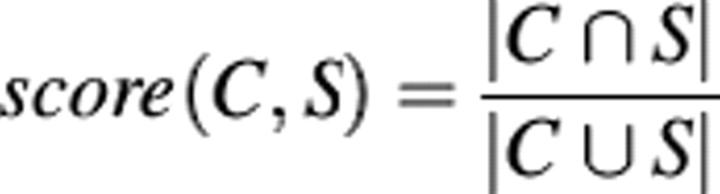 |
Note that a score of one means C = S, and a score of zero means C ∩ S = {Ø}. Finally, we find the highest scoring cluster for each InterPro annotation. Figure 1 shows an area plot describing the distribution of the scores for the highest scoring cluster of each InterPro annotation. Remarkably, we find that ProtoNet is able to produce clusters that are extremely consistent with the InterPro classification (mean score 0.85), even though ProtoNet uses only BLAST e-values and is completely unsupervised. Furthermore, ProtoNet shows high consistency with manual and semiautomatic classifications as well. For more results, see Kaplan et al. (2004) and Shachar and Linial (2004).
Figure 1.

An area plot of the scores of the highest-scoring clusters for a total of 3184 InterPro entries. The horizontal line represents the average score (0.85). Only InterPro entries that are assigned by InterPro to at least 10 proteins were considered in order to avoid counting trivial cases of success (if all InterPro entries are considered, the average score is 0.91). Calculation was performed on the clustering of the SwissProt database, as described in Kaplan et al. 2004 (also, see text).
One unique aspect of ProtoNet is that it is an unsupervised method. Supervised methods are given a training set upon which they learn a pattern and then use it to perform prediction. Therefore supervised methods are only able to detect predefined. In contrast, ProtoNet's unsupervised approach detects previously unknown families and previously unknown relationships between families. The following detailed example of GAS1 demonstrates this point.
GAS1 (growth arrest sequence 1) is a tumor suppressor that prevents DNA synthesis by blocking the entry of cells into the S phase (Mullor and Ruiz i Altaba 2002). During embryogenesis GAS1 is differentially expressed and its expression has been associated with cell death during limb development, while in the cerebellum GAS1 was shown to act as a positive growth regulator (Marques and Fan 2002). The molecular function of GAS1 in vivo remains elusive. A routine BLAST search using the human or mouse GAS1 protein sequence as the query sequence fails to detect any significant hits to any protein groups other than GAS1 proteins from related species. InterPro also fails to detect a connection to any protein families. However, the ProtoNet 4.0 classification tree suggests a relationship between GAS1 and a large family of GFRα, the GPI (glycosyl-phosphatidyl-inositol) coreceptor for glial cell line–derived neurotrophic factor (GDNF) and its related factors. Examining the ProtoNet hierarchy, we find a cluster that combines GAS1 from vertebrates, worms, and insects with GDNF receptors from avians, rodents, and primates (Fig. 2). Interestingly, proteins belonging to the GFRα family consistently emerge by a BLAST search, yet with a score that is below any statistical significance (Fig. 2; www.protonet.cs.huji.ac.il). Submission of GAS1 to the Meta-server for fold recognition (Ginalski and Rychlewski 2003) substantiated the connection between GAS1 and GFRα and identified Protein Data Bank 1q8dA (108 amino acids from rat GFRα1) as a parent model with very high confidence (e-value of 6 × 10−24). Additional evidence for the functional connectivity between GAS1 and GFRα substantiated our study (Furman et al. 2006).
Figure 2.

Graphical representation of the BLAST e-value matrix data of ProtoNet cluster A429475, consisting of 12 proteins. The color of the cells in the matrix codes the significance of the e-value, from dark gray (highly significant e-value of nearly zero) to white (nonsignificant e-value of ≥100). The cluster combines GRFα (marked Q9Z2A3 and Q9Z2A2 from mouse, GFR2_CHICK, GFR2_HUMAN, GFR2_MOUSE, GFR4_CHICK, Q920Y3 from mouse, and Q9VDM9 from Drosophila) that are coreceptors for the GDNF family of ligands and the GAS1 homologs (marked GAS1_HUMAN, GAS1_MOUSE, GAS1_CAEEL, and GB11290). Note that the honey bee GAS1 homolog (GB11290) serves as an intermediate to connect these apparently unrelated protein families.
Using ProtoNet to infer annotation
Given that the protein clusters and the hierarchies that are produced by the ProtoNet system are highly coherent with other classifications (InterPro [Mulder et al. 2002], SCOP [Hubbard et al. 1999], GOA [Camon et al. 2004], and ENZYME [Bairoch 2000]), these can be used in order to annotate a new sequence. When provided with a new sequence, it is localized to an existing cluster. Once it is localized, we can learn about its functionality from its relative position in the hierarchy. To do this, we first assign to each cluster the annotations of its member proteins, which adhere to the following two conditions: (1) the annotation is shared by at least 75% of the proteins in the cluster and (2) the annotation achieves a P-value <0.001 under the assumption that the annotations are distributed hypergeometrically. These two requirements ensure that only annotations that are statistically significant and represent a majority of the proteins of the cluster will be assigned to the cluster. Furthermore, these requirements provide a secondary measure of caution to prevent clusters that are not biologically coherent due to methodical flaws (i.e., mixed groups of functionally unrelated proteins) from being used to infer annotations. Once the clusters are assigned annotations, the new sequence is assigned the annotations of the cluster to which it belongs and the annotations of all of the cluster's parents in the hierarchy. By doing this, robustness is used in order to avoid most of the pitfalls noted previously. One pitfall that is difficult to overcome is the issue of correctly inferring the function of paralogs that evolved into having a new function. Such sequences might be misclassified in our method, but this is inevitable regardless of the method used.
The aforementioned procedure was applied for >10,000 unannotated predicted proteins from the honey bee genome. A ProtoNet-like approach including ∼200,000 sequences was applied (www.protobee.cs.huji.ac.il), and for ∼75% of the honey bee proteins, some biological annotation was successfully assigned (N. Kaplan and M. Linial, unpubl.).
Looking back at the example of local similarity, if the proteins of a cluster are varied biologically but share a local region of similarity (and therefore some functional features), only the annotations that are shared by the proteins of the cluster will be assigned to the cluster. This can greatly reduce the chance of excessive transfer of annotations and transfer of incorrect annotations (provided that the incorrect annotations are isolated incidences and do not represent the majority of cases in the database). In addition to the high sensitivity/specificity results in comparison with other method and the threshold relativity that the clustering method is able to take into account, it seems that this method succeeds in avoiding many of the common pitfalls of local alignment searches.
Discussion
The concept underlying automatic function prediction is using experimental biological knowledge on a small set of proteins to correctly predict the function of a large set of sequences (“one for all”). Several new approaches for automatic function prediction were introduced recently in order to advance beyond the shortcomings of simple local alignment searches (Godzik 2003; Edgar and Sjolander 2004; Yang 2004; Han et al. 2005). While the relative performance of these methods is difficult to benchmark, it is clear that they are all superior to the naïve approach. In this work, we present an annotation inference method based on the ProtoNet hierarchical organization. The method is unique in two important aspects: its unsupervised hierarchy construction and its use of robustness in order to overcome annotation errors. While in terms of specificity we expect ProtoNet to be slightly inferior to advanced supervised methods, it seems that in terms of sensitivity, the unsupervised approach allows detection of extremely faint functional relationships that are otherwise undetectable (Shachar and Linial 2004). The use of robustness (“all for one”) in annotation inference helps avoid annotation errors by adding a perspective of relativity to the BLAST e-values and the functional annotations, putting them in the context of the whole protein database.
An interesting advantage of ProtoNet over the naïve local similarity search approach is that any kind of annotation can be assigned to the new sequence. This means that any data that are available on the underlying database of proteins are available for use in annotation. By using UniProt as its underlying database, ProtoNet is able to assign InterPro, UniProt keywords, GO, ENZYME, and SCOP annotations. This not only offers a wider and constantly-growing range of available annotations but also overcomes inconsistencies between different sources.
It is worth mentioning that much work has been done on automatic functional annotation. An approach that is related to the one presented in this article is prediction by phylogenomic methods, using the evolutionary context of a sequence for function prediction (Engelhardt et al. 2005). The use of the evolutionary context is analogous to the use of the classification hierarchy in this work.
One problem that remains partially unaddressed by ProtoNet is the problem of multiple domains. Since a protein often consists of several domains, it can be viewed as belonging to several protein families. In ProtoNet, proteins are the basic entities. As a result of this, every protein appears once and can therefore belong to several families only if they contain each other. This issue is irresolvable in the current scheme. However, this issue is addressed in a related work called EVEREST (www.everest.cs.huji.ac.il), in which protein domains are the basic entities that are clustered.
While local similarity searches usually give a statistical evaluation of the results, it is often very difficult to deduce from this evaluation what biological similarity exists amongst the query protein and the matches found. This is especially true for borderline or even clearly insignificant statistical values. As ProtoNet uses a clustering method, it is unable to provide a good statistical evaluation of the results. However, since the statistical evaluation simply acts as a mean for evaluating validity of prediction quantitatively, ProtoNet provides several alternative measures that are related to the structure and localization of the protein in the tree. These measurements can be used to assess the validity of the classification of any query protein.
Acknowledgments
Special thanks to the ProtoNet team for support at all stages of ProtoNet development. N.K. is supported by SCCB, the Sudarsky Center for Computational Biology in the Hebrew University of Jerusalem. This study was supported by the EU Framework VI DIAMONDS consortium.
Footnotes
Reprint requests to: Michal Linial, CCB, The Sudarsky Center for Computational Biology, Department of Biological Chemistry, Life Science Institute, The Hebrew University, Jerusalem 91904, Israel; e-mail: michall@cc.huji.ac.il; fax: 972-2-6585448.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062185706.
Abbreviations: GAS1, growth arrest sequence 1; GDNF, glial-cell-line-derived neurotrophic factor; GFR, GDNF family receptor; GPI, glycosyl phosphatidylinositol; HMM, hidden Markov model; PSSM, position-specific scoring matrix.
References
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A. 2000. The ENZYME database in 2000 Nucleic Acids Res. 28 304–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A., Apweiler R., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M.et al. 2005. The Universal Protein Resource (UniProt) Nucleic Acids Res. 33 D154–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P., Jensen L.J., von Mering C., Ramani A.K., Lee I., Marcotte E.M. 2004. Protein interaction networks from yeast to human Curr. Opin. Struct. Biol. 14 292–299. [DOI] [PubMed] [Google Scholar]
- Camon E., Barrell D., Lee V., Dimmer E., Apweiler R. 2004. The Gene Ontology Annotation (GOA) Database—An integrated resource of GO annotations to the UniProt Knowledgebase In Silico Biol. 4 5–6. [PubMed] [Google Scholar]
- Edgar R.C. and Sjolander K. 2004. COACH: Profile–profile alignment of protein families using hidden Markov models Bioinformatics 20 1309–1318. [DOI] [PubMed] [Google Scholar]
- Engelhardt B.E., Jordan M.I., Muratore K.E., Brenner S.E. 2005. Protein function prediction by Bayesian phylogenomics PLoS Comput. Biol. 1 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman O., Glick E., Segovia J., Linial M. 2006. Is GAS1 a co-receptor of the GDNF family of ligands? Trends Pharmacol. 2 72–79. [DOI] [PubMed] [Google Scholar]
- Ginalski K. and Rychlewski L. 2003. Detection of reliable and unexpected protein fold predictions using 3D-Jury Nucleic Acids Res. 31 3291–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godzik A. 2003. Fold recognition methods Methods Biochem. Anal. 44 525–546. [DOI] [PubMed] [Google Scholar]
- Han S., Lee B.C., Yu S.T., Jeong C.S., Lee S., Kim D. 2005. Fold recognition by combining profile–profile alignment and support vector machine Bioinformatics 21 2667–2673. [DOI] [PubMed] [Google Scholar]
- Hubbard T.J., Ailey B., Brenner S.E., Murzin A.G., Chothia C. 1999. SCOP: A structural classification of proteins database Nucleic Acids Res. 27 254–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T. and Swindells M.B. 2002. Getting the most from PSI-BLAST Trends Biochem. Sci. 27 161–164. [DOI] [PubMed] [Google Scholar]
- Kaplan N. and Linial M. 2005. Automatic detection of false annotations via binary property clustering BMC Bioinformatics 6–46. [DOI] [PMC free article] [PubMed]
- Kaplan N., Friedlich M., Fromer M., Linial M. 2004. A functional hierarchical organization of the protein sequence space BMC Bioinformatics 5–196. [DOI] [PMC free article] [PubMed]
- Kaufman L. and Rousseeuw P. In Finding groups in data: An introduction to cluster analysis . 1990. John Wiley and Sons, New York.
- Krause A., Stoye J., Vignron M. 2005. Large scale hierarchical clustering of protein sequences BMC Bioinformatics 6–15. [DOI] [PMC free article] [PubMed]
- Kriventseva E.V., Fleischmann W., Zdobnov E.M., Apweiler R. 2001. CluSTr: A database of clusters of SWISS-PROT+TrEMBL proteins Nucleic Acids Res. 29 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linial M. 2003. How incorrect annotations evolve: The case of short ORFs Trends Biotechnol. 21 298–300. [DOI] [PubMed] [Google Scholar]
- Marques G. and Fan C.M. 2002. Growth arrest specific gene 1: A fuel for driving growth in the cerebellum Cerebellum 1 259–263. [DOI] [PubMed] [Google Scholar]
- McGinnis S. and Madden T.L. 2004. BLAST: At the core of a powerful and diverse set of sequence analysis tools Nucleic Acids Res. 32 W20–W25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Biswas M., Bradley P., Bork P., Bucher P.et al. 2002. InterPro: An integrated documentation resource for protein families, domains and functional sites Brief. Bioinform. 3 225–235. [DOI] [PubMed] [Google Scholar]
- Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L.et al. 2005. InterPro, progress and status in 2005 Nucleic Acids Res. 33 D201–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullor J.L. and Ruiz i Altaba A. 2002. Growth, hedgehog and the price of GAS Bioessays 24 22–26. [DOI] [PubMed] [Google Scholar]
- Sasson O. “The protein metric space: A study in clustering.” Ph.D. thesis 2005. School of Computer Science and Engineering, The Hebrew University of Jerusalem, Israel.
- Sasson O., Vaaknin A., Fleischer H., Portugaly E., Bilu Y., Linial N., Linial M. 2003. ProtoNet: Hierarchical classification of the protein space Nucleic Acids Res. 31 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar O. and Linial M. 2004. A robust method to detect structural and functional remote homologues Proteins 57 531–538. [DOI] [PubMed] [Google Scholar]
- Skolnick J., Fetrow J.S., Kolinski A. 2000. Structural genomics and its importance for gene function analysis Nat. Biotechnol. 18 283–287. [DOI] [PubMed] [Google Scholar]
- Sonnhammer E.L. and Koonin E.V. 2002. Orthology, paralogy and proposed classification for paralog subtypes Trends Genet. 18 619–620. [DOI] [PubMed] [Google Scholar]
- Yang Z.R. 2004. Biological applications of support vector machines Brief. Bioinform. 5 328–338. [DOI] [PubMed] [Google Scholar]