

Abstract
Although neither kinase-dead human epidermal growth factor receptor (HER)3 nor orphan HER2 can be activated by HER-related ligands on their own, the formation of HER2/HER3 heterodimers creates the most mitogenic and transforming receptor complex within the HER (erbB) family of transmembrane receptor tyrosine kinases. The incorporation of markers such as HER3 transactivation, HER2/HER3 dimer, or others that may provide information regarding the level of HER pathway engagement has been demonstrated to allow identification of patients who respond to or escape HER-targeted therapies. Pioneering studies showed that high expression of kinase-dead HER3 can predict early escape from the anti-HER2 monoclonal antibody trastuzumab. Also, the growth-inhibitory effects of HER1/2 tyrosine kinase inhibitors (TKIs) were previously found to be attenuated in the presence of heregulin, which is a high-affinity combinatorial ligand for HER3. All of these concepts are being revisited with respect to the efficacy of HER family TKI therapies; in particular, HER3 signalling buffered against incomplete inhibition of HER2 kinase activity has been suggested to be the mechanism that allows HER2 over-expressing breast cancer cells to escape HER TKIs. It remains to be elucidated whether reactivation of HER3 signalling can also account for the poor efficacy of HER TKIs in treating breast carcinomas that contain low overall levels of HER2 receptors. However, it appears that regardless of the mechanism that triggers the formation of oncogenic HER2/HER3 heterodimers (HER2 over-expression or overall low HER2 but high levels of the HER3 ligand heregulin), HER3 transphosphorylation is a common response of breast cancer cells upon treatment with current inhibitors of the HER receptor tyrosine kinase network. Because kinase-inactive HER3 is not presently an amenable target for forthcoming HER TKIs, molecular approaches that can efficiently block heregulin-triggered HER3 transactivation or nucleocytoplasmic trafficking of heregulin might offer novel strategies with which to manage HER-driven breast cancer disease.
The notion that breast cancer disease can be viewed as a biological process that is driven by overactive human epidermal growth factor receptor (HER)1/2 receptor tyrosine kinases (RTKs) has led to development of various anti-HER tyrosine kinase agents. Several of these have undergone clinical trials, including low-molecular-weight inhibitors with highly selective and reversible tyrosine kinase inhibiting properties [1-5]. Unfortunately, following much clinical and basic science research, we have learned that simple measurement of overactive HER tyrosine kinases does not predict HER TKI efficacy. Initial phase II studies [3,4] demonstrated that the HER1 tyrosine kinase inhibitor (TKI) gefitinib did not have high efficacy in heavily pretreated populations of patients with metastatic breast cancer, particularly after chemotherapy. With few exceptions, clinical studies of the HER1 TKI gefitinib in breast cancer have demonstrated poor clinical responses and a disease control rate of approximately 10%. Indeed, tumour responses induced by HER1 TKIs are infrequent and transient, with sensitive patients rapidly developing secondary resistance. Therefore, molecular criteria for predicting sensitivity to HER TKIs are needed to allow appropriate use of these agents and to facilitate planning of future research.
Failure to reverse breast cancer progression despite apparent inhibition of HER1 and HER2 kinase functions in cell-based assays as well as in patient tissues and tumours is an enigma that is not yet resolved [6]. Although it is kinase defective, the HER family member HER3 can be phosphorylated by HER1 or HER2, and HER3 can couple with the pro-survival phosphatidylinositol-3-OH kinase (PI[3]K)/Akt pathway directly whereas HER1 and HER2 cannot [5] (Figure 1). Based on these findings, Sergina and coworkers [6] recently suggested that HER3, and consequently the PI(3)K/Akt pathway, evade inhibition by current HER family TKIs via a compensatory shift in the HER3 phosphorylation/dephosphorylation equilibrium [6] (Figure 2). In their hands, Akt-mediated negative feedback signalling appeared to promote increased membrane HER3 (driving the phosphorylation reaction) and to reduce HER3 phosphatase activity (impeding the dephosphorylation reaction). They therefore concluded that the biological marker used to assess the efficacy of HER TKIs should be transphosphorylation of HER3, rather than autophosphorylation of HER1 or HER2 [6].
Figure 1.
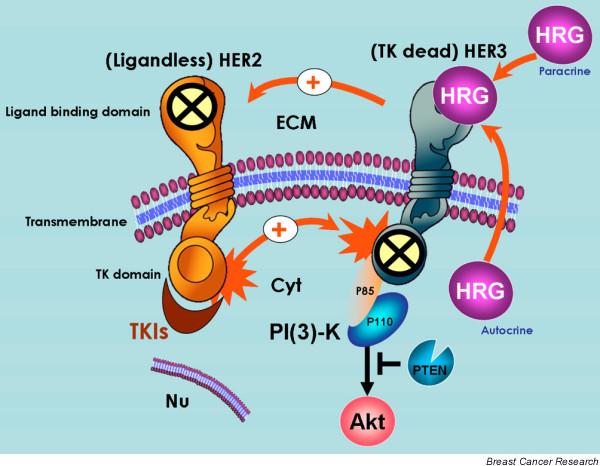
Mechanisms of HER2/HER3 activation in breast cancer cells. It is generally accepted that spontaneous homodimerization and activation of human epidermal growth factor receptor (HER)2 occurs in breast cancer cells with HER2 gene amplification. Another potential mechanism of HER2 phosphorylation is transactivation by ligand (autocrine/paracrine heregulin)-bound HER3. Although HER2 does not bind any of the HER ligands directly (including heregulin), its catalytic activity potently amplifies signalling by HER-containing heterodimers via increasing ligand binding affinity or receptor recycling and stability. On the other hand, although it is kinase defective, HER3 can be phosphorylated by HER2. Phosphorylated HER3 can couple to the phosphatidylinositol-3-OH kinase (PI [3]K)/Akt pathway directly, whereas HER2 cannot. Therefore, neither orphan (ligandless) HER2 nor tyrosine kinase (TK) dead HER3 can be activated by HER-related ligands on their own. However, the formation of ligand-independent and ligand-dependent HER2/HER3 heterodimers creates the most mitogenic and transforming receptor complex within the HER family of transmembrane receptor tyrosine kinases (RTKs). In HER2 over-expressing breast cancer cells, HER tyrosine kinase inhibitors (TKIs) inhibit basal phosphorylation of HER3 and its association with HER2 and with PI(3)K. Cyt, cytoplasm; ECM, extracellular milieu; Nu, nucleus.
Figure 2.
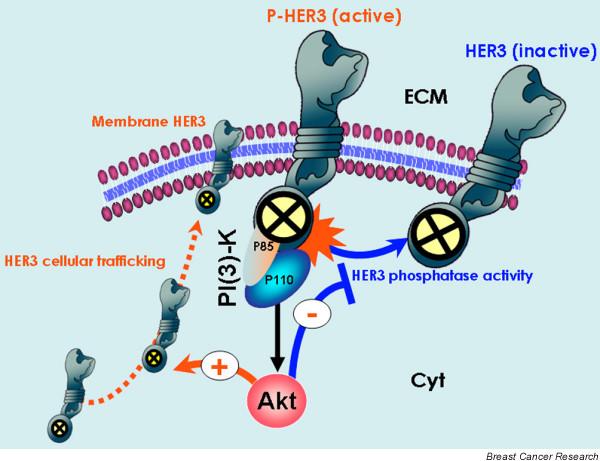
Biological markers to assess the efficacy of HER TKIs (I): Akt-regulated HER3 phosphorylation-dephosphorylation equilibrium. The study conducted by Sergina and coworkers [6] suggests that the biological marker used to assess the efficacy of human epidermal growth factor receptor (HER) tyrosine kinase inhibitors (TKIs) should be transphosphorylation of HER3 rather than autophosphorylation of HER1 or HER2 [6]. They reported that HER3 and phosphatidylinositol-3-OH kinase (PI [3]K)/Akt signalling is not effectively inhibited by current HER TKIs. Through Akt-mediated negative feedback signalling, a compensatory shift occurs in the HER3 phosphorylation-dephosphorylation equilibrium, driven by increased HER3 membrane expression (phosphorylation reaction) and by reduced HER3 phosphatase activity (dephosphorylation reaction). Despite significant inhibition of HER2 activity, the reactivation of HER3 signalling to a new steady-state HER3 phosphorylation level requires much higher concentrations of HER TKIs because the un-inhibited HER3 phosphorylation state is significantly higher in this new steady state. Therefore, although kinase-inactive HER3 is not a direct target of HER TKIs, HER3 substrate resistance appears to undermine their efficacy significantly.
Although neither kinase-dead HER3 nor orphan HER2 can be activated by HER-related ligands on their own, the formation of ligand-independent and ligand-dependent HER2/HER3 heterodimers creates the most mitogenic and transforming receptor complex within the HER family of transmembrane RTKs [1,5,7-9]. Moreover, inhibition of HER3 transphosphorylation or its preferential cross-talk with HER2 is known to be required for the antitumour effect of HER2 signalling inhibitors [10-12] (Figure 1). High expression of HER3 was previously shown to predict early escape from HER-targeted therapies using, for instance, the anti-HER2 monoclonal antibody trastuzumab [13]. However, incorporation of markers such as HER3 transactivation, HER2/HER3 dimer, or others that may provide information regarding the level of HER pathway engagement and so allow identification of patients who respond to or escape HER-targeted therapies has already been proposed [12,13]. We previously demonstrated, in breast cancer cells exhibiting enhanced HER3 signalling, that specific anti-HER2 strategies (namely trastuzumab) efficiently interfere with the lateral signalling between HER3 and HER2, shutting down the ability of HER3/HER2 heterodimers to function as an 'oncogenic unit' [11].
All of these findings are now being revisited with respect to the efficacy of HER family TKI therapy, with HER3 signalling buffered against incomplete inhibition of HER2 kinase activity being suggested to be the mechanism that allows HER2 over-expressing breast cancer cells to escape from HER TKIs (Figure 2). Using HER2 over-expressing breast cancer cells, Sergina and coworkers [6] similarly reported that TKI refractory HER3 phosphorylation is due to HER2 because it can be suppressed by anti-HER2 small interfering RNA transfection. It should be noted that Sergina and coworkers employed mono-specific HER1 reversible TKIs (for instance, gefitinib and erlotinib) and pan-HER irreversible inhibitors (for example, CI-1033 [canertinib]). However, these HER TKIs may be suboptimal for management of HER2 over-expressing breast carcinomas [14,15]. Moreover, many so-called HER specific inhibitors (including gefitinib, erlotinib and canertinib) have been found to be rather promiscuous kinase inhibitors, binding several non-HER kinases at similar affinities to their target HER1 [14,15]. Whether HER3 transphosphorylation can allow HER2 over-expressing breast cancer cells to escape bi-specific HER1/HER2 reversible TKIs such as GW572016 (lapatinib; the most specific inhibitor, binding its intended targets, namely HER1 and HER2, with high affinity) remains to be elucidated.
Another important issue that was not addressed in the study conducted by Sergina and coworkers [6] is whether reactivation of HER3 signalling can also account for the inefficacy of HER TKIs against HER1-positive breast carcinomas that contain low overall levels of HER2 receptors, which represent a subset of extremely aggressive tumours [2,3,5]. In fact, we recently reported that two apparently distinct breast cancer populations, one over-expressing HER2 with low levels of the HER3 transactivator heregulin and the other over-expressing heregulin but not HER2, indeed employ the same signalling pathway (HER3 → PI[3]K/Akt) to control breast cancer cell responses to anti-HER2 therapeutics [11,12]. Because epidermal growth factor (EGF)-related ligands activate different HER receptors, and most tumours of epithelial origin express multiple HER receptors and co-express one or more of the HER-related ligands, multiple HER receptor combinations might be active in a tumour. This characteristic should influence tumour response to a HER-targeted therapeutic; therefore, the efficacy of anticancer drugs that block a single HER receptor may be compromised by the presence of EGF-related ligands. In this regard, Motoyama and colleagues [16] conducted pioneering work in demonstrating that the growth-inhibitory effects of HER1/2 TKIs are significantly attenuated in the presence of exogenous heregulin, which is a high-affinity combinatorial ligand for the HER3 and HER4 receptors in breast cancer cells. Accordingly, the antitumour actions expected to result from pharmacological blockade of HER2/HER3 dimer can be entirely mimicked by specifically blocking the autocrine expression of heregulin. In vitro, anti-heregulin strategies (antisense cDNA heregulin transfection and structural deletions that impair the autocrine activity of heregulin) significantly block HER3/HER2 transactivation and deactivate anti-apoptotic PI(3)K/Akt signalling [11,17,18]. In vivo, heregulin-induced HER3/HER2 transactivation, in the absence of HER2 over-expression, is sufficient for generation of adenocarcinomas while favouring the metastatic spread of breast cancer cells in mice [19]. Furthermore, blockade of the heregulin/HER3 signalling pathway reduces tumour formation and size and suppresses metastasis [20].
Within the context of HER-targeting inhibitors, it has been observed that breast cancer cells with acquired resistance to the anti-HER2 monoclonal antibody trastuzumab express higher endogenous levels of EGF-related ligands as compared with parental, trastuzumab-sensitive cells [12]. Moreover, Schaefer and colleagues recently demonstrated that the growth-inhibiting effects of the pan-HER TKIs CI-1033 and PD158780 indeed correlate with expression levels of the HER ligand heregulin [21]. Hence, those investigators identified an autocrine growth stimulation loop that may represent an interesting target for new therapeutic strategies in HER TKI refractory tumours. In our laboratory, when breast cancer cells intrinsically resistant to gefitinib (HER2-negative MCF-7 cells) were engineered to exhibit constitutively an autocrine heregulin loop (MCF-7 cells were forced to over-express heregulin endogenously), they exhibited further a twofold to threefold increase in the 50% inhibitory concentration of gefitinib. This heregulin-enhanced HER TKI refractory phenotype was accompanied by persistent 'HER3 superphosphorylation', an enormous upregulation of the PI(3)K p85 regulatory subunit, and downstream hyperactivation of Akt (Menendez JA, Lupu R, unpublished data). Therefore, the impact of HER2 on both the biological behaviour of breast cancer and responses to HER-targeting therapies is better appreciated when one considers not only the presence of HER2 over-expression but also the presence of active forms of other HER receptors (especially HER3), the expression levels of HER3/HER4 ligands and the activation status of downstream transduction cascades [11,12,22].
That TKI-induced forward shift in HER3 steady-state phosphorylation is driven by HER3 re-localization to the plasma membrane and that it can be suppressed by inhibitors of vesicular trafficking might also relate to the HER3 ligand heregulin. Offterdinger and coworkers [23] previously demonstrated that heregulin not only profoundly affects HER3 function (in an autocrine manner) but also further regulates (in an intracrine manner) HER3 subcellular distribution. This affect occurs via induction of nucleolar and nuclear export of HER3, anchoring it to non-nuclear structures, and consequently leading to a slow enrichment of HER3 in the cytoplasm/membrane compartments and its deprivation in the nucle(ol)us [23] (Figure 3). Sergina and coworkers [6] observed that the HER3 pool, largely within intracellular compartments (including cell nuclei), exhibits significantly increased expression at the plasma membrane upon gefitinib treatment.
Figure 3.
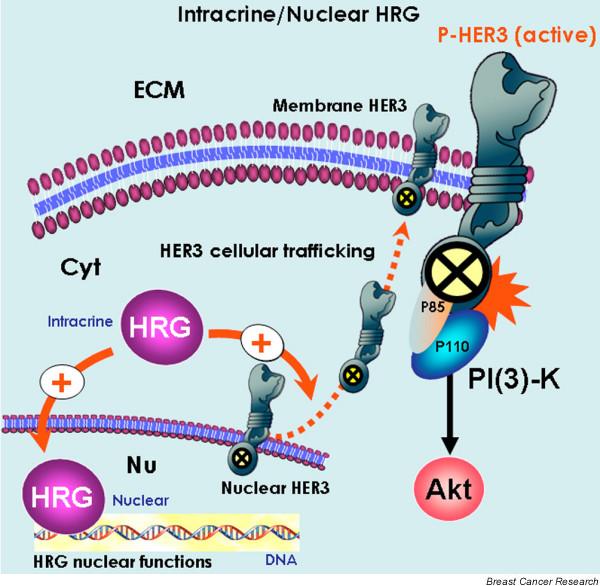
Biological markers to assess the efficacy of HER TKIs (II): intracrine/nuclear functions of the HER3 ligand heregulin. The intrinsic or acquired ability of breast cancer cells to specifically regulate the expression and/or the subcellular compartmentalization of the human epidermal growth factor receptor (HER)3 ligand heregulin might represent a previously unrecognized mechanism that regulates the efficacy if HER tyrosine kinase inhibitors (TKIs). In one hand, heregulin has been found to promote slow enrichment of HER3 in the cytoplasm/membrane compartments and its deprivation in the nucle(ol)us [23]. This intracrine mechanism of action may significantly increase HER3 membrane expression and, therefore, the HER3 phosphorylation reaction. On the other hand, a 'direct mode' that involves active transport of heregulin to the cell nucleus, and its association with and regulation of target genes or nuclear proteins may add even more complexity to the breast cancer 'HER-TKIs' scenario, because it might bypass the antiproliferative effects of HER-directed TKIs without need for the activation of specific HER receptor tyrosine kinase members and/or HER network-driven activation of downstream signalling cascades [24-28].
Similarly to HER3 silencing in HER2 over-expressing breast cancer cells, we recently reported that experimental ablation of heregulin by small interfering RNA triggers potent pro-apoptotic activity in otherwise cytostatic gefitinib in low HER3 expressing MDA-MB-231 cells. This surprising finding suggests that a causal connection may also exist between intracrine heregulin and breast cancer cell responses to HER TKIs. This hypothesis was partially validated by monitoring gene expression profiles and subcellular localization of HER-related ligands before and after gefitinib treatment. Using real-time reverse transcription polymerase chain reaction, Ferrer-Soler and coworkers [24] characterized gefitinib-resistant and gefitinib-sensitive breast cancer cell lines according to mRNA expression of EGF-related ligands before and after gefitinib exposure. When expression data were normalized relative to ribosomal RNA (18S RNA) as the endogenous control gene, HER1 over-expressing MDA-MB-468 cells (the main HER1-positive breast cancer model of gefitinib resistance) were found to exhibit significantly up-regulated (by up to eight times) levels of the HER3 ligand heregulin. Conversely, expression of heregulin was completely abolished in HER2 over-expressing SKBR3 cells, which are exquisitely sensitive to gefitinib. A second finding was that gefitinib-resistant MDA-MB-468 cells exhibited a striking nuclear accumulation of heregulin following gefitinib exposure. Conversely, gefitinib treatment significantly reduced dot-like structures containing heregulin in the nuclei of gefitinib-sensitive SKBR3 cells [24]. Third, prevention of heregulin transport to the nucleus by deleting its nuclear localization signal [25,26] not only obviated the ability of heregulin to enhance resistance to gefitinib but also further disrupted HER2/HER3 cross-talk in MCF-7 breast cancer cells (Menendez JA, Lupu R, unpublished observations).
Thus, changes in heregulin subcellular compartmentalization modify the activation status of the HER3/HER2 dimer (and, in turn, efficacy of HER TKIs), probably through alterations in the nucleocytoplasmic shuttling or the endosomal sorting of HER RTKs (Figure 3). Moreover, intracrine/nuclear heregulin might also bypass the antiproliferative effects of HER-directed TKIs without need for activation of specific HER RTK members or HER network-driven activation of downstream signalling cascades. This 'direct mode', involving active transport of the HER3 ligand heregulin to the cell nucleus and its association with and regulation of target genes or nuclear proteins, warrants further studies because it might demonstrate even greater complexity of the breast cancer 'HER TKIs' scenario [27,28].
It is unclear at this time how to select a population of breast cancer patients who are most likely to be sensitive to HER TKIs. The study conducted by Sergina and coworkers [6] suggests that the biological marker used to assess the efficacy of HER TKIs should be transphosphorylation of HER3, rather than autophosphorylation of HER1 or HER2. Certainly, regardless of the mechanism that triggers the formation of HER2-containing heterodimers (HER2 over-expression or overall low HER2 but high levels of the autocrine/paracrine HER3 ligand heregulin), HER3 transphosphorylation appears to be a common response of breast cancer cells upon pharmacological disturbance of the HER RTK network. On the other hand, virtually nothing is known about the paracrine/autocrine/intracrine actions of HER ligands that are involved in the efficacy of HER TKIs. Auspiciously, two up-to-date reports have begun to shed light on the clinical implications of the HER3 ligand heregulin as a previously unrecognized modulator of the effectiveness of anti-HER therapies. First, de Alava and coworkers [29] recently demonstrated that over-expression of transmembrane heregulin significantly correlated with a longer event-free survival and overall survival in patients with low or normal HER2 expression who were treated with the mono-specific HER2 inhibitor trastuzumab. However, this was not the case in patients with HER2 over-expression. Second, Hutcheson and colleagues [30] recently reported that heregulin efficiently overcomes the cell growth and invasion inhibitory effects of the mono-specific HER1 inhibitor gefitinib in tamoxifen-resistant breast cancer cells through promotion of HER2/HER3 heterodimerization and activation of the PI(3)K/Akt signalling pathway.
Because HER3 is kinase inactive, it is not a direct target of TKIs and cannot at present be targeted readily with drug treatment [31]. Alternatively, future therapies aimed at avoiding ligand-triggered HER3 transactivation, alongside a better understanding of the therapeutic relevance of paracrine/autocrine/intracrine expression and nucleocytoplasmic trafficking of heregulin, may offer novel approaches with which to manage HER-driven breast cancer disease.
Abbreviations
EGF = epidermal growth factor; HER = human epidermal growth factor receptor; PI(3)K = phosphatidylinositol-3-OH kinase; RTK = receptor tyrosine kinase; TKI = tyrosine kinase inhibitor.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Javier A Menendez, Email: jmenendez@ico.scs.es.
Ruth Lupu, Email: r-lupu@northwestern.edu.
Acknowledgements
The authors are supported by funding from INSTITUTO de SALUD CARLOS III (Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria -FIS-, Spain, Grants CP05-00090, PI06-0778, and RD06-0020-0028) and BCTR0600894 from the Susan G Komen Breast Cancer Foundation to Javier A Menendez, and the Extramural Funding of the National Institute of Health, RO1CA116623 and the Breast Cancer Auxillary Program of the Evanston Northwestern Healthcare to Ruth Lupu.
References
- Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Fukuoka M, Baselga J. Gefitinib-a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:956–965. doi: 10.1038/nrc1506. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV, Darzynkiewicz Z. Why Iressa failed: Toward novel use of kinase inhibitors (outkook) Cancer Biol Ther. 2003;2:137–140. doi: 10.4161/cbt.2.2.286. [DOI] [PubMed] [Google Scholar]
- Agrawal A, Gutteridge E, Gee JM, Nicholson RI, Robertson JF. Overview of tyrosine kinase inhibitors in clinical breast cancer. Endocr Relat Cancer. 2005;12:S135–S144. doi: 10.1677/erc.1.01059. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995;14:4267–4275. doi: 10.1002/j.1460-2075.1995.tb00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999;18:2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF III, Hynes NE. The erbB2/erbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3-kinase and Akt is required for antibody-mediated effects of p27, cyclin D1, and antitumor action. Cancer Res. 2002;62:4132–4141. [PubMed] [Google Scholar]
- Menendez JA, Mehmi I, Lupu R. Trastuzumab in combination with heregulin-activated Her-2 (erbB-2) triggers a receptor-enhanced chemosensitivity effect in the absence of Her-2 overexpression. J Clin Oncol. 2006;24:3735–3746. doi: 10.1200/JCO.2005.04.3489. [DOI] [PubMed] [Google Scholar]
- Arteaga CL. Can trastuzumab be effective against tumors with low HER2/Neu (ErbB2) receptors? J Clin Oncol. 2006;24:3722–3725. doi: 10.1200/JCO.2006.06.5268. [DOI] [PubMed] [Google Scholar]
- Smith BL, Chin D, Maltzman W, Crosby K, Hortobagyi GN, Bacus SS. The efficacy of herceptin therapies is influenced by the expression of other erbB receptors, their ligands and the activation of downstream signalling proteins. Br J Cancer. 2004;91:1190–1194. doi: 10.1038/sj.bjc.6602090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M. et al. A small molecule-interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Spector N Xia W El-Hariry I Yarden Y Bacus S HER2 therapy. Small molecule HER-2 tyrosine kinase inhibitors Breast Cancer Res 20079205. 10.1186/bcr165217338834 [DOI] [Google Scholar]
- Motoyama AB, Hynes NE, Lane HA. The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res. 2002;62:3151–3158. [PubMed] [Google Scholar]
- Atlas E, Bojanowski K, Mehmi I, Lupu R. A deletion mutant of Heregulin increases the sensitivity of breast cancer cells to chemotherapy without promoting tumorigenicity. Oncogene. 2003;22:3441–3451. doi: 10.1038/sj.onc.1206410. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Mehmi I, Lupu R. Heregulin-triggered Her-2/neu signaling enhances nuclear accumulation of p21WAF1/CIP1 and protects breast cancer cells from cisplatin-induced genotoxic damage. Int J Oncol. 2005;26:649–659. [PubMed] [Google Scholar]
- Atlas E, Cardillo M, Mehmi I, Zahedkargaran H, Tang C, Lupu R. Heregulin is sufficient for the promotion of tumorigenicity and metastasis of breast cancer cells in vivo. Mol Cancer Res. 2003;1:165–175. [PubMed] [Google Scholar]
- Tsai MS, Shamon-Taylor LA, Mehmi I, Tang CK, Lupu R. Blockage of heregulin expression inhibits tumorigenicity and metastasis of breast cancer. Oncogene. 2003;22:761–768. doi: 10.1038/sj.onc.1206130. [DOI] [PubMed] [Google Scholar]
- Schaefer KL, Brachwitz K, Braun Y, Diallo R, Wai DH, Zahn S, Schneider DT, Kuhnen C, Vollmann A, Brockhoff G. et al. Constitutive activation of neuregulin/ERBB3 signaling pathway in clear cell sarcoma of soft tissue. Neoplasia. 2006;8:613–622. doi: 10.1593/neo.06238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste L, Montero JC, Esparis-Ogando A, Pandiella A. Activation of ErbB2 by overexpression or by transmembrane neuregulin results in differential signalling and sensitivity to herceptin. Cancer Res. 2005;65:6801–6810. doi: 10.1158/0008-5472.CAN-04-4023. [DOI] [PubMed] [Google Scholar]
- Offterdinger M, Schofer C, Weipoltshammer K, Grunt TW. cerbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol. 2002;157:929–939. doi: 10.1083/jcb.200109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Soler L, Vazquez-Martin A, Brunet J, Menendez JA, De Llorens R, Colomer R. An update of the mechanisms of resistance to EGFR-tyrosine kinase inhibitors in breast cancer: gefitinib (Iressa™)-induced changes in the expression and nucleo-cytoplasmic trafficking of HER-ligands (review) Int J Mol Med. 2007;20:3–10. [PubMed] [Google Scholar]
- Golding M, Ruhrberg C, Sandle J, Gullick WJ. Mapping nucleolar and spliceosome localization sequences of neuregulin1-3. Exp Cell Res. 2004;299:110–118. doi: 10.1016/j.yexcr.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Breuleux M, Schoumacher F, Rehn D, Kung W, Mueller H, Eppenberger U. Heregulins implicated in cellular functions other than receptor activation. Mol Cancer Res. 2006;4:27–37. doi: 10.1158/1541-7786.MCR-05-0016. [DOI] [PubMed] [Google Scholar]
- Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M, Vadlamudi RK, Kumar R. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001;3:30–37. doi: 10.1038/35050532. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan P, Varambally S, Tomlins SA, Ray S, Huang S, Chinnaiyan AM, Harari PM. Enhancing the antitumor activity of ErbB blockade with histone deactylase (HDAC) inhibition. Int J Cancer. 2006;118:1041–1050. doi: 10.1002/ijc.21465. [DOI] [PubMed] [Google Scholar]
- de Alava E, Ocana A, Abad M, Montero JC, Esparis-Ogando A, Rodriguez CA, Otero AP, Hernandez T, Cruz JJ, Pandiella A. Neuregulin expression modulates clinical response to trastuzumab in patients with metastatic breast cancer. J Clin Oncol. 2007;25:2656–2663. doi: 10.1200/JCO.2006.08.6850. [DOI] [PubMed] [Google Scholar]
- Hutcheson IR, Knowlden JM, Hiscox SE, Barrow D, Gee JM, Robertson JF, Ellis IO, Nicholson RI. Heregulin beta 1 drives gefitinib-resistant growth and invasion in tamoxifen-resistant MCF-7 breast cancer cells. Breast Cancer Res. 2007;9:R50. doi: 10.1186/bcr1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Moasser MM. Targeting HER proteins in cancer therapy and the role of non-target HER3. Br J Cancer. 2007;97:453–457. doi: 10.1038/sj.bjc.6603910. [DOI] [PMC free article] [PubMed] [Google Scholar]