
Figure 1.
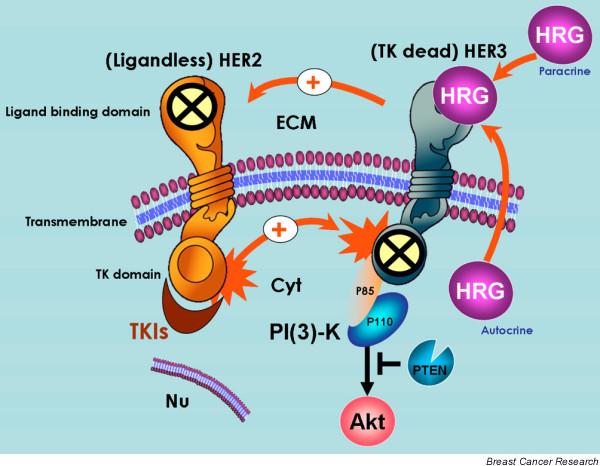
Mechanisms of HER2/HER3 activation in breast cancer cells. It is generally accepted that spontaneous homodimerization and activation of human epidermal growth factor receptor (HER)2 occurs in breast cancer cells with HER2 gene amplification. Another potential mechanism of HER2 phosphorylation is transactivation by ligand (autocrine/paracrine heregulin)-bound HER3. Although HER2 does not bind any of the HER ligands directly (including heregulin), its catalytic activity potently amplifies signalling by HER-containing heterodimers via increasing ligand binding affinity or receptor recycling and stability. On the other hand, although it is kinase defective, HER3 can be phosphorylated by HER2. Phosphorylated HER3 can couple to the phosphatidylinositol-3-OH kinase (PI [3]K)/Akt pathway directly, whereas HER2 cannot. Therefore, neither orphan (ligandless) HER2 nor tyrosine kinase (TK) dead HER3 can be activated by HER-related ligands on their own. However, the formation of ligand-independent and ligand-dependent HER2/HER3 heterodimers creates the most mitogenic and transforming receptor complex within the HER family of transmembrane receptor tyrosine kinases (RTKs). In HER2 over-expressing breast cancer cells, HER tyrosine kinase inhibitors (TKIs) inhibit basal phosphorylation of HER3 and its association with HER2 and with PI(3)K. Cyt, cytoplasm; ECM, extracellular milieu; Nu, nucleus.