

Summary
The biological role of a natural inhibitor of cysteine peptidases (designated ICP) of Leishmania has been investigated by genetic manipulation of the parasite. Null mutants grew normally in vitro, were as infective to macrophages in vitro as wild-type parasites, but had reduced infectivity to mice. Mutants re-expressing ICP from a single gene gave partial restoration of virulence in vivo, whereas mutants over-expressing ICP secreted the inhibitor and showed markedly reduced virulence in mice. Promastigotes of the null mutants had similar cysteine peptidase activities as the wild-type parasites, suggesting that ICP is not required for the expression or processing of the enzymes. The only proteins found to bind to ICP in promastigote cell lysates were fully processed forms of CPA and CPB, showing that ICP does not bind in abundance either to zymogens of the cysteine peptidases or other leishmanial proteins. However, only a small proportion of ICP co-localised with CPA and CPB in the promastigote (in the endoplasmic reticulum and Golgi) and the majority of ICP resided in vesicles that are apparently distinct from endosomes and the multivesicular tubule (MVT)-lysosome. These data suggest that ICP has a role other than modulation of the activity of the parasite's own cysteine peptidases and their normal trafficking to the MVT-lysosome via the flagellar pocket. The finding that ICP partially co-localised with an endocytosed cysteine peptidase leads us to postulate that ICP has a role in protection of the parasite against the hydrolytic environment of the sandfly gut and/or the parasitophorous vacuole of host macrophages.
Keywords: cysteine peptidase inhibitor, ICP, chagasin, lysosomes, endosomes, Leishmania
INTRODUCTION
Leishmania mexicana contains three types of Clan CA cysteine peptidases (CPs), the cathepsin L-like CPA and CPB and the cathepsin B-like CPC. CPB occurs as multiple isoenzymes encoded by a tandem array of CPB genes (Mottram et al., 1997) and is an important virulence factor (Mottram et al., 1996; Alexander et al., 1998). The CPBs are most abundant in the amastigote stage of the life cycle that occurs in the mammalian host, where they are located in large lysosomes called megasomes (Pupkis et al., 1986), whereas in metacyclic promastigotes (the infective stage in the sandfly vector) they are thought to be present in an unusual lysosomal compartment designated the multivesicular tubule (MVT) (Mullin et al., 2001); CPB is expressed at a very low level in procyclic promastigotes (the multiplicative stage in the sandfly vector). Intracellular trafficking of leishmanial CPB in metacyclic promastigotes is unusual in being primarily via the flagellar pocket (Brooks et al., 2000b), with targeting signals residing in the pro-domain (Huete-Pérez et al., 1999). Onward transfer from the flagellar pocket to the lysosomes appears to require removal of the pro-domain, presumably with activation of the enzyme (Brooks et al., 2000b). The flagellar pocket of Leishmania and other trypanosomatids is the only external membrane of the parasites that is available for vesicular trafficking and represents a unique intersection of the secretory and endocytic pathways (McConville et al., 2002b). Indeed, between the flagellar pocket and endoplasmic reticulum (ER)/nucleus are many membranous compartments involved in these trafficking pathways. Considerable progress has been made over the last decade in distinguishing different organelles that comprise these pathways in the related trypanosomatid Trypanosoma brucei and elucidating some of their functions (Morgan et al., 2002a; Morgan et al., 2002b). Less is known of the situation in Leishmania, but it is to be expected that similar pathways also occur (McConville et al., 2002b) although the precise composition and dynamics of the system is likely to differ from that of T. brucei as the two parasites live in different environments and, notably, only Leishmania resides intracellularly in macrophages.
The actions of mammalian lysosomal CPs (cathepsins) are controlled in part by endogenous tight-binding CP inhibitors from the cystatin superfamily (Grzonka et al., 2001; Abrahamson et al., 2003). Natural inhibitors of CPs were reported in Leishmania (Irvine et al., 1992), but cystatins could not be detected and the parasite's genome indeed apparently lacks such genes (http://www.genedb.org/genedb/leish/index.jsp). However, efforts to identify natural CP inhibitors in trypanosomatids led to the discovery in T. cruzi of chagasin, a potent inhibitor of the parasite's major lysosomal CP known as cruzipain (Monteiro et al., 2001). Subsequent database mining and sequence analyses revealed apparent chagasin homologues in the genomes of various bacteria and unicellular eukaryotes (Rigden et al., 2002; Sanderson et al., 2003). The homologues in Leishmania and T. brucei, named ICP for inhibitor of cysteine peptidase, were shown indeed to be potent and specific inhibitors of several clan CA CPs such as CPB of L. mexicana and mammalian cathepsin L; mammalian cathepsin B was also inhibited, but with a reduced efficiency (Sanderson et al., 2003). ICP was found to be expressed at highest levels in the procyclic promastigote stage of the life cycle, which is in contrast to CPB that is expressed at highest levels in the amastigote.
Nevertheless, nothing was known about the natural target or biological roles of ICPs. One possibility seemed to be that it interacts with and controls activity of leishmanial CPs during the enzyme's trafficking to the lysosomal network. In order to gain greater understanding of ICP function in Leishmania, we have used gene targeting to create parasite lines that either lack or over-express ICP. We show here that the major CPs of Leishmania are correctly processed, trafficked and activated in the ICP null mutants. Moreover, ICP does not substantially co-localise with endogenous leishmanial CPs. The data presented lead us to hypothesize that the principal role of ICP is in protection of the parasite against host, rather than parasite, CPs.
Results
Targeted deletion of the L. mexicana ICP gene and generation of complemented cell lines
Targeted deletion of the diploid L. mexicana ICP locus was achieved by homologous recombination. The two alleles were sequentially replaced after parasite transfection with linearised targeting constructs pGL792 and pGL896, containing selectable markers between ICP 5′ and 3′ flanking regions (Fig. 1A). The transfection for the first allele, with the pGL792 construct, yielded a population of parasites resistant to blasticidin. This population was used for the second round of transfections with the pGL896 (hygromycin) construct and four clones were obtained (designated Δicp). Two clones were analysed by Southern blot (Δicp clone 1 and Δicp clone 3), one of which is shown in Fig 1B. Genomic DNA digested by Age I/Nae I and hybridised with an ICP-specific probe revealed a 1.55 kb product containing the gene in wild-type (WT) parasites, but not in Δicp (Fig. 1B). When hybridised with probes derived from the antibiotic selectable marker genes, BLA and HYG, the DNA obtained after two rounds of targeting yielded DNA fragments of 3.5 kb and 4.2 kb, respectively, which were not present in the wild-type DNA (Fig. 1B). This shows the replacement of the two alleles of the ICP gene by the BLA and HYG genes. Two different types of Δicp cell lines re-expressing ICP were generated, one in an integrative vector, which should provide a constant level of gene expression similar to that in the wild-type parasite (designated Δicp:: PRRNA ICP) and one in an episomal vector, which can result in high expression levels due to multiple copies being present (designated Δicp[pXG ICP]). The Age I/Nae I-digested DNA from Δicp:: PRRNA ICP was analysed by Southern blot with a probe specific to the PAC gene conferring resistance to puromycin. The probe labelled a DNA fragment of ∼ 4.7 kb, which is what was expected from the restriction map of the initial construct (Fig. 1A). The ICP probe also labelled that same fragment, indicating that the ICP gene was re-integrated (Fig. 1B). The DNA from Δicp[pXG ICP] was digested by Age I/Nae I and analysed by Southern blot. The NEO and ICP probes revealed the presence of the predicted 1.9 kb DNA fragment as expected (Fig. 1B).
Fig. 1. Targeted replacement of the ICP gene.

A. Schematic representation of the ICP locus and the plasmid constructs used for gene replacement, re-integration and over-expression. ORFs are shown as arrows, intergenic and flanking DNA sequences are shown as boxes. Restriction enzymes used for the different constructs are shown (see text), as well as the expected sizes of the AgeI/NaeI fragments revealed by the Southern blot. DHFR, dihydrofolate reductase gene; BLA, blasticidin resistance gene; HYG, hygromycin resistance gene; NEO, neomycin resistance gene, PAC, puromycin resistance gene.
B. Southern blot analysis. Genomic DNA was digested with AgeI and NaeI, separated on a 1% agarose gel, blotted onto a nylon membrane and hybridised with 32P-labelled DNA probes. The hybridisation probes were fragments containing the entire coding region of their respective genes. lane 1, wild-type L. mexicana; lane 2, Δicp ; lane 3, Δicp:: PRRNA ICP; lane 4, Δicp[pXG ICP]. The positions of the molecular size markers are shown on the right.
To assess the expression of the ICP protein, a Western blot was performed with total lysates from early stationary phase promastigotes of the different cell lines, immunoblotted with affinity-purified anti-ICP antibodies (Sanderson et al., 2003). The ICP protein was not detected in extracts from Δicp clone 1 (Fig. 2A, Δicp) and a similar result was observed for clone 3 (data not shown). This confirms that the protein is no longer expressed by these mutants, whereas wild-type cells at the same growth stage did express ICP (Fig. 2A, WT). Δicp[pXG ICP] and Δicp::PRRNA ICP lines showed a successful restoration of ICP expression (Fig. 2A). However, whereas the level of expression yielded by the integrative construct was broadly comparable to that of the wild-type parasites, that resulting from episomal expression in pXG was about 5-fold higher in procyclic promastigotes when 25 μg ml−1 of neomycin was used. The ratio increased to 20-fold in metacyclic promastigotes, in which expression of native ICP is lower (Sanderson et al., 2003).
Fig. 2. Protein expression.
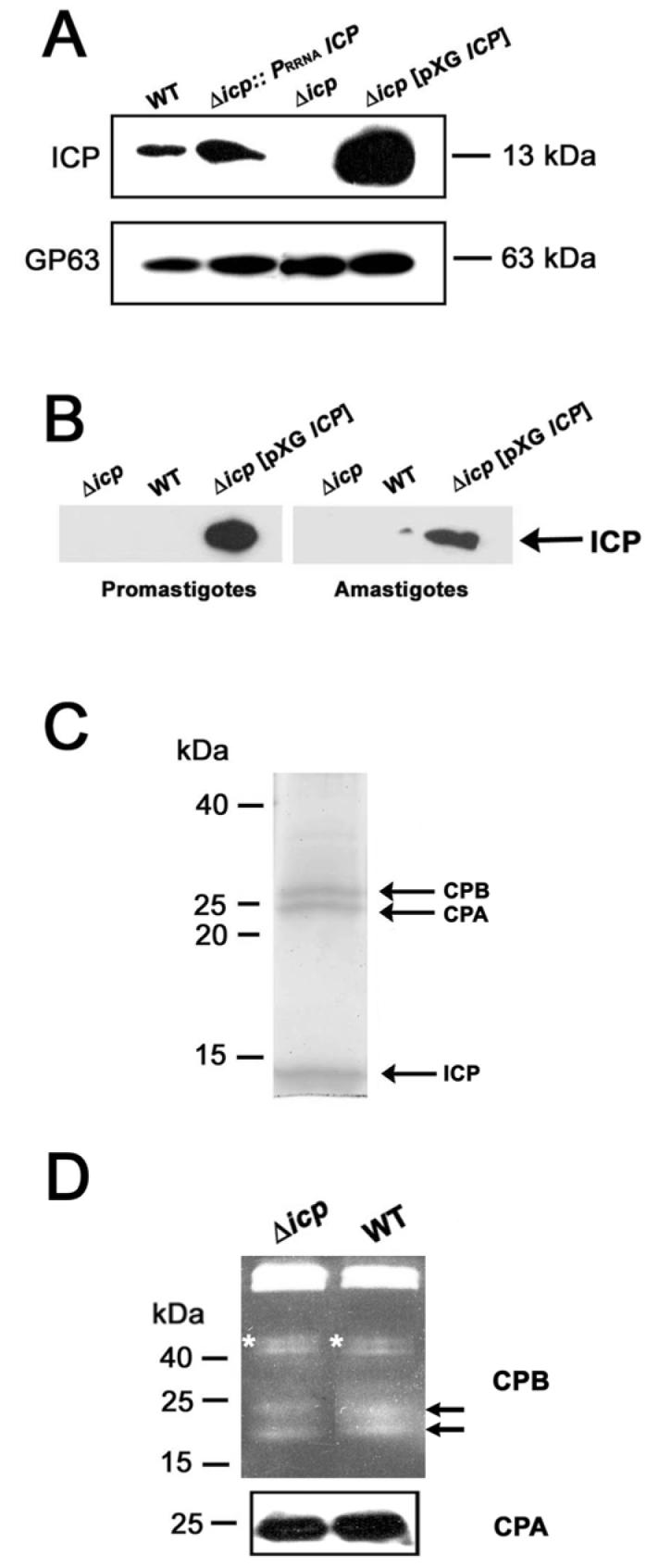
A. Whole-cell lysates prepared from stationary-phase promastigotes of wild-type (WT), Δicp, Δicp:: PRRNAICP and Δicp[pXG ICP] were analysed using 12% (w/v) SDS-PAGE and immunoblotted with affinity-purified polyclonal antibodies raised against ICP, and monoclonal antiserum raised against gp63 to confirm equivalent protein loading. The equivalent of approximately 107 parasites was loaded per lane, molecular masses are indicated on the right.
B. Proteins were isolated from spent medium from cultures containing ∼108 promastigotes or axenic amastigotes of wild-type (WT), Δicp, and Δicp[pXG ICP] parasites. Proteins were analysed by Western blot using affinity-purified anti-ICP antibody.
C. Immunoprecipitation of ICP and its associated proteins from L. mexicana promastigote lysates. The affinity-purified anti-ICP antibodies were coupled to a protein A/G Sepharose column and used to purify protein complexes from stationary-phase promastigotes lysates. Approximately 5 ×109 cells were used from wild-type Leishmania. Proteins identified by mass spectrometry are indicated with arrows. Molecular masses are shown on the left.
D. Analysis of CPA and CPB. Lysates of 107 stationary phase promastigotes were used for gelatin SDS-PAGE to detect CPB activities (upper panel) and Western blotting using anti-CPA antibody (lower panel). Molecular masses are indicated on the left. Asterisks and arrows mark the positions of precursor CPB and mature CPB, respectively.
ICP is secreted by promastigotes of the over-expressing line
One hypothesis is that ICP could act on extracellular CPs as an inhibitor, we thus wanted to assess if that protein was secreted in normal culture conditions by the parasite. We performed immunoprecipitation experiments where anti-ICP antibody was applied to spent medium from both late log/early stationary phase promastigote and axenic amastigote cultures of wild-type, Δicp and Δicp[pXG ICP] lines. An antibody directed against an intracellular protein, CRK3 (Grant et al., 1998), was used on the same fractions as a control for cell integrity and yielded no signal (data not shown). As shown in Fig. 2B, the Δicp[pXG ICP] cell line secreted readily detectable levels of ICP, but no traces of the protein were found for wild-type parasites, or the negative control Δicp. However, bearing in mind that Δicp[pXG ICP] has far greater internal amounts of ICP than wild-type parasites (Fig. 2A), it remains a possibility that wild-type parasites are secreting a small amount of ICP that is not detectable by this method.
ICP interacts only with the CPA and CPB cysteine peptidases in promastigote lysates
Immunoprecipitation experiments were undertaken in order to identify possible intracellular partners of ICP. Total cell extracts of late log phase promastigotes of wild-type and Δicp, as a control, were prepared and used for immunoprecipitation with affinity-purified anti-ICP antibodies. The final elution fractions were analysed by SDS-PAGE with SYPRO Ruby staining (Fig. 2C). The protein profile obtained for wild-type promastigotes contained only three major proteins. Their electrophoretic mobilities corresponded to 13, 25 and 28 kDa. (Fig. 2C, arrows). Similar analysis of Δicp, resulted in no proteins being detected (data not shown). To identify the immunoprecipitated proteins, the corresponding bands were excised from the gel, subjected to trypsin digestion and the generated peptides analyzed by MALDI mass spectrometry. The observed peptide masses were compared with those obtained from the virtual tryptic digest of the whole NCBI non-redundant (nr) database (http://www.ncbi.nlm.nih.gov/). This approach led to the unambiguous identification of the 13 kDa protein as ICP, the 25 kDa protein as CPA and the 28 kDa protein as CPB.
All three L. mexicana proteins identified by mass spectrometry analysis have the correct predicted sizes and their identities were confirmed by Western blot analysis with specific antibodies (data not shown). We determined the stoichiometry of ICP, CPA and CPB, using a PhosphoImager. After normalisation for the size of the proteins, the data indicated that for 2 molecules of ICP there was 1.10 (± 0.01) molecule of CPA and 0.94 (± 0.15) molecule of CPB. It is known that ICP has a high affinity for cathepsin L-like CPs (Sanderson et al., 2003) and the association observed could reflect either interaction between the enzymes and ICP in the living cell or binding subsequent to cell lysis when the compartmentation that occurs in the living cell is disrupted. Subsequent analyses (see below) are suggestive of the latter being the case. The results of this experiment do show, however, that ICP does not occur in Leishmania mainly bound to any other protein, although binding in small amounts to other proteins cannot be formally excluded.
L. mexicana precursor and mature CPB can be visualized by gelatin SDS-PAGE (Brooks et al., 2000b). We used this technique to analyse cell lysates of wild-type and Δicp stationary phase promastigotes for CPB activity (Fig. 2D). No differences were observed in precursor (Fig. 2D, asterisk) or mature (arrowed) CPB in the two cell lines. In addition, Western blot analysis showed that equivalent levels of CPA were present in both wild-type and Δicp lines (Fig. 2D). This shows that ICP is not required for the correct processing or activation of CPs in Leishmania.
Growth, differentiation and infectivity
Under standard in vitro culture conditions, there was no significant difference in the growth kinetics between wild-type, Δicp, Δicp[pXG ICP] and Δicp::PRRNA ICP promastigotes. In populations of stationary phase cells grown under the same culture conditions, Δicp and wild-type parasites contained similar proportions of infective metacyclic (about 22 %) and non-infective procyclic promastigotes (data not shown). The promastigote forms of Δicp were also able to differentiate in vitro into amastigote-like forms with similar kinetics to the wild-type parasites.
Late stationary-phase promastigotes and amastigote-like forms of the different cell lines were assessed in vitro for their ability to infect and survive in peritoneal macrophages explanted from BALB/c mice. After seven days incubation, there were no significant differences between the mean number of macrophages infected with wild-type parasites, Δicp or Δicp[pXG ICP] (Fig. 3A). These results show that parasite invasion and survival inside macrophages is not apparently affected by the absence or over-expression of the ICP protein.
Fig. 3. ICP null mutants are infective to macrophages and mice.
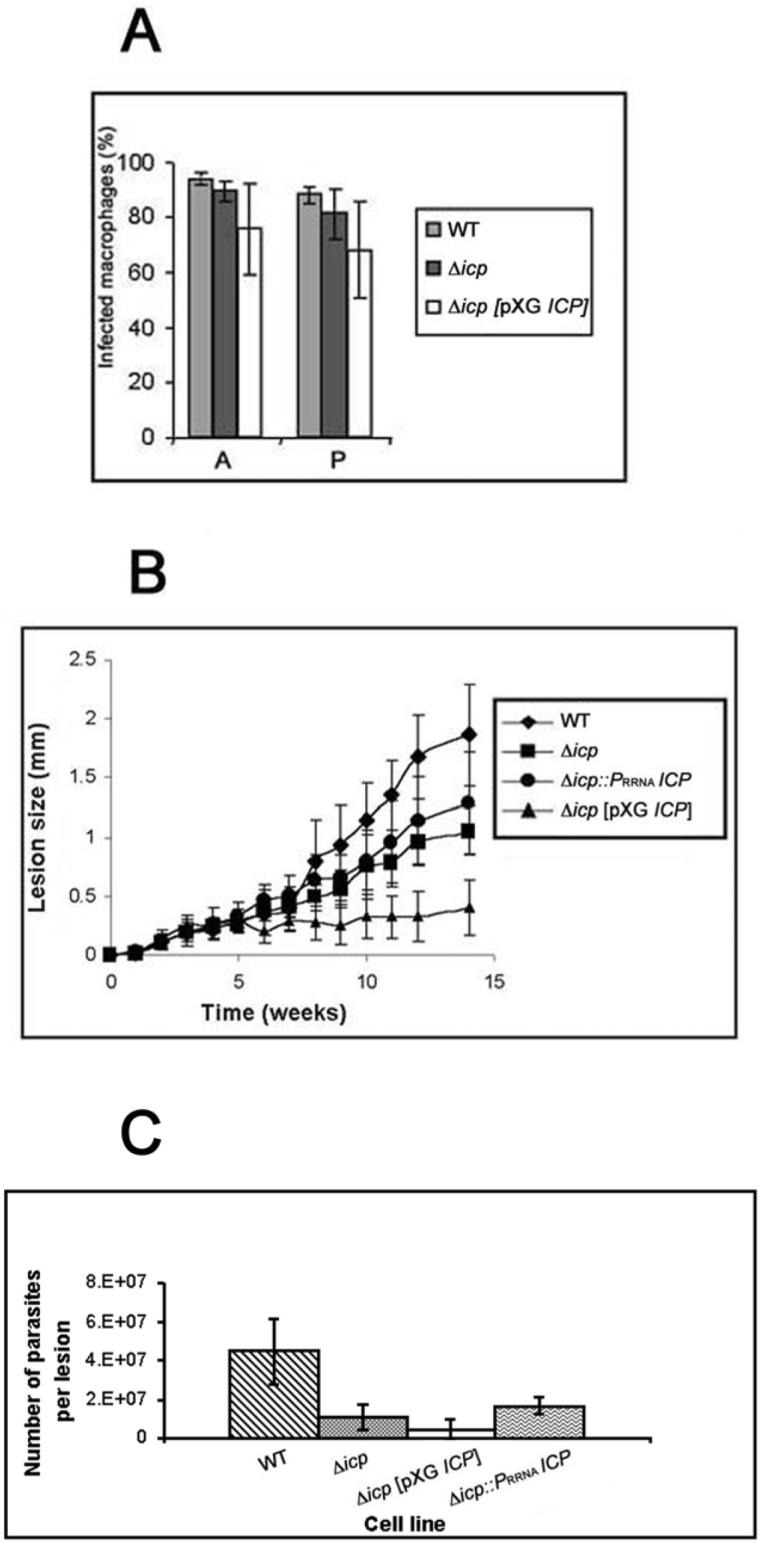
A. In vitro macrophage infectivity assays. Wild-type (WT) and transgenic stationary-phase L. mexicana promastigotes (P), and axenic amastigotes (A), were incubated with mouse peritoneal macrophages at a ratio of 5:1 and the parasite load determined by counting the number of infected macrophages after 7 days of incubation. The data show the percentage of infected macrophages ± SD from triplicate infections.
B. Infection of BALB/c mice with wild-type and transgenic stationary-phase L. mexicana promastigotes. Mice were challenged with 5 × 105 in the left hind footpad. The swelling caused by the respective cell lines was recorded. Data shown (mean lesion diameter ± SD from groups of 5 mice) are representative of three experiments done with two independent clones for each line.
C. Parasite loads in footpad lesions from infected BALB/c mice. Footpad lesions were dissected at the end of the infection experiment (week 12), tissue was disrupted in PBS and the number of parasites was estimated microscopically by counting.
The ability of the transgenic parasites to cause lesions in BALB/c mice was also investigated. An equal number (5 × 105) of stationary phase promastigotes was injected into the footpad of susceptible mice and lesions development was monitored. The Δicp clones were less efficient in establishing and maintaining an infection than were wild-type parasites (Fig. 3B). Nevertheless, the Δicp cell lines clearly were infective. Adding back the ICP gene with the integrative construct led to an increased infectivity compared with Δicp, although lesion development was still significantly slower than the wild-type levels (Fig. 3B). Interestingly, over-expressing ICP did not restore lesion development to wild-type levels, on the contrary the cell line generated only small lesions. Lesions were produced over the first few weeks but there was little or no subsequent growth (Fig. 3B). Amastigotes were recovered from the lesions after 14 weeks and inoculated into medium for transformation to promastigotes in vitro and also analysed by Western blot for ICP expression. Δicp, Δicp[pXG ICP] and Δicp::PRRNA ICP amastigotes were able to differentiate back to promastigotes in vitro, with similar kinetics to wild-type parasites, and Δicp[pXG ICP] and Δicp::PRRNA ICP were found to express ICP (data not shown). The amastigote load in the lesions was also assessed by disrupting the whole lesions in a defined volume of PBS and determining the parasite numbers. This showed that there was a correlation between amastigote numbers and lesion sizes (Fig. 3C).
Localisation of ICP, CPA and CPB
The results from the immunoprecipitation experiments detailed above showed that ICP interacts with CPA and CPB in lysates of Leishmania, but to assess whether these interactions occurred in living cells it was necessary to investigate the respective cellular localisations of each of the proteins. Immunofluorescence experiments were performed on early stationary phase promastigotes with CPA-, CPB- and ICP-specific antibodies. Punctate patterns of fluorescence were observed for all three antibodies, indicative of a vesicular localisation, with the most intense staining in the region between the nucleus and kinetoplast (Fig. 4). Some co-localisation was observed between ICP and CPA/CPB (arrowed in Fig. 4A), however the majority of ICP was found in structures lacking the CPs. CPA and CPB had a large degree of co-localisation (Fig. 4B).
Fig. 4. Localisation of ICP, CPA and CPB.
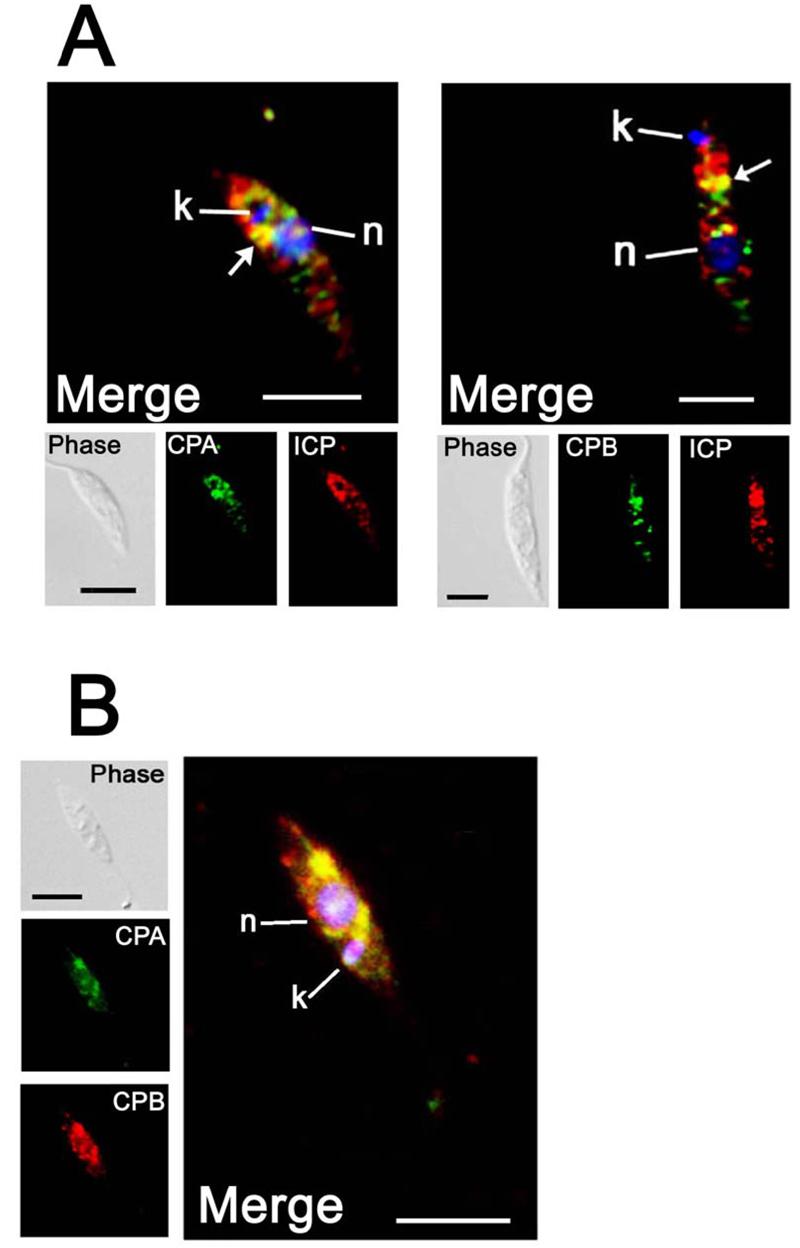
A. Immunofluorescence analysis of early stationary-phase promastigotes with purified rabbit anti-ICP antibodies (red) and rabbit anti-CPA or anti-CPB antibodies (green). B. Immunofluorescence analysis of stationary-phase promastigotes with purified rabbit anti-CPB antibodies (red) and anti-CPA antibodies (green). Primary antibodies were labelled directly with the desired fluorophore using the Zenon labelling kit (Molecular Probes) and the fluorescence images have been deconvolved using the Volume Deconvolution module of the Openlab Software (Improvision). . Merged images are magnified, with DAPI-stained DNA (blue) of the nucleus (n) and the kinetoplast (k), the scale bar represents 10 μm. The co-localised signal is yellow, highlighted by arrows in A.
ICP, CPA and CPB do not co-localise with Concanavalin A
The region between the nucleus and kinetoplast is where most of the components of the secretory pathway and the endosomal/lysosomal system are localised. We thus performed co-localisation experiments with molecular markers of organelles thought to be involved in these pathways. We first used fluorescently labelled Concanavalin A (Con A), that has been shown in Leishmania to co-localise with Rab7-positive endosomes (Denny et al., 2002). The rate of endocytosis of the promastigote form of Leishmania is quite low compared with that of the bloodstream form of T. brucei (Ghedin et al., 2001; Mullin et al., 2001; McConville et al., 2002b), therefore we allowed the labelled Con A to be internalised for 2 h before fixation of the cells. FITC-labelled Con A was located in a compartment next to the nucleus, probably corresponding to a sub-compartment of the endosomal system. In addition, some of the Con A was retained in the flagellar pocket (Fig. 5A). Labelling with specific antibodies showed that the vesicles containing the CPs were in some cases closely associated with the organelles containing the Con A, yet distinct from them (as demonstrated in Fig 5A). ICP was shown to be located in another distinct compartment, as it did not co-localise with the organelle labelled by Con A (Fig. 5A).
Fig. 5. Co-localisation with endocytosed markers.
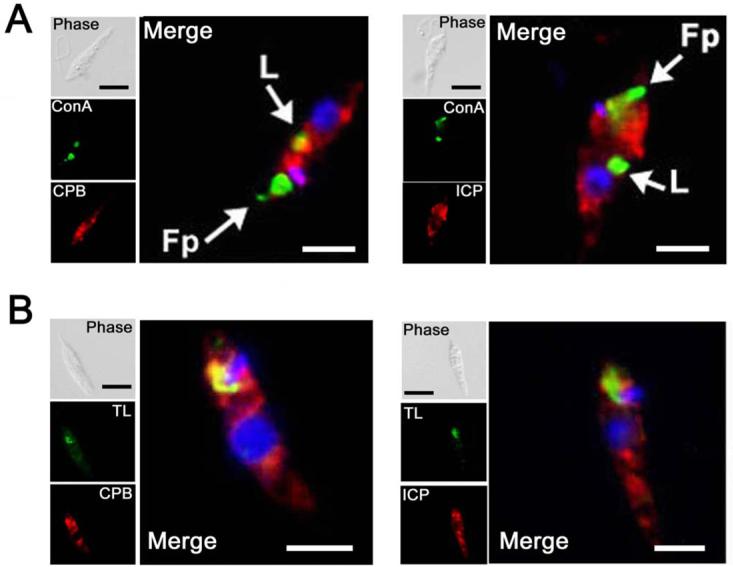
A. Promastigotes were exposed to FITC-conjugated Con A (green) at 4° C for 10 min prior to a chase for 2 h at 25° C to permit accumulation in the flagellar pocket (Fp, arrowed) and late endosomes (L, arrowed). Cells were fixed and stained with anti-CPB or anti-ICP rabbit antibodies, followed by Texas Red-conjugated goat anti-rabbit antibody (red).
B. FITC-conjugated tomato lectin (TL) was added to promastigotes at 4° C for 10 min prior to a chase for 30 min at the same temperature to permit accumulation in a specific compartment (green). Cells were fixed and stained with anti-CPB or anti-ICP rabbit antibodies, followed by Texas Red-conjugated goat anti-rabbit antibody (red). Merged images are magnified, with DAPI-stained DNA (blue) and co-localised signal, where present, is yellow. The scale bar represents 10 μm.
CPB has been reported to be targeted to the lysosomes through the flagellar pocket (Brooks et al., 2000b) and the ICP homologue in T. cruzi (known as chagasin) has been detected in that compartment by electron microscopy (Monteiro et al., 2001). It is noteworthy that neither of these proteins was strongly detected in the flagellar pocket in these studies (Fig. 5A), although this does not rule out a transient presence of the proteins in the pocket.
CPA and CPB but not ICP are located in a compartment containing proteins with N-linked glycans containing poly-N-acetyl lactosamine
We also used tomato lectin, which recognises linear poly-N-acetyl lactosamine (pNAL) in the N-linked glycan of some proteins within the early endosomal system of T. brucei (Nolan et al., 1999; Pal et al., 2002), to investigate if pNAL-containing proteins exist in a similar compartment in Leishmania. Both CPA and CPB co-localised with pNAL-containing proteins close to the kinetoplast (Fig. 5B, CPA, data not shown). In contrast, ICP was found in close proximity to, but clearly distinct from, the tomato lectin-reactive compartment (Fig. 5B).
CPA, CPB and ICP are present in the ER and Golgi
Proteins destined for the cell surface or lysosomes of Leishmania are initially assembled in the endoplasmic reticulum (ER), before being trafficked to the Golgi (McConville et al., 2002). To test if ICP is located in the ER, we performed co-localisation studies with an antibody raised against a resident protein of the ER, serine palmitoyltransferase subunit 2, LmLCB2 (Denny et al., 2004). As shown on Fig. 6A, CPB and ICP co-localised with LmLCB2 in an area located between the nucleus and the kinetoplast, suggesting that the ER is one organelle where ICP co-localises with the CPs. We also used a marker for the Golgi, a GFP fusion with the T. brucei GRIP domain protein (McConville et al., 2002a), to show that some ICP and CPB are located within this organelle (Fig 6B). These data suggest that both ICP and the CPs traffic through the ER and Golgi and then into separate vesicles and onwards to different organelles.
Fig. 6. Co-localisation with ER and Golgi markers.
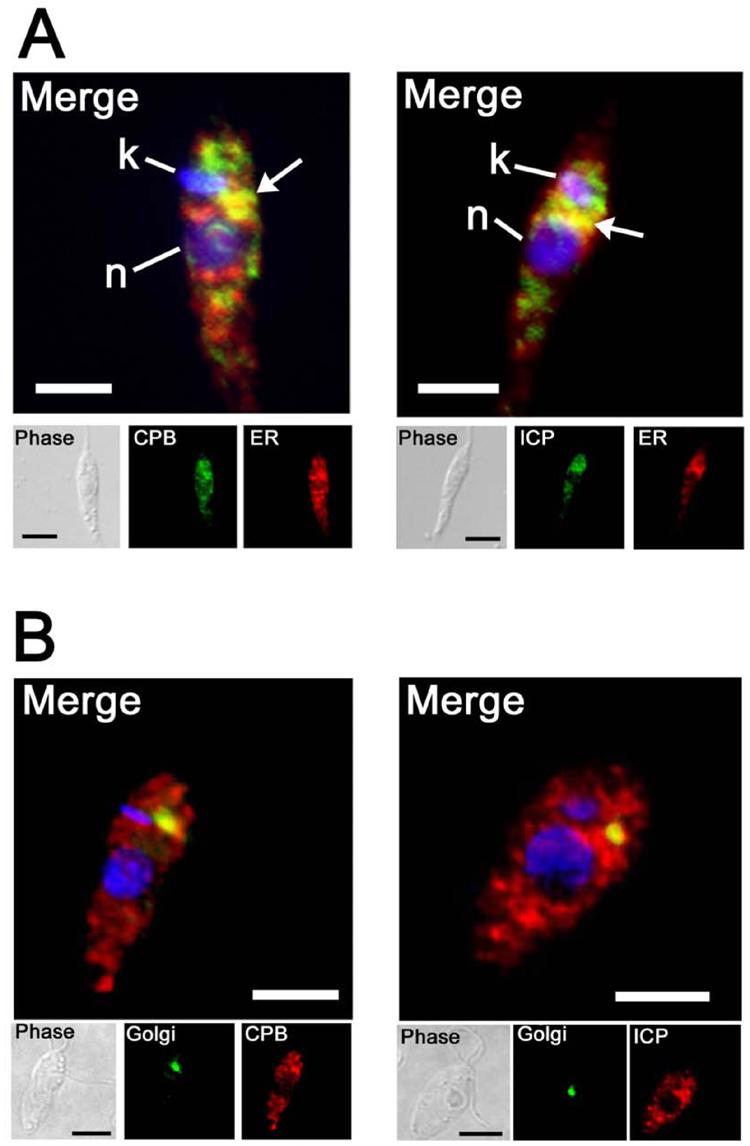
A. Early stationary-phase promastigotes were treated for microscopy and stained with mouse anti-LmLCB2 (an ER protein), followed by a goat anti-mouse FITC-conjugate antibody (green), and with anti-CPB or anti-ICP rabbit antibodies, followed by a goat anti-rabbit Texas Red-conjugate antibody (red). Merged images are shown with DAPI-stained DNA (blue) of the nucleus (n) and the kinetoplast (k), the scale bar represents 10 μm. The co-localised signal is yellow, highlighted by arrows.
B. Log-phase promastigotes expressing GFP-fused GRIP protein (trans-Golgi resident protein) were fixed and stained with mouse anti-GFP antibody, followed by a goat anti-mouse FITC-conjugate antibody (green), and with anti-CPB or anti-ICP rabbit antibodies, followed by Texas Red-conjugated goat anti-rabbit antibody (red). Merged images are magnified, with DAPI-stained DNA (blue) and co-localised signal is yellow. The scale bar represents 10 μm.
Internalisation of a cysteine peptidase into L. mexicana promastigotes
To assess if ICP could act to inhibit an internalised CP, L. mexicana promastigotes were incubated with biotinylated papain and then anti-ICP antibodies and fluorophore-conjugated streptavidin used to perform localisation experiments. In general, the internalised papain showed a reticulated pattern distributed throughout the cell body (Fig. 7). There was partial co-localisation between papain and ICP largely, but not solely, located around the nucleus (Fig. 7A, arrows). In some cases, a strong co-localisation could be observed next to the nucleus in the posterior part of the cell (Fig. 7B, arrow). However, there remained multiple unidentified ICP-containing vesicles in both the anterior and posterior parts of the cell that did not co-localise with endocytosed papain.
Fig. 7. Internalisation of papain.
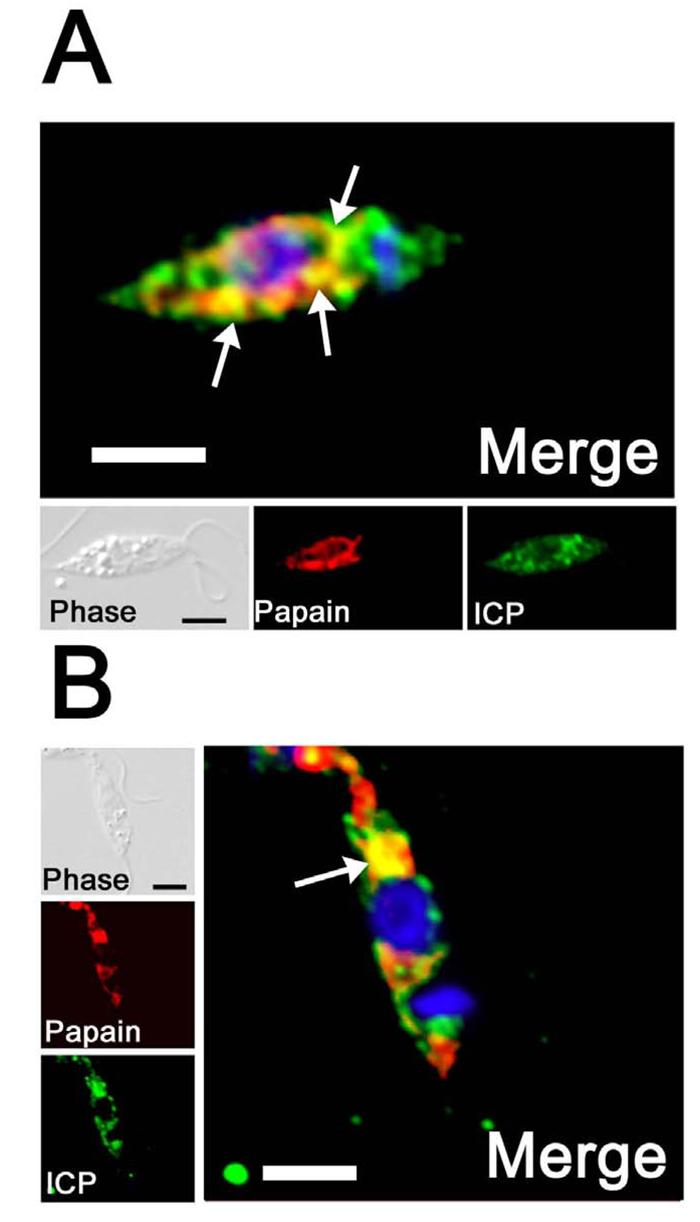
Log phase promastigotes were incubated for 10 mins with 1 μg of papain previously treated with sulfo-NHS-SS-biotin. Surface biotin was removed by reducing the disulphide bonds using glutathione. Cells were treated for fluorescence and internal papain was revealed by using Alexa Fluor 594-conjugated streptavidin (red). Cells were also stained with anti-ICP antibodies, followed by FITC-conjugated goat anti-rabbit antibody (green). The fluorescence images have been deconvolved using the Volume Deconvolution module of the Openlab Software (Improvision). Merged images are magnified, with DAPI-stained DNA (blue) of the nucleus and the kinetoplast. Co-localised signal is yellow and some is highlighted with arrows. The scale bar represents 10 μm.
Discussion
Targeted gene deletion of the ICP gene was accomplished by gene replacement of its two alleles and re-expressing and over-expressing lines were also produced. These genetic manipulations did not have noticeable effects upon the viability of the parasites themselves, as the Δicp and other genetically modified promastigotes were all able to grow normally in vitro, differentiate into amastigotes, and infect and survive within mouse macrophages in vitro (Fig. 3A). The situation in vivo appears to be more complex. The Δicp cell line itself has reduced ability to produce lesions in BALB/c mice (Fig. 3B), a phenotype that was partially complemented by re-integration of a single copy of the ICP gene to produce the Δicp:: PRRNA ICP line. The data presented (Fig. 2A) suggest that ICP levels are moderately similar in wild-type and Δicp:: PRRNA ICP promastigotes, however this may not be the case in lesion amastigotes. Indeed, ICP is down-regulated in wild-type amastigotes (Sanderson et al., 2003), yet the pRIB vector is likely to provide constitutive (and thus higher) expression (Misslitz et al., 2000). The effect of higher expression can be seen with the Δicp[pXG ICP] line, which showed markedly reduced infectivity to mice (Fig. 3B) although it was fully able to infect and survive in macrophages in vitro (Fig. 3A). Thus expression levels of ICP might be important for the function of the protein and may explain why full complementation was not achieved with the Δicp:: PRRNA ICP line. Δicp[pXG ICP] secreted appreciable amounts of ICP in both the promastigote and amastigote stages (Fig. 2B) and ICP could have modulated the immune response mounted in the mice, thus leading to reduced infectivity in the animals. It has been reported that mice treated with a natural CP inhibitor, cystatin, promoted a protective response against Leishmania infection and a switch from a predominately Th2 to a Th1 response (Das et al., 2001), so ICP might be acting in a similar way. This possibility is under investigation.
CPA and CPB were found to be the major proteins bound to ICP in L. mexicana lysates. The binding of ICP with the enzymes is not surprising, as ICP has been shown to bind with high affinity to cathepsin L-like Clan CA CPs, exemplified by CPA and CPB, but with a 1000-fold lower affinity to cathepsin B (Sanderson et al., 2003). Consistent with this is the failure to detect an interaction between CPC, a cathepsin B-like enzyme of L. mexicana (Bart et al., 1995), and ICP in the pull-down experiments. Quantification of ICP, CPA and CPB levels bound to the column showed a molar ratio of ∼2:1:1, suggesting that all of the ICP molecules were bound to either CPA or CPB. As the majority of ICP is located in a distinct subcellular compartment to CPA and CPB (Fig. 4), it is likely that most of the binding of the CPs by ICP occurs after lysis of the cells and breakdown of the compartmentation. Thus the finding that CPA and CPB are the only proteins bound in abundance by ICP indicates that the inhibitor is not bound in large amounts to any other protein in living cells and also that it is in lower abundance than CPA and CPB. The only location where both the CPs and ICP were detected were the ER and Golgi compartments (Fig. 6), organelles in which the CPs are thought to be present in their zymogen form. However, the sizes of CPA and CPB immunoprecipitated with ICP are those of the mature forms of the proteins (24 and 27 kDa, respectively). This suggests that ICP does not interact directly with the precursor forms of the CPs in the ER or Golgi. This correlates with the known mechanism by which the pro-domain of cysteine peptidases prevents activation of the CPs in these compartments (Groves et al., 1998). ICP, however, may function in these compartments to inhibit inappropriately activated CPA or CPB and thus protect the compartments from proteolytic damage.
The location and mechanism by which CPA and CPB zymogen is processed to the mature enzyme is still not known, but there is some evidence to suggest that it could occur in the flagellar pocket (Brooks et al., 2000b). The proregion also facilitates the trafficking itself (Huete-Pérez et al., 1999). In general, proteins assembled in the ER can be delivered to the lysosomes by two distinct routes. There is a direct intracellular route involving multivesicular bodies (MVBs) for the transport of proteins, and an indirect route via the flagellar pocket and the early endosomes, prior to the targeting to the lysosomes (for reviews see (Waller and McConville, 2002; McConville et al., 2002b)). We reported previously that Leishmania CPs appear to be trafficked through the indirect route, as unprocessed CPB accumulated in the flagellar pocket of a L. mexicana line deficient in both CPA and CPB in which active site-mutated CPB was re-expressed (Brooks et al., 2000b). Moreover, these data suggested that the zymogens reach that compartment before being processed. Similarly, unprocessed CPs accumulated in the flagellar pocket of L. major promastigotes when they were incubated with specific CP inhibitors (Selzer et al., 1999). These observations imply that the maturation of the CPs within the flagellar pocket is a necessary prerequisite for onward transfer of the mature enzyme, presumably via the endosomal pathway. Our finding that, in late log phase promastigotes, CPA and CPB partially co-localise with tomato lectin, a putative endosomal marker (Fig. 6), would support this hypothesis. A question that remains is how the enzymes' activities are controlled when they are in these organelles, which seemed a possible role for ICP. However, the findings of this study show that this is not the case.
The finding that ICP is trafficked through the ER and Golgi suggests that this is the route by which the protein reaches the vesicles in which the majority of the inhibitor is found. These vescicles are distributed through the cell body, occurring particularly between the flagellar pocket and nucleus and in the posterior end of the cell. These vesicles do not co-localise with putative markers of the endosomal system, such as Con A or tomato lectin (Fig. 5). The finding that the over-expressed ICP is being secreted in high amounts by the parasite hints that the vesicles may be components of the secretory pathway or at the intersection of the secretory and endosomal systems.
We have performed our immuno-localisation experiments on late log phase or early stationary phase promastigotes, so that we studied cells in which both ICP and the CPs are expressed. We found that CPA and CPB are present in the early endosomal compartment in that stage of the parasite, but not in a compartment labelled by endocytosed Con A. Con A uptake has been used previously in Leishmania to characterise Rab7-positive compartments (Denny et al., 2002), and Rab7 is a marker for the late endosomes/lysosomes and has been shown to partially co-localise with CPB in axenic amastigotes (Denny et al., 2002). However, the endocytic compartment is quite complex, made of structurally and functionally distinct regions (Gruenberg, 2001), and as yet has been only poorly characterised in Leishmania at a molecular level, which prevents the precise identification of the ConA-labelled compartment.
CPA and CPB labelled structures between the nucleus and the kinetoplast, but there was also a more dispersed vesicular labelling that is distinct from ICP (Fig. 4). CPB has been proposed to be present in the MVT-lysosome in stationary phase promastigotes (Mullin et al., 2001). It is likely that this compartment is disrupted by the fixation process used for immunofluoresence, as it has been documented previously (Mullin et al., 2001), which would result in the appearance of vesicles. Indeed, in our hands endocytosed markers which have been to shown to be labeling the MVT-lysosome in live Leishmania parasites, for instance bodipy ceramide or FM 4-64 (Mullin et al., 2001), did not give any consistent labeling after the treatment we used for antibody incubation for co-localisation experiments (data not shown). Thus our data are consistent with CPA and CPB being present in the MVT-lysosome.
The finding that ICP partially co-localises with endocytosed papain provides an intriguing insight into a potential role for ICP in the host-parasite interaction. Leishmania carry out active endocytosis of host proteins (Singh et al., 2003) and they live in a highly hydrolytic environment rich in host peptidases in both the parasitophorous vacuole of the mammalian macrophage as amastigotes (Alexander et al., 1999) and in the digestive tract of the insect host as promastigotes (Sacks, 2001). The significant co-localisation of endocytosed biotinylated papain with ICP, particularly in, but not confined to, a perinuclear region (Fig. 7), support the hypothesis that ICP could protect the parasite from the potentially toxic effects of endocytosed active cysteine peptidases. It is apparent, however, that ICP is found also in vesicles distinct from endocytosed papain; these may be ICP stores ready for release into other subcellular compartments. Although we have been unable to detect secretion of ICP in wild type parasites, significant levels of secretion was detected in the cell line over-expressing ICP. A low level of ICP secretion under in vivo conditions, or release from dying parasites, may aid survival through modulating the action of host peptidases or even the host's immune response. Release of CPB from parasites into the extracellular milieu of a mouse lesion has been documented (Ilg et al., 1994) and this is thought to contribute to the survival and also the pathogenicity of the parasite (Mottram et al., 2004).
Thus the data arising from this study leads us to formulate the hypothesis that ICP has a role in the host-parasite interaction by modulating the activity of host CPs, either after endocytosis of the enzymes or in the parasite's environment. The results show that ICP is not required for the expression, trafficking or processing of the major CPs of Leishmania, CPA or CPB. We cannot rule out that ICP acts to modulate the parasite's own CPs, for instance to protect the parasite from inappropriate activation of its own CPs within the ER or Golgi. Further studies in this area will provide information on the contribution of ICP function to the parasite's survival within its hosts and the new markers that this study has provided will be useful in unraveling the details of the endosomal and secretory pathways of Leishmania..
Experimental procedures
Parasites
L. mexicana (MNYC/BZ/M379) promastigotes were grown in modified Eagle's medium (designated complete HOMEM medium) with 10% (v/v) heat-inactivated foetal calf serum at 25°C, as described previously (Mottram et al., 1997). Amastigote-like forms (axenic amastigotes) were grown in vitro in Schneider's Drosophila medium (Life Technologies, Inc.) with 20% (v/v) heat-inactivated foetal calf serum at pH 5.5 and 32°C in the presence of 5% (v/v) CO2 (Bates et al., 1992). The required antibiotics were added to the cultures of the ICP null mutants of L. mexicana and derived cell lines typically as follows: hygromycin B (Sigma) at 50 μg ml−1; phleomycin (Cayla, France) at 10 μg ml−1; puromycin (Calbiochem) at 10 μg ml−1; blasticidin S (Calbiochem) at 10 μg ml−1; and neomycin (G418, Geneticin, Life Technologies) at 25 μg ml−1. Concentrations up to 500 μg ml−1 were used as required.
Generation of a L. mexicana ICP null mutant cell line
The 602 bp 5′ flank fragment of ICP was generated by PCR with primers NT115 and NT116 (see Table 1 for all oligonucleotides used in this study) on L. mexicana genomic DNA, digested with HindIII and SalI and inserted into HindIII/SalI-digested plmcpb2-hyg (Mottram et al., 1996) to give plmicp-hyg5. The 3′ fragment was generated by PCR using primers NT117 and NT118. The resulting 504 bp fragment was digested by SmaI and BglII and cloned into plmicp-hyg5 to give pGL896. The cassette used for transfection was released by HindIII/BglII digestion. pGL792 plasmid, used for the replacement of the second ICP allele, was generated from plasmid pGL896 by replacing the SpeI/BamHI cassette containing the hygromycin resistance gene by a SpeI/BamHI cassette containing the blasticidin S deaminase gene and obtained from plasmid pGL437 (Brooks et al., 2000a).
Table 1.
Primers used in this study.
| NT115 | 5′-GGAAGCTTCACGCCATCCGTATGATGG-3′ |
| NT116 | 5′-TCGTCGACCGTTGCACCCGTGTCTGTG-3′ |
| NT117 | 5′-GACCCGGGAAAACGGAAGAACGCAGAGG-3′ |
| NT118 | 5′-GCAGATCTCGTGTGGGGCTTCTGAGCTGG-3′ |
| OL1140 | 5′-GTGCAACTCGAGATGATCGCCCCGCTCAGT-3′ |
| OL1195 | 5′-CGTTTTAGATCTCTACTTCACGTTGAGGTG -3′ |
| OL1138 | 5′-AGCATACCCGGGATGCATCACCATCACCATCACATCGCCCCGCTCAGTGTG-3′ |
| OL1139 | 5′-CGTTTTGGTACCCTACTTCACGTTGAGGTG-3′ |
| OL1096 | 5′-CCGAGCACTAGTATGAAAAAGCCTGAACTC-3′ |
| OL1097 | 5′-ACTAGAGGATCCCTATTCCTTTGCCCTCGG-3′ |
| OL494 | 5′-ACTAGTATGGCCAAGCCTTTGTCT-3′ |
| OL495 | 5′-GGATCCTTAGCCCTCCCACACATA-3′ |
Generation of L. mexicana re-expressing cell lines
For the re-expression experiments, a copy of ICP was inserted in the pRIB expression vector bearing the puromycin resistance gene (Garami and Ilg, 2001). ICP was obtained by PCR from L. mexicana genomic DNA with primers OL1140 and OL1195 containing BglII and BamHI sites, respectively. The 350 bp fragment obtained was digested by XhoI/BglII and cloned into XhoI/BglII digested pRIB, yielding plasmid pGL892. The integration cassette from that plasmid was excised by digestion with PacI and PmeI before transfection. That fragment possesses 5′ and 3′ flanking regions for chromosomal integration at a ribosomal locus, which is known to be expressed in both promastigotes and amastigotes (Misslitz et al., 2000). In addition, the ICP gene was expressed in the pXG episomal vector (Ha et al., 1996). An N-terminal-poly-histidine-tagged version of the ICP gene was produced by PCR using the primers OL1138 and OL1139. The resulting 360 bp fragment was digested by SmaI/BamHI and ligated into pXG plasmid, previously digested by the same enzymes, to give the pGL895 plasmid. L. mexicana wild type promastigotes were electroporated with 20 μg of the gene replacement cassettes or 10 μg of episome and transfectants were selected with the appropriate antibiotics as previously described (Mottram et al., 1996).
Southern Blot analysis of transfectants
DNA was isolated from the transfectants with the DNeasy kit (Qiagen). 3 μg were digested by AgeI amd NaeI, electrophoresed through a 0.8% agarose gel, and blotted onto Hybond C Super (Amersham Pharmacia). A 350 bp ICP-specific probe was generated from a PCR fragment obtained with primers OL1140 and OL1195. A probe specific for the hygromycin resistance gene was obtained from a 1026 bp fragment synthesised by PCR with primers OL1096 and OL1097. A probe specific for the blasticidin S deaminase gene was generated from a 390 bp fragment produced by PCR with primers OL494 and OL495. Fragments were labelled with a random priming kit (Stratagene) and the blot was hybridised at 65°C overnight. Washes were for 15 min at 65°C with 2× SSC/0.1% SDS and then twice with 0.2× SSC/0.1% SDS.
Macrophage infections
Peritoneal exudate macrophages (PEMs) were extracted from peritoneal lavage of female BALB/c mice and infected at a ratio 5:1 with stationary phase promastigotes or amastigotes-like forms obtained in vitro (Frame et al., 2000). After 7 days incubation at 32°C, cells were fixed, stained with Giemsa stain and parasite loads determined microscopically by counting 200 PEMs.
Mouse infections
Groups of five mice were inoculated in the footpad with 5 × 105 stationary-phase L. mexicana promastigotes resuspended in 0.02 ml of phosphate-buffered saline (PBS), pH 7.4. The resultant lesions were monitored over a 12 weeks period.
Antibodies and immunoblotting
Affinity-purified antiserum specific for L. mexicana ICP was generated as described previously (Sanderson et al., 2003). Western blots were performed as described previously (Brooks et al., 2000b) with a 1:1000 dilution of affinity-purified anti-ICP antibody followed by a 1:5000 dilution of the appropriate horseradish peroxidase-conjugated secondary antibody. The West-Pico chemiluminescence detection system (Pierce) was used to visualise antigens.
Purification of the ICP-protein complexes and identification of their components
Immunoprecipitation was carried out with anti-ICP purified antibody. Briefly, the cell pellets (∼5.109 stationary phase promastigotes) were resuspended in 500 μl solubilisation buffer (PBS, pH 7.4, 0.2 % Nonidet P-40) and briefly sonicated. The lysate was centrifuged at 12,000 × g for 15 min and the resultant supernatant was put in contact with protein A/G-Sepharose beads to which 200 μg of purified anti-ICP antibody had been previously covalently linked using the Seize X immunoprecipitation kit (Pierce). The immunoprecipitation was then carried out according to the protocol published by the manufacturer. The proteins were precipitated with trichloroacetic acid, separated by SDS-PAGE and stained with SYPRO Ruby (Bio-Rad). The proteins of interest were cut and digested in-gel with trypsin and the tryptic peptides were analyzed by microcapillary LC-MS with automated switching to MS/MS mode for peptide fragmentation and sequence analysis (Gygi et al., 1999). The collision-induced dissociation (CID) spectra were compared on both a generic and a Leishmania-specific database, with the MASCOT program (http://www.matrixscience.com) (Perkins et al., 1999). The stoichiometry of the different components was determined on a SDS-PAGE gel after SYPRO Ruby staining, using a PhosphoImager, and the densitometry of each band was assessed with the ImageQuant software (Molecular Dynamics). Stoichiometry determinations for the various proteins was conducted by comparing the weighted signal strength of each protein with that of ICP (Malone et al., 2001). Results were normalised using the predicted molecular mass of the different proteins.
Gelatin SDS-PAGE analysis
CP activity was assayed by gelatin SDS-PAGE as previously described (Robertson and Coombs, 1990). Coomassie blue stain was used to visualize the hydrolysis of 0.2% gelatin co-polymerised in a 12% polyacrylamide separating gel.
Immunofluorescence
For immunofluorescence analyses, log or early stationary phase promastigotes were fixed with 2% (v/v) formaldehyde in PBS (0.15 M NaCl/5 mM potassium phosphate, pH 7.4) for 30 min at 25°C. The solution was adjusted to 0.1% Triton X-100 and was incubated for 10 min; then glycine (0.1 M) was added for 10 min to neutralise active aldehyde groups. After centrifugation, the cells were resuspended in PBS and allowed to adhere to glass slides until completely dry. The slides were incubated for 30 min with rabbit affinity-purified anti-ICP, affinity-purified anti-CPB (Brooks et al., 2000b) or anti-CPA (Mottram et al., 1992) antibodies and mouse anti-LmLCB2 antibody (Denny et al., 2004), followed by Texas Red-conjugated goat anti-rabbit secondary antibody diluted 1:100 (Molecular Probes) and fluorescein isothiocyanate-conjugated goat anti-mouse secondary antibody diluted 1:100 (Sigma). When co-localisation with two primary antibodies from the same species was needed, they were directly labelled with the desired fluorophore using the Zenon kit from Molecular Probes according to the protocol supplied by the manufacturer. Cells were viewed with a Zeiss UV microscope, and images were captured by an Orca-ER camera (Hammamatsu) and Openlab software v 3.1.3 with deconvolution module (Improvision).
Fluorescent staining of cells
For labelling of the flagellar pocket and the late endosomal compartment, 107 L. mexicana promastigotes were harvested by centrifugation and washed in serum-free HOMEM for 10 min. They were resuspended in 100 μl of serum-free HOMEM and incubated with 1 μl of FITC-conjugated Concanavalin A (5 mg ml−1 solution; Molecular Probes) for 10 min at 4°C. They were then washed for 10 min in 1 ml of serum-free HOMEM, resuspended in 100 μl of the same medium and chased for 2 h at 25°C before being fixed and prepared for microscopy. For labelling of another endosomal compartment, the same protocol as above was followed except that 1 μl of FITC-conjugated tomato lectin (1.3 mg ml−1 solution; Sigma) and a 30 min chase were used.
Papain-labelling and uptake experiment
1 mg of papain (from Papaya latex, Sigma) was biotinylated with Sulfo-NHS-SS-biotin (Pierce) according to the protocol from the manufacturer. L. mexicana promastigotes were incubated with 1 μg of biotinylated papain in serum-free HOMEM medium for 10 min. After two washes in PBS, any sulfo-NHS-SS-biotin present on the cell surface was cleaved by 15 min of incubation on ice with serum-free HOMEM, 50 mM glutathione, pH 9.0. After two additional washes in PBS, cells were prepared for fluorescence as described above. Internalised biotinylated papain was detected by incubating the fixed cells for 30 min with 1 μl of Alexa Fluor 594-conjugated streptavidin (80 μg ml−1; Molecular Probes). A control was also made with fluorophore-conjugated streptavidin without prior incubation with biotinylated papain to assess background labelling.
Extracellular detection of ICP
10 ml of spent medium from cultured parasites were collected, filter-sterilised, and the proteins were precipitated with TCA. The medium was mixed with pre-chilled 25% (w/v) TCA (final concentration 6% w/v), chilled on ice for 15 min and centrifuged at 10000 g for 10 min. The pellet was resuspended and washed twice with 0.3 ml of acetone, and then dried at 100 °C for 5 min. Precipitated proteins were then resuspended in 200 μl of SDS-PAGE loading buffer and were assessed for the presence of ICP by Western blotting with ICP-specific polyclonal serum (Sanderson et al., 2003).
Acknowledgments
We thank Paul Denny for the provision of antibodies raised against LmLCB2 and Malcolm McConville for the gift of the pXGRIP-GFP plasmid. We are also grateful to Maurice Dixon and David Laughland for their help in infectivity studies, Richard Burchmore for his help with mass spectrometry, Gareth Westrop for technical support and Ana Paula Lima for useful discussions on ICP function. This work was supported by the Medical Research Council.
References
- Abrahamson M, Alvarez-Fernandez M, Nathanson CM. Cystatins. Biochemical Society Symposium. 2003;70:179–199. doi: 10.1042/bss0700179. [DOI] [PubMed] [Google Scholar]
- Alexander J, Coombs GH, Mottram JC. Leishmania mexicana cysteine proteinase-deficient mutants have attenuated virulence for mice and potentiate a T Helper 1 (TH1) response. J Immunol. 1998;161:6794–6801. [PubMed] [Google Scholar]
- Alexander J, Satoskar AR, Russell DG. Leishmania species: models of intracellular parasitism. J Cell Sci. 1999;112:2993–3002. doi: 10.1242/jcs.112.18.2993. [DOI] [PubMed] [Google Scholar]
- Bart G, Coombs GH, Mottram JC. Isolation of lmcpc, a gene encoding a Leishmania mexicana cathepsin B-like cysteine proteinase. Mol Biochem Parasitol. 1995;73:271–274. doi: 10.1016/0166-6851(95)00113-f. [DOI] [PubMed] [Google Scholar]
- Bates PA, Robertson CD, Tetley L, Coombs GH. Axenic cultivation and characterization of Leishmania mexicana amastigote-like forms. Parasitology. 1992;105:193–202. doi: 10.1017/s0031182000074102. [DOI] [PubMed] [Google Scholar]
- Brooks DR, McCulloch R, Coombs GH, Mottram JC. Stable transformation of trypanosomatids through targeted chromosomal integration of the selectable marker gene encoding basticidin S deaminase. FEMS Microbiol Lett. 2000a;186:287–291. doi: 10.1111/j.1574-6968.2000.tb09119.x. [DOI] [PubMed] [Google Scholar]
- Brooks DR, Tetley L, Coombs GH, Mottram JC. Processing and trafficking of cysteine proteases in Leishmania mexicana. J Cell Sci. 2000b;113:4035–4041. doi: 10.1242/jcs.113.22.4035. [DOI] [PubMed] [Google Scholar]
- Das L, Datta N, Bandyopadhyay S, Das PK. Successful therapy of lethal murine visceral leishmaniasis with cystatin involves up-regulation of nitric oxide and a favorable T cell response. J Immunol. 2001;166:4020–4028. doi: 10.4049/jimmunol.166.6.4020. [DOI] [PubMed] [Google Scholar]
- Denny PW, Lewis S, Tempero JE, Goulding D, Ivens AC, Field MC, Smith DF. Leishmania RAB7: characterisation of terminal endocytic stages in an intracellular parasite. Mol Biochem Parasitol. 2002;123:105–113. doi: 10.1016/s0166-6851(02)00133-0. [DOI] [PubMed] [Google Scholar]
- Denny PW, Goulding D, Ferguson MAJ, Smith DF. Sphingolipid-free Leishmania are defective in membrane trafficking, differentiation and infectivity. Mol Microbiol. 2004;52:313–327. doi: 10.1111/j.1365-2958.2003.03975.x. [DOI] [PubMed] [Google Scholar]
- Frame MJ, Mottram JC, Coombs GH. Analysis of the roles of cysteine proteinases of Leishmania mexicana in the host-parasite interaction. Parasitology. 2000;121:367–377. doi: 10.1017/s0031182099006435. [DOI] [PubMed] [Google Scholar]
- Garami A, Ilg T. The role of phosphomannose isomerase in Leishmania mexicana glycoconjugate synthesis and virulence. J Biol Chem. 2001;276:6566–6575. doi: 10.1074/jbc.M009226200. [DOI] [PubMed] [Google Scholar]
- Ghedin E, Debrabant A, Engel JC, Dwyer DM. Secretory and endocytic pathways converge in a dynamic endosomal system in a primitive protozoan. Traffic. 2001;2:175–188. doi: 10.1034/j.1600-0854.2001.020304.x. [DOI] [PubMed] [Google Scholar]
- Grant KM, Hassan P, Anderson JS, Mottram JC. The crk3 gene of Leishmania mexicana encodes a stage-regulated cdc2-related histone H1 kinase that associates with p12cks1. J Biol Chem. 1998;273:10153–10159. doi: 10.1074/jbc.273.17.10153. [DOI] [PubMed] [Google Scholar]
- Groves MR, Coulombe R, Jenkins J, Cygler M. Structural basis for specificity of papain-like cysteine protease proregions toward their cognate enzymes. Protein-Struct Funct Genet. 1998;32:504–514. [PubMed] [Google Scholar]
- Gruenberg J. The endocytic pathway: a mosaic of domains. Nat Rev Mol Cell Biol. 2001;2:721–730. doi: 10.1038/35096054. [DOI] [PubMed] [Google Scholar]
- Grzonka Z, Jankowska E, Kasprzykowski F, Kasprzykowska R, Lamkiewicz L, Wiczk W, Wieczerzak E, Ciarkowski J, Drabik P, Janowski R, Kozak M, Jaskolski M, Grubb A. Structural studies of cysteine proteases and their inhibitors. Acta Biochim Pol. 2001;48:1–20. [PubMed] [Google Scholar]
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha DS, Schwarz JK, Turco SJ, Beverley SM. Use of the green fluorescent protein as a marker in transfected Leishmania. Mol Biochem Parasitol. 1996;77:57–64. doi: 10.1016/0166-6851(96)02580-7. [DOI] [PubMed] [Google Scholar]
- Huete-Pérez JA, Engel JC, Brinen LS, Mottram JC, McKerrow JH. Protease trafficking in two primitive eukaryotes is mediated by a prodomain protein motif. J Biol Chem. 1999;274:16249–16256. doi: 10.1074/jbc.274.23.16249. [DOI] [PubMed] [Google Scholar]
- Ilg T, Fuchs M, Gnau V, Wolfram M, Harbecke D, Overath P. Distribution of parasite cysteine proteinases in lesions of mice infected with Leishmania mexicana amastigotes. Mol Biochem Parasitol. 1994;67:193–203. doi: 10.1016/0166-6851(94)00126-x. [DOI] [PubMed] [Google Scholar]
- Irvine JW, Coombs GH, North MJ. Cystatin-like cysteine proteinase inhibitors of parasitic protozoa. FEMS Microbiol Lett. 1992;96:67–72. doi: 10.1016/0378-1097(92)90458-z. [DOI] [PubMed] [Google Scholar]
- Malone JP, Radabaugh MR, Leimgruber RM, Gerstenecker GS. Practical aspects of fluorescent staining for proteomic applications. Electrophoresis. 2001;22:919–932. doi: 10.1002/1522-2683()22:5<919::AID-ELPS919>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- McConville MJ, Ilgoutz SC, Teasdale RD, Foth BJ, Matthews A, Mullin KA, Gleeson PA. Targeting of the GRIP domain to the trans-Golgi network is conserved from protists to animals. Eur J Cell Biol. 2002a;81:485–495. doi: 10.1078/0171-9335-00268. [DOI] [PubMed] [Google Scholar]
- McConville MJ, Mullin KA, Ilgoutz SC, Teasdale RD. Secretory pathway of trypanosomatid parasites. Microbiol Mol Biol Rev. 2002b;66:122–154. doi: 10.1128/MMBR.66.1.122-154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misslitz A, Mottram JC, Overath P, Aebischer T. Targeted integration into a rRNA locus results in uniform and high level expression of transgenes in Leishmania amastigotes. Mol Biochem Parasitol. 2000;107:251–261. doi: 10.1016/s0166-6851(00)00195-x. [DOI] [PubMed] [Google Scholar]
- Monteiro ACS, Abrahamson M, Lima APCA, Vannier-Santos MA, Scharfstein J. Identification, characterization and localization of chagasin, a tight-binding cysteine protease inhibitor in Trypanosoma cruzi. J Cell Sci. 2001;114:3933–3942. doi: 10.1242/jcs.114.21.3933. [DOI] [PubMed] [Google Scholar]
- Morgan GW, Hall BS, Denny PW, Carrington M, Field MC. The kinetoplastida endocytic apparatus. Part I: a dynamic system for nutrition and evasion of host defences. Trends Parasitol. 2002a;18:491–496. doi: 10.1016/s1471-4922(02)02391-7. [DOI] [PubMed] [Google Scholar]
- Morgan GW, Hall BS, Denny PW, Field MC, Carrington M. The endocytic apparatus of the kinetoplastida. Part II: machinery and components of the system. Trends Parasitol. 2002b;18:540–546. doi: 10.1016/s1471-4922(02)02392-9. [DOI] [PubMed] [Google Scholar]
- Mottram JC, Robertson CD, Coombs GH, Barry JD. A developmentally regulated cysteine proteinase gene of Leishmania mexicana. Mol Microbiol. 1992;6:1925–1932. doi: 10.1111/j.1365-2958.1992.tb01365.x. [DOI] [PubMed] [Google Scholar]
- Mottram JC, Souza AE, Hutchison JE, Carter R, Frame MJ, Coombs GH. Evidence from disruption of the lmcpb gene array of Leishmania mexicana that cysteine proteinases are virulence factors. Proc Natl Acad Sci USA. 1996;93:6008–6013. doi: 10.1073/pnas.93.12.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottram JC, Frame MJ, Brooks DR, Tetley L, Hutchison JE, Souza AE, Coombs GH. The multiple cpb cysteine proteinase genes of Leishmania mexicana encode isoenzymes which differ in their stage-regulation and substrate preferences. J Biol Chem. 1997;272:14285–14293. doi: 10.1074/jbc.272.22.14285. [DOI] [PubMed] [Google Scholar]
- Mottram JC, Coombs GH, Alexander J. Cysteine peptidases as virulence factors of Leishmania. Curr Opin Microbiol. 2004; 7(4):375–381. doi: 10.1016/j.mib.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Mullin KA, Foth BJ, Ilgoutz SC, Callaghan JM, Zawadzki JL, McFadden GI, McConville MJ. Regulated degradation of an endoplasmic reticulum membrane protein in a tubular lysosome in Leishmania mexicana. Mol Biol Cell. 2001;12:2364–2377. doi: 10.1091/mbc.12.8.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan DP, Geuskens M, Pays E. N-linked glycans containing linear poly-N-acetyllactosamine as sorting signals in endocytosis in Trypanosoma brucei. Curr Biol. 1999;9:1169–1172. doi: 10.1016/S0960-9822(00)80018-4. [DOI] [PubMed] [Google Scholar]
- Pal A, Hall BS, Nesbeth DN, Field HI, Field MC. Differential endocytic functions of Trypanosoma brucei Rab5 isoforms reveal a glycosylphosphatidylinositol-specific endosomal pathway. J Biol Chem. 2002;277:9529–9539. doi: 10.1074/jbc.M110055200. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Pupkis MF, Tetley L, Coombs GH. Leishmania mexicana: amastigote hydrolases in unusual lysosomes. Exp Parasitol. 1986;62:29–39. doi: 10.1016/0014-4894(86)90005-6. [DOI] [PubMed] [Google Scholar]
- Rigden DJ, Mosolov VV, Galperin MY. Sequence conservation in the chagasin family suggests a common trend in cysteine proteinase binding by unrelated protein inhibitors. Protein Sci. 2002;11:1971–1977. doi: 10.1110/ps.0207202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CD, Coombs GH. Characterization of 3 groups of cysteine proteinases in the amastigotes of Leishmania mexicana mexicana. Mol Biochem Parasitol. 1990;42:269–276. doi: 10.1016/0166-6851(90)90170-q. [DOI] [PubMed] [Google Scholar]
- Sacks DL. Leishmania-sand fly interactions controlling species-specific vector competence. Cell Microbiol. 2001;3:189–196. doi: 10.1046/j.1462-5822.2001.00115.x. [DOI] [PubMed] [Google Scholar]
- Sanderson SJ, Westrop GD, Scharfstein J, Mottram JC, Coombs GH. Functional conservation of a natural cysteine peptidase inhibitor in protozoan and bacterial pathogens. FEBS Lett. 2003;542:12–16. doi: 10.1016/s0014-5793(03)00327-2. [DOI] [PubMed] [Google Scholar]
- Selzer PM, Pingel S, Hsieh I, Ugele B, Chan VJ, Engel JC, Bogyo M, Russell DG, Sakanari JA, McKerrow JH. Cysteine protease inhibitors as chemotherapy: Lessons from a parasite target. Proc Natl Acad Sci U S A. 1999;96:11015–11022. doi: 10.1073/pnas.96.20.11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SB, Tandon R, Krishnamurthy G, Vikram R, Sharma N, Basu SK, Mukhopadhyay A. Rab5-mediated endosome-endosome fusion regulates hemoglobin endocytosis in Leishmania donovani. EMBO J. 2003;22:5712–5722. doi: 10.1093/emboj/cdg557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller RF, McConville MJ. Developmental changes in lysosome morphology and function Leishmania parasites. Int J Parasitol. 2002;32:1435–1445. doi: 10.1016/s0020-7519(02)00140-6. [DOI] [PubMed] [Google Scholar]