

Abstract
Recent studies suggested that modification of the membrane contact site of vitamin K-dependent proteins may enhance the membrane affinity and function of members of this protein family. The properties of a factor VII mutant, factor VII-Q10E32, relative to wild-type factor VII (VII, containing P10K32), have been compared. Membrane affinity of VII-Q10E32 was about 20-fold higher than that of wild-type factor VII. The rate of autoactivation VII-Q10E32 with soluble tissue factor was 100-fold faster than wild-type VII and its rate of activation by factor Xa was 30 times greater than that of wild-type factor VII. When combined with soluble tissue factor and phospholipid, activated factor VII-Q10E32 displayed increased activation of factor X. Its coagulant activity was enhanced in all types of plasma and with all sources of tissue factor tested. This difference in activity (maximum 50-fold) was greatest when coagulation conditions were minimal, such as limiting levels of tissue factor and/or phospholipid. Because of its enhanced activity, factor VII-Q10E32 and its derivatives may provide important reagents for research and may be more effective in treatment of bleeding and/or clotting disorders.
Factor VII, a protein that requires vitamin K for biosynthesis, is a central component in the initiation of coagulation. In complex with tissue factor (TF), the active form of factor VII, factor VII(a), can activate blood clotting factors IX and X (1). Involvement in the initial steps of blood coagulation makes factor VII an attractive target for study. In addition, direct activation of factor X by factor VII(a) effectively bypasses the coagulation step that involves factors VIII(a) and IX(a). Deficiency in factors VIII and IX form the basis of hemophilia A (2) and B (3), respectively. These bleeding disorders are major health concerns in the United States (4). Factor VII deficiency, a rare autosomal recessive disorder, also can cause serious bleeding complications. Wild-type recombinant factor VII(a) can effectively treat patients with factors VIII, IX, and VII deficiencies (5–7). Thus, the possibility of creating a factor VII protein with enhanced biological activity may be useful for therapeutic application as well as for research.
An attractive target for enhanced function of vitamin K-dependent plasma proteins is the membrane contact site. This family of proteins displays a broad range of affinities for acidic phospholipid membranes, despite a high degree of sequence homology in the γ-carboxyglutamic acid (Gla) domain (8, 9). Although the purpose(s) of such diverse affinities are unclear, they may serve to balance pro- and anti-coagulation arms of hemostasis (10) and/or alter the kinetic mechanism of individual steps of these reactions. Recent studies revealed a correlation between the amino acids at positions 11, 33, and 34 (bovine prothrombin numbering) and overall membrane affinity (8). Mutation of these amino acids may produce proteins with higher membrane affinity and increased function. Indeed, substitution of His-10 in bovine protein C resulted in a protein with enhanced membrane affinity and biological function (10).
This study was initiated to determine whether factor VII function could be enhanced by mutation of the membrane binding site. A possible optimum mutant was generated, consisting of factor VII in which Pro-10 and Lys-32 were replaced by Gln and Glu, respectively. Glu-32 was converted to Gla during biosynthesis. The mutant displayed a much higher membrane affinity than the wild-type protein and had enhanced activity in several factor VII-dependent processes such as autoactivation, factor X activation, and blood coagulation. Similar modified proteins may find important uses in research and, conceivably, in the treatment of coagulation disorders.
MATERIALS AND METHODS
Factor VII-Q10E32.
Mutant factor VII was generated from wild-type factor VII cDNA (11). The P10Q mutation and the K32E mutation were introduced into the wild-type factor VII cDNA by a PCR strategy, which, in the process, eliminated a mutation-diagnostic XmaIII restriction enzyme site. Four PCR primers were designed that would prime synthesis of two mutant fragments, one from MluI to BglII, and the other from BglII to SstII. These primers were used under standard PCR cycling conditions (GENEAMP, Perkin–Elmer) to prime fragment synthesis using 1 ng of the wild-type factor VII cDNA as template. The resulting fragments were gel-purified and digested with MluI and BglII or BglII and SstII. The two fragments then were ligated into the factor VII cDNA in the expression vector Zem219b (11) from which the corresponding wild-type sequence had been removed as a MluI–SstII fragment. The mutated fragments were sequenced in their entirety to confirm the P10Q and K32E substitutions, as well as to eliminate the possibility of other PCR-induced sequence changes.
Transfection, Selection, and Purification.
Baby hamster kidney cells were grown in DMEM supplemented with 10% fetal calf serum and penicillin-streptomycin. Subconfluent cells were transfected with the factor VII expression plasmid by using Lipofectamine (GIBCO/BRL) essentially according to the manufacturer’s recommendations. Two days posttransfection, cells were trypsinized and diluted to selective medium containing 1 μM methotrexate. Stably transfected baby hamster kidney cells were subsequently cultured in serum-free DMEM supplemented with penicillin-streptomycin, 5 μg/ml of vitamin K, and 1 μM methotrexate, and conditioned media collected for the isolation of VII-Q10E32 by two applications of immunoaffinity chromatography using a calcium-dependent mAb (CaFVII22) coupled to Affi-Gel 10 (12). The purified mutant factor VII exhibited a single band on SDS/PAGE, with no evidence of factor VII(a) in the preparation. The pure factor VII mutant exhibited a specific activity of 1,400–2,800 factor VII units/mg depending on its dilution in the single-stage factor VII assay, with higher dilutions yielding higher specific activity. Protein concentrations were determined by the Bradford assay (13) using BSA as the standard.
Factor VII, Factor X, TF, and Thromboplastin.
Soluble recombinant human TF apoprotein TF1–218 (sTF; ref. 14) and human brain thromboplastin (TP) (15) were prepared by published methods. Single-chain recombinant human factor VII was provided by Anders Pedersen (Novo-Nordisk, Copenhagen). Full-length recombinant TF apoprotein was provided by Gordon Vehar (Genentech). Relipidation of full-length TF was performed as described previously (16). Bovine factor X and Xa were prepared as described (17) except that purified factor X activating enzyme was used.
Gla Analysis.
Gla content of VII-Q10E32 and wild-type VII was determined by amino acid analysis after base hydrolysis (20 hr at 115°C in 2 N of KOH in polypropylene containers sealed under Argon atmosphere). The hydrolysates were neutralized with perchloric acid, KClO4 was removed by centrifugation, and the pH was adjusted to 9.0 with 0.1 M bicarbonate/carbonate buffer. The amino acids were modified with dimethylaminoazobenzenesulfonyl chloride (Sigma; 2.7 mM in 67% CH3CN) at 75°C for 20 min. Derivatized amino acids were diluted with 0.05 M sodium phosphate buffer (pH 7.0) and separated by reverse-phase HPLC (0.4 × 25 cm Vydac C18 column) using the method described by Beckman. Buffers A (10 mM citrate buffer, pH 6.5) and B (30% buffer A in 70% CH3CN) were used in the following program: 0–6 min, 24% buffer B; 6–21 min, a gradient of 24–51% B; 21–27 min, a gradient of 51–86% B. The flow rate was 1.0 ml/min. Gla/Asp/Glu standards (Sigma) eluted at 13.1/16.5/17.2 min, respectively. Gla and Glu content were estimated from amino acid standards that had been subjected to hydrolysis and dabsylation procedures. Direct calculation of Gla content from the amount of digested protein [quantitated by ɛ280 = 1.29/(cm·g/liter); ref. 18] gave values of 12.0 for factor VII-Q10E32 and 10.7 for wild-type factor VII, with 15% SDs. A more accurate estimation of Gla content was obtained by comparison with Glu content of the protein hydrolysate. Assuming 38 Glu plus Gln per factor VII-Q10E32 and 37 per wild-type factor VII, values for Gla were 10.7 ± 0.8 VII-Q10E32 (n = 5) and 9.6 ± 0.9 for factor VII (n = 4). Theoretical values were 11 and 10 for full carboxylation of Glu within residues 1–44 of factor VII-Q10E32 and factor VII, respectively (8).
High-resolution ion exchange chromatography also indicated full carboxylation. A number of Gla-containing proteins and peptides can be separated on the basis of a single charge difference. Examples include factor X1 and factor X2 (19), prothrombin residues 1–156 and residues 4–156 (20), and prothrombin peptides 1–42 and 1–45 (21). Proteins were applied to a Mono Q column (Pharmacia, HR 5/5) and eluted with a linear gradient of 50 mM Tris, 0.1 M NaCl, pH 7.5 to Tris-0.75M NaCl at a flow rate of 1 ml/min. The gradient was started at 24 min and ended at 54 min. Factor VII eluted as a symmetrical peak at 31.8 min and the VII-Q10E32 eluted at 34.2 min. Mild chymotryptic digest released residues 1–44 of factor VII, which eluted at 34.4 min for wild-type factor VII and 36.5 min for VII-Q10E32. Thus, ion exchange chromatographic properties supported the conclusion of full carboxylation of the mutant, including Glu-32.
Phospholipid Vesicle Preparation.
Small unilamellar phospholipid vesicles (SUVs) were prepared by published procedures (22). Briefly, the phospholipids (Sigma) were mixed in organic solvent, which was evaporated by a stream of nitrogen. The dried phospholipids (6 mg/ml) were resuspended in 3 ml of 50 mM Tris, 100 mM NaCl, pH 7.5 buffer. The solution was sonicated periodically for 3 min on ice using a Heat System-Ultrasonics W-385 sonicator. The sonicated solution was applied to a column of Sephrose 4B and fractions containing SUVs were pooled. Phospholipid concentrations were determined by using the phosphorous assay and a phospholipid-to-phosphorous weight ratio of 25 (23).
Activation of Factor VII-Q10E32 and Wild-Type Factor VII.
Activated factor VII-Q10E32 (VII(a)-Q10E32) was obtained by autoactivation (37°C, 20 min) in a mixture containing 7 μM VII-Q10E32, 0.7 μM sTF, 5 mM calcium and phospholipid (phosphatidylserine (PS)/phosphatidylcholine (PC), 25:75, 0.1 g/g protein). Alternatively, VII(a)-Q10E32 was formed by bovine factor Xa cleavage of VII-Q10E32 (1:100 weight ratio, incubation for 1 hr at 37°C in 5 mM calcium and PS/PC SUVs). Wild-type factor VII(a) was a homogeneous, recombinant protein provided by Novo-Nordisk. Two preparations of recombinant factor VII(a) were used; one consisted of a commercial, lyophilized product and another that had not been lyophilized. The latter protein was further purified on FPLC Mono-Q and showed a specific activity of 80,000 units/mg, calibrated with normal human pooled plasma (purchased from George King, Biomedical, Overland Park, KS).
Membrane Binding.
Protein-membrane binding was measured by light scattering (24). Binding is expressed as the ratio, M2/M1, where M2 is the molecular weight of the protein-membrane complex and M1 is the vesicles alone. If phospholipid and protein concentrations are known, the molar concentrations of bound [P/PL] and free protein [P] can be estimated. These values and the maximum protein binding capacity [P/PLMAX] of the vesicles can be used to calculate the dissociation constant (Kd) for protein-membrane interaction by the relationship in Eq. 1.
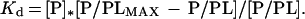 |
1 |
Curve fitting of the membrane-binding data was performed by using the computer program kaleidagraph to estimated dissociation constants.
Measurement of Factor VII Activation.
Factor VII autoactivation was conducted in 50 mM Tris/100 mM NaCl, pH 7.5 buffer containing VII-Q10E32 or wild-type VII, 100 nM sTF, PS/PC SUVs (25:75, 22 μg/ml), 1 mg/ml of BSA, and 5 mM calcium. The formation of VII(a) was estimated at various time intervals by the addition of 0.15 mM H-d-isoleucyl-l-prolyl-l-arginine-p-nitroaniline-dihydrochloride (S-2288; ɛ = 1 × 104 M−1 cm−1; Kabi Pharmacia Hepar, Franklin, OH), and the rate of substrate hydrolysis was monitored by absorbance at 405 nm. For the wild-type factor VII preparation, some factor VII(a) (0.05 mol/mol VII) was added to the reaction mixture. Alternatively, factor Xa-catalyzed activation of factor VII was performed by a similar technique. Briefly, 30 nM wild-type factor VII or VII-Q10E32 was incubated with 0.1 nM bovine factor Xa and PS/PC SUVs (25:75; 1.5 μg/ml) in 1.2 ml of 50 mM Tris/100 mM NaCl/5 mM calcium/1 mg/ml BSA, pH 7.5 buffer. Aliquots (0.25 ml) were removed at selected time intervals and assayed for S-2288 amidase activity after incubation with 50 nM sTF. The initial amidolytic activity of the VII-Q10E32 preparation was less than 4% of the fully active VII(a)-Q10E32.
Factor X Activation by Factor VII(a).
Bovine factor X (100 nM) was activated in 50 mM Tris buffer (pH 7.5) containing 100 mM NaCl, 5 mM calcium, various amounts of phospholipid (PS/PC, 25:75), and 1 mg/ml BSA at 22.5°C. Factor VII(a) [0.06 nM of VII(a)-Q10E32 or 0.6 nM wild-type VII(a)] was added at zero time. Factor Xa production was determined by removing 0.2-ml aliquots from the reaction mixture and mixing with 0.2 ml of buffer that contained 10 mM EDTA and 0.4 mM N-benzoyl-l-isoleucyl-l-glutamyl-glycyl-l-arginine-p-nitroaniline hydrochloride (S-2222; Kabi Pharmacia Hepar) at 1- and 3-min incubations. The amount of factor Xa generated was calculated from absorbance change and the extinction coefficient of p-nitroaniline (1 × 104M−1 cm−1) and a velocity of 33 sec−1 for substrate hydrolysis by purified bovine Xa under the conditions of this assay.
Coagulation Assays.
Blood clotting assays were performed at 37°C by using the tilt-tube method to detect clot formation. Factor VII-deficient human plasma (60 μl; Sigma) was allowed to equilibrate at 37°C for 1 min. Wild-type VII or VII-Q10E32, 50 nM sTF plus phospholipid vesicles, and/or different types of TP [rabbit TP-high sensitivity (TP-HS), Sigma; rabbit TP, Sigma; human TP (described above)] in 50 mM Tris/100 mM NaCl, pH 7.5 buffer (0.1 ml) were added to the plasma. Finally, 0.1 ml of 25 mM CaCl2 was added to initiate the reaction. Some preparations of commercial thromboplastin contained calcium and were the final reagent used to initiate coagulation. Calcium-containing preparations were diluted with standard buffer containing 10 mM calcium. Factor VIII-deficient human plasma (Sigma) and normal human plasma (University of Minnesota Blood Bank) also were used to analyze coagulation potency. Coagulation time was assessed for samples containing various concentrations of zymogen and activated forms of wild-type VII or VII-Q10E32. Data were analyzed by the linear relationship between log (clotting time) vs. log [VII(a)].
Analysis of the Circulatory Lifetime of Factor VII-Q10E32.
Factor VII-Q10E32 (36 μg) was injected into anesthetized (sodium nembutol) Sprague–Dawley rats weighing 325–350 g at zero time. Injection was through the juggler vein, into which a cannula had been inserted. At selected time intervals, 0.9 ml of blood was withdrawn into 0.1 ml of 0.1 M citrate from the carotid artery, into which a cannula had been inserted. The concentration of factor VII-Q10E32 in the circulation was estimated from the clotting time of factor VII-deficient plasma. Briefly, 1 ml of a 1:10 dilution of the rat plasma was added to 60 μl of factor VII-deficient plasma. A solution of TP-HS (1:100 dilution in buffer containing 10 mM calcium) was added to start the reaction. Coagulation was assessed as described above, and the amount of VII-Q10E32 in the plasma was determined by comparison to a standard curve prepared from pure protein. The amount of factor VII activity in rat plasma was determined from a sample withdrawn before injection of the protein and was subtracted as background. At completion of the experiment, the animals were euthanized by excess sodium nembutol. Animal experimentation was conducted by protocol 95070003 as described by the University of Minnesota Animal Care Committee.
RESULTS
Membrane Interaction by Factor VII-Q10E32.
Fig. 1A shows the binding of wild-type VII(a) and factor VII-Q10E32 to SUVs of 25% PS. Binding of VII-Q10E32 followed theoretical values, indicating that free protein levels were essentially zero. The wild-type protein displayed much lower affinity than the mutant protein. Although dissociation constants for VII-Q10E32 could not be estimated, affinity was clearly higher than that of bovine factor X (Fig. 1A), one of the highest affinity vitamin K-dependent proteins (Kd = 40 nM for vesicles of 20% PS at 2 mM calcium; ref. 8).
Figure 1.

Comparison of membrane interaction by wild-type protein and Q10E32 mutant factor VII. (A) Binding to PS/PC SUVs (25:75, 25 μg/ml) at 5 mM calcium. Wild-type VII(a) (○), VII-Q10E32 (•), and bovine factor X (□) were added to the levels shown. (B) Binding to PS/PC SUVs (10:90, 25 μg/ml). Conditions, proteins, and symbols are the same as in A. The amount of membrane-bound protein is expressed as M2/M1, where M2 is the molecular weight of the protein-vesicle complex and M1 is that of the vesicles alone. The dashed line represents the theoretical values, if all of the added protein bound to the membrane. All reactions were quantitatively reversed by addition of 10 mM EDTA. Data show average values and SDs for three titrations.
Binding of wild-type VII(a), VII-Q10E32, and factor X to membranes of lower PS content (10% PS SUVs) demonstrated the same pattern of affinities (Fig. 1B). Less than quantitative binding of VII-Q10E32 allowed an affinity constant to be estimated from the relationship in Eq. 1. Assuming a maximum protein binding capacity to be 1.0 g of protein/g of phospholipid for all proteins, the data gave approximate Kd values of 4.3 μM for wild-type factor VII, 0.52 μM for bovine factor X, and 0.22 μM for VII-Q10E32. Thus, factor VII-Q10E32 had greatly improved membrane interaction relative to normal factor VII and exhibited one of the strongest membrane interactions among the vitamin K-dependent proteins.
Factor VII-Q10E32 Activation.
An initial step in coagulation involves the activation of factor VII. One pathway is autoactivation of factor VII by factor VII(a) in the presence of either ionic surfaces (25) or TF (12). Autoactivation of factor VII in the presence of sTF and phospholipid vesicles showed that VII-Q10E32 was a much better substrate than wild-type factor VII (Fig. 2A). For example, an amidase activity of 3 μM S-2288 per min was attained in 48 sec for 36 nM VII-Q10E32, but required approximately 100 min for 50 nM wild-type factor VII. The low rates of autoactivation by wild-type factor VII(a) were consistent with values reported previously (26). Although not studied in detail, the mutant also was more rapidly autoactivated in the presence of relipidated human single-chain TF (data not shown).
Figure 2.

Activation of wild-type factor VII and VII-Q10E32. (A) Autoactivation by soluble TF. Assay conditions are described in Materials and Methods. Amidase activity (S-2288) of factor VII(a) generated by a solution of wild-type factor VII [50 nM; ○ containing 2.5 nM factor VII(a)] or VII-Q10E32 (36 nM; •) are shown. (B) Factor Xa-catalyzed activation. Assay conditions and concentrations (30 nM VII or VII-Q10E32 and 0.1 nM factor Xa) are given in Materials and Methods. Symbols are the same as in A and the rate of S-2288 hydrolysis, determined in an aliquot of the solution, is plotted. Full activation gave a velocity of 7 μM S2288 per min in both reactions. Values shown are the average and SD of three experiments.
Conversion of factor VII to factor VII(a) is catalyzed by several other proteases, such as factor Xa (27, 28). Again, VII-Q10E32 showed a faster rate of activation by Xa than the normal factor VII (Fig. 2B). The high rate of autoactivation during the 15-sec incubation with sTF precluded precise evaluation of rate constants for factor Xa activation of VII-Q10E32 at the first time point. Nevertheless, increased activity at later time points showed that factor Xa did activate VII-Q10E32. The rate was much faster than factor Xa activation of wild-type VII. For example, after 30 min, the wild-type factor VII had not attained the level of activity seen for the mutant at 1 min.
Factor X Activation by sTF/VII(a).
Once formed, factor VII(a) in complex with TF can activate factor X and factor IX by limited proteolysis (29). Fig. 3 compares the ability of wild-type factor VII(a) and VII(a)-Q10E32 to generate factor Xa in the presence of sTF and varying amounts of phospholipid vesicles. The mutant enzyme was more efficient than the wild-type factor VII(a) and the difference was most pronounced at low phospholipid concentrations. At high phospholipid concentration (0.2 mg/ml), there was only a 2-fold difference in the rate of factor X activation (data not shown). This finding was expected from the fact that high membrane concentration will cause a greater portion of the wild-type factor VII(a) to bind to the membrane (26). In any event, superior function of VII(a)-Q10E32 would be greatest at low phospholipid exposure.
Figure 3.

Factor X activation by wild-type and mutant VII(a). The activation of 0.1 μM factor X with 100 nM sTF and varying amounts of phospholipid vesicles were determined as described in Materials and Methods. Results for wild-type factor VII(a) (○) and VII(a)-Q10E32 (•) are given for a concentration of 0.06 nM. Values shown are from triplicate experiments.
Procoagulant Activity of VII-Q10E32.
Coagulation by wild-type factor VII(a) and VII(a)-Q10E32 was assessed with several types of plasma and sources of TF. In all cases, VII-Q10E32 demonstrated higher activity than wild-type factor VII. The largest difference was observed when TF levels were low (1:100 dilution of TP-HS) and activation of the zymogen was necessary. At a clotting time of 40 sec, VII-Q10E32 gave 50-fold higher activity than wild-type factor VII (Fig. 4A). In fact, zymogen VII-Q10E32 was approximately equal to the activity of wild-type factor VII(a). With sTF and phospholipid vesicles, the difference was less pronounced (about 5-fold) for the factor VII(a) species (Fig. 4B). Results similar to those in Fig. 4B were obtained with a preparation of human TP added at full strength and at a 1:10 dilution (data not shown). Furthermore, factor VII(a)-Q10E32 showed approximately a 4-fold higher activity than the wild-type factor VII(a) with factor VIII-deficient plasma (data not shown). In normal human plasma, factor VII(a)-Q10E32 with sTF (100 nM) displayed similar superiority to wild-type protein. Although the mutant was superior under all conditions tested, the difference to wild-type factor VII(a) appeared much greater when coagulation conditions were limiting.
Figure 4.

Coagulation of factor VII-deficient plasma by zymogens and the activated forms of VII and VII-Q10E32. Clotting assays are described in Materials and Methods. (A) Clotting with limiting TF. Wild-type factors VII (○) and VII(a) (□) and VII-Q10E32 (•) were added along with a 1:100 dilution of rabbit-TP-HS. (B) Clotting with soluble TF and phospholipid vesicles. Normal VII(a) (○) and VII(a)-Q10E32 (•) were added to the levels shown along with 50 nM sTF and PS/PC vesicles (10:90; 75 μg/ml). Final protein concentrations in the assay are reported in picomolar. Time to form a clot was recorded. Clotting times are the average of triplicate experiments.
Circulation Lifetime of VII-Q10E32.
Circulatory lifetimes and behavior are important features of reagents that may be used in vivo. Injection of 100–110 μg/kg of VII-Q10E32 mutant into anesthetized rats did not result in obvious abnormal coagulation. A control animal that received surgery, but no VII-Q10E32, showed that the amount of background activity from rat factor VII did not change in 100 min. Fig. 5 shows the concentration of factor VII-Q10E32 in rat circulation as a function of time. The lifetime of the mutant appeared normal, with approximately 50% of the activity remaining after 100 min. This result is similar and, in some cases, longer than the circulation time of other vitamin K-dependent proteins and enzymes (30, 31).
Figure 5.

Circulatory time of factor VII-Q10E32. The procedures for injection of factor VII-Q10E32 and sample removal are outlined in Materials and Methods. At the times shown, blood was withdrawn from the carotid artery and the amount of factor VII activity was assessed in human factor VII-deficient plasma by using a 1:100 dilution of rabbit TP-HS. Background levels of factor VII activity in an untreated animal were about 5 ± 0.5 nM VII at both 0 and 100 min after surgery. The concentration of factor VII-Q10E32 in the plasma was determined from a standard curve created from pure factor VII-Q10E32. The data show the results from two animals.
DISCUSSION
Vitamin K-dependent proteins with enhanced function may be important agents for research and biomedical applications. This study applied the approach of improving the function of human factor VII by substitution of specific amino acids to enhance membrane binding affinity. The principal goal was to show that factor VII with increased membrane affinity could be obtained and to determine the impact of this change on several blood coagulation reactions. The mutation sites were selected from predictions that the presence of Pro-10 and the lack of Gla-32 resulted in low membrane affinity (8). The mutant, which was selected and produced, VII-P10Q, K32E, displayed full carboxylation of the Gla domain.
Factor VII-Q10E32 showed several improvements over wild-type factor VII. It had enhanced binding to acidic phospholipid membranes, it gave a much higher rate of autoactivation, and it was more rapidly activated by factor Xa. The active form of the protein, VII(a)-Q10E32, was more effective in factor X activation than wild-type VII(a). Procoagulant activities of factor VII(a)-Q10E32 were greater than wild-type protein under all conditions tested. The improvements were most pronounced for minimal coagulation stimuli. Although the exact basis for enhanced activity is not known, the most likely explanation is increased protein–protein (enzyme-substrate) interaction on the membrane surface.
Earlier predictions suggested that both the Q10 and E32 sites should contribute to improved membrane binding (8). For bovine protein C, a Pro-10His mutant showed a 10-fold impact of this residue (10). The greater enhancement of VII-Q10E32 suggested significant influence of E32. If corroborated by further study, single mutations would make it possible to create a family of proteins with a range of membrane affinities and, presumably, a range of specific enzyme activities. A family of proteins should provide a more accurate determination of the membrane binding site and the role of membrane contact in blood coagulation.
An exciting possibility for improved factor VII proteins is their use in treatment of disease. Recombinant factor VII offers an agent to bypass factors IX and VIII and is useful in certain types of factor VIII and IX deficiencies (6). Increased biological activity of VII-Q10E32 may allow use of smaller doses or less frequent administration. Other uses may be for thrombocytopenia, patients with impaired platelet production, and other situations where wild-type factor VII(a) can shorten the bleeding time (6). Furthermore, active-site inactivated factor VII(a) has been reported to prevent thrombosis in rabbits (32) and analogous derivatives of the VII(a)-Q10E32 may be more potent.
Overall, this study has shown the feasibility of enhancing the function of a vitamin K-dependent protein by manipulation of the membrane binding site of the Gla domain. Success of this strategy with both protein C (10) and factor VII (this study) suggest that it may be applied to virtually any vitamin K-dependent plasma proteins. Proteins with enhanced function offer additional avenues to study blood clotting reactions and may provide more effective reagents for treatment of bleeding and clotting disorders.
Acknowledgments
This study was supported in part by Grant 17472 (to G.L.N.) from the University of Minnesota Graduate School and by Grant HL35246 (to W.K.) from the National Institutes of Health. A.M.S. was supported in part by the Arnold H. Johnson Fellowship.
ABBREVIATIONS
- Gla
γ-carboxyglutamic acid
- VII(a)
wild-type factor VII(a)
- SUV
small unilamellar vesicle
- TF
tissue factor
- sTF
soluble recombinant human TF apoprotein TF1–218
- S-2288
H-d-isoleucyl-l-prolyl-l-arginine-p-nitroaniline-dihydrochloride
- TP
brain thromboplastin
- TP-HS
rabbit TP-high sensitivity
- PS
phosphatidylserine
- PC
phosphatidylcholine
References
- 1.Furie B, Furie B C. New Engl J Med. 1992;326:800–806. doi: 10.1056/NEJM199203193261205. [DOI] [PubMed] [Google Scholar]
- 2.Sadler J E, Davie E W. In: The Molecular Basis of Blood Diseases. Stamatoyannopoulos G, Nienhuis A W, Leder P, Majerus P W, editors. Philadelphia: Saunders; 1987. pp. 575–630. [Google Scholar]
- 3.Hedner U, Davie E W. In: Hemostasis and Thrombosis: Basic Principles and Practice. Colman R W, Hirsh J, Marder V J, Salzman E W, editors. Philadelphia: Lippincott; 1987. pp. 39–47. [Google Scholar]
- 4.Lusher J M, Warrier I. Hematol Oncol Clin North Am. 1992;6:1021–1033. [PubMed] [Google Scholar]
- 5.Bauer, K. A. (1996) Haemostasis 26, Suppl. 1, 155–158. [DOI] [PubMed]
- 6.Glazer S, Hedner U, Falch J F. Adv Exp Med Biol. 1995;386:163–174. doi: 10.1007/978-1-4613-0331-2_15. [DOI] [PubMed] [Google Scholar]
- 7.Hedner, U. (1996) Haemostasis 26, Suppl. 1, 102–108. [DOI] [PubMed]
- 8.McDonald J F, Shah A M, Schwalbe R A, Kisiel W, Dahlbäck B, Nelsestuen G L. Biochemistry. 1997;36:5120–5127. doi: 10.1021/bi9626160. [DOI] [PubMed] [Google Scholar]
- 9.Nelsestuen G L, Kisiel W, DiScipio R G. Biochemistry. 1978;17:2134–2138. doi: 10.1021/bi00604a017. [DOI] [PubMed] [Google Scholar]
- 10.Shen L, Shah A M, Dahlbäck B, Nelsestuen G L. Biochemistry. 1997;36:16025–16031. doi: 10.1021/bi971730v. [DOI] [PubMed] [Google Scholar]
- 11.Petersen L C, Boel E, Johannessen M, Foster D C. Biochemistry. 1990;29:3451–3457. doi: 10.1021/bi00466a005. [DOI] [PubMed] [Google Scholar]
- 12.Nakagaki T, Foster D C, Berkner K L, Kisiel W. Biochemistry. 1991;30:10819–10824. doi: 10.1021/bi00109a001. [DOI] [PubMed] [Google Scholar]
- 13.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Petersen L C, Spreecher C A, Foster D C, Blumberg H, Hamamoto T, Kisiel W. Biochemistry. 1996;35:266–272. doi: 10.1021/bi951501d. [DOI] [PubMed] [Google Scholar]
- 15.Nawroth P P, Kisiel W, Stern D M. Thromb Res. 1986;44:625–637. doi: 10.1016/0049-3848(86)90164-7. [DOI] [PubMed] [Google Scholar]
- 16.Kazama Y, Pastuszyn A, Wildgoose P, Hamamoto T, Kisiel W. J Biol Chem. 1993;268:16231–16240. [PubMed] [Google Scholar]
- 17.Pletcher C H, Nelsestuen G L. J Biol Chem. 1983;258:1086–1091. [PubMed] [Google Scholar]
- 18.Kisiel W, Davie E W. Biochemistry. 1975;14:4928–4934. doi: 10.1021/bi00693a023. [DOI] [PubMed] [Google Scholar]
- 19.Morita T, Jackson C M. J Biol Chem. 1986;261:4008–4014. [PubMed] [Google Scholar]
- 20.Weber D J, Berkowitz P, Panek M G, Huh N W, Pedersen L G, Hiskey R G. J Biol Chem. 1992;267:4564–4569. [PubMed] [Google Scholar]
- 21.Pollock J S, Sheppard A J, Weber D J, Olson D L, Klapper D G, Pedersen L G, Hiskey R G. J Biol Chem. 1988;263:14216–14223. [PubMed] [Google Scholar]
- 22.Huang C. Biochemistry. 1969;8:344–352. doi: 10.1021/bi00829a048. [DOI] [PubMed] [Google Scholar]
- 23.Chen P S, Toribara T Y, Warner H. Anal Chem. 1956;28:1756–1758. [Google Scholar]
- 24.Nelsestuen G L, Lim T K. Biochemistry. 1977;16:4164–4171. doi: 10.1021/bi00638a005. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen A H, Lund-Hansen T, Bisgaard-Frantzen H, Olsen F, Petersen L C. Biochemistry. 1989;28:9331–9336. doi: 10.1021/bi00450a013. [DOI] [PubMed] [Google Scholar]
- 26.Fiore M M, Neuenschwander P F, Morrissey J H. J Biol Chem. 1994;269:143–149. [PubMed] [Google Scholar]
- 27.Radcliffe R, Nemerson Y. J Biol Chem. 1976;251:4797–4802. [PubMed] [Google Scholar]
- 28.Rao L V, Rapaport S I. Proc Natl Acad Sci USA. 1988;85:6687–6691. doi: 10.1073/pnas.85.18.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Østerud B, Rapaport S I. Proc Natl Acad Sci USA. 1977;74:5260–5264. doi: 10.1073/pnas.74.12.5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelsestuen G L, Suttie J W. Biochem Biophys Res Commum. 1971;45:198–203. doi: 10.1016/0006-291x(71)90069-6. [DOI] [PubMed] [Google Scholar]
- 31.Thomsen M K, Diness V, Nilsson P, Rasmussen S N, Taylor T, Hedner U. Thromb Haemost. 1993;70:458–464. [PubMed] [Google Scholar]
- 32.Arnlijots B, Ezban M, Hedner U. J Vasc Surg. 1997;25:341–346. doi: 10.1016/s0741-5214(97)70356-2. [DOI] [PubMed] [Google Scholar]